
Dielectric Spectroscopy Studies of Conformational Relaxation Dynamics in Molecular Glass-Forming Liquids




Abstract
:1. Introduction
2. Method
3. Results
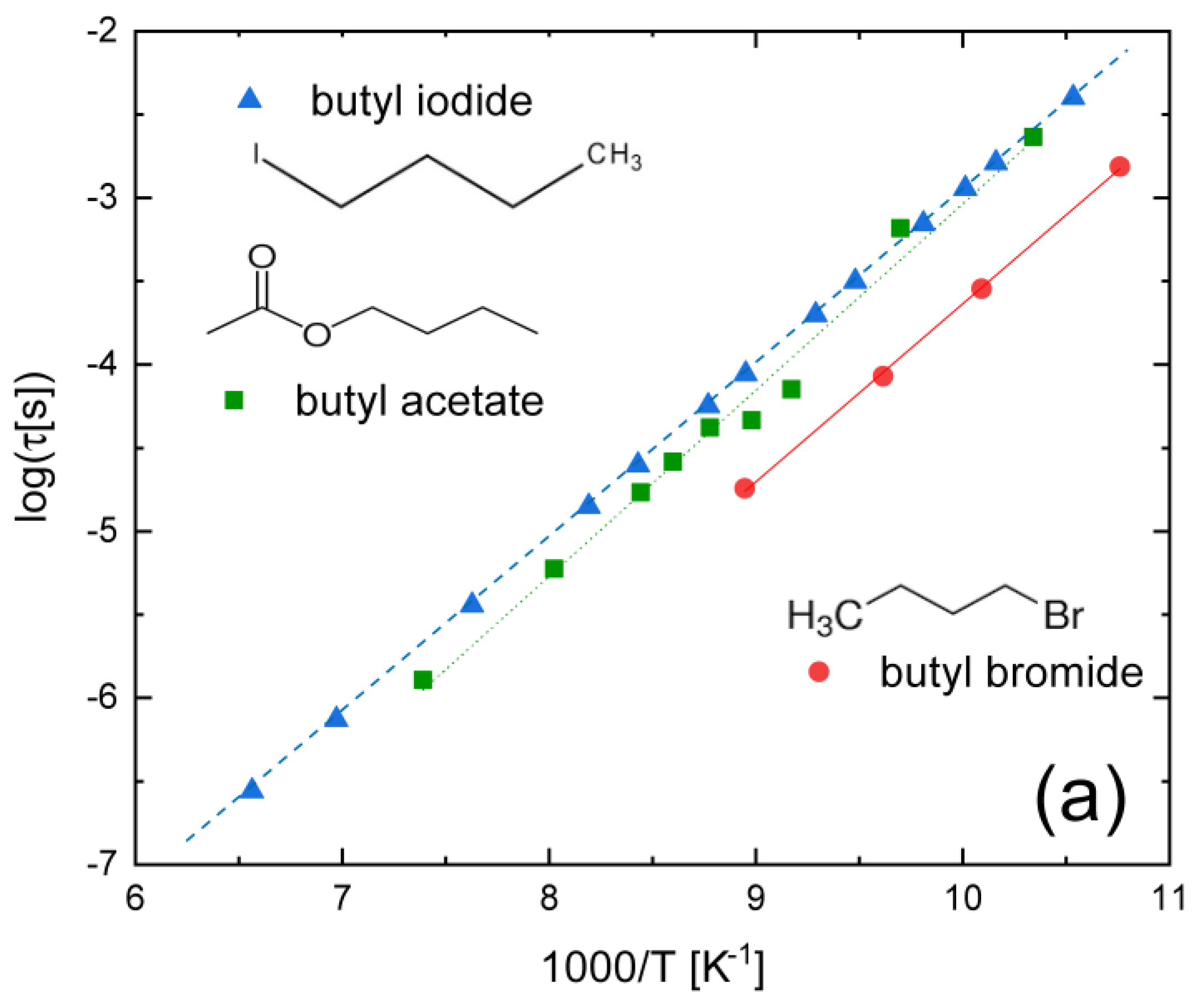
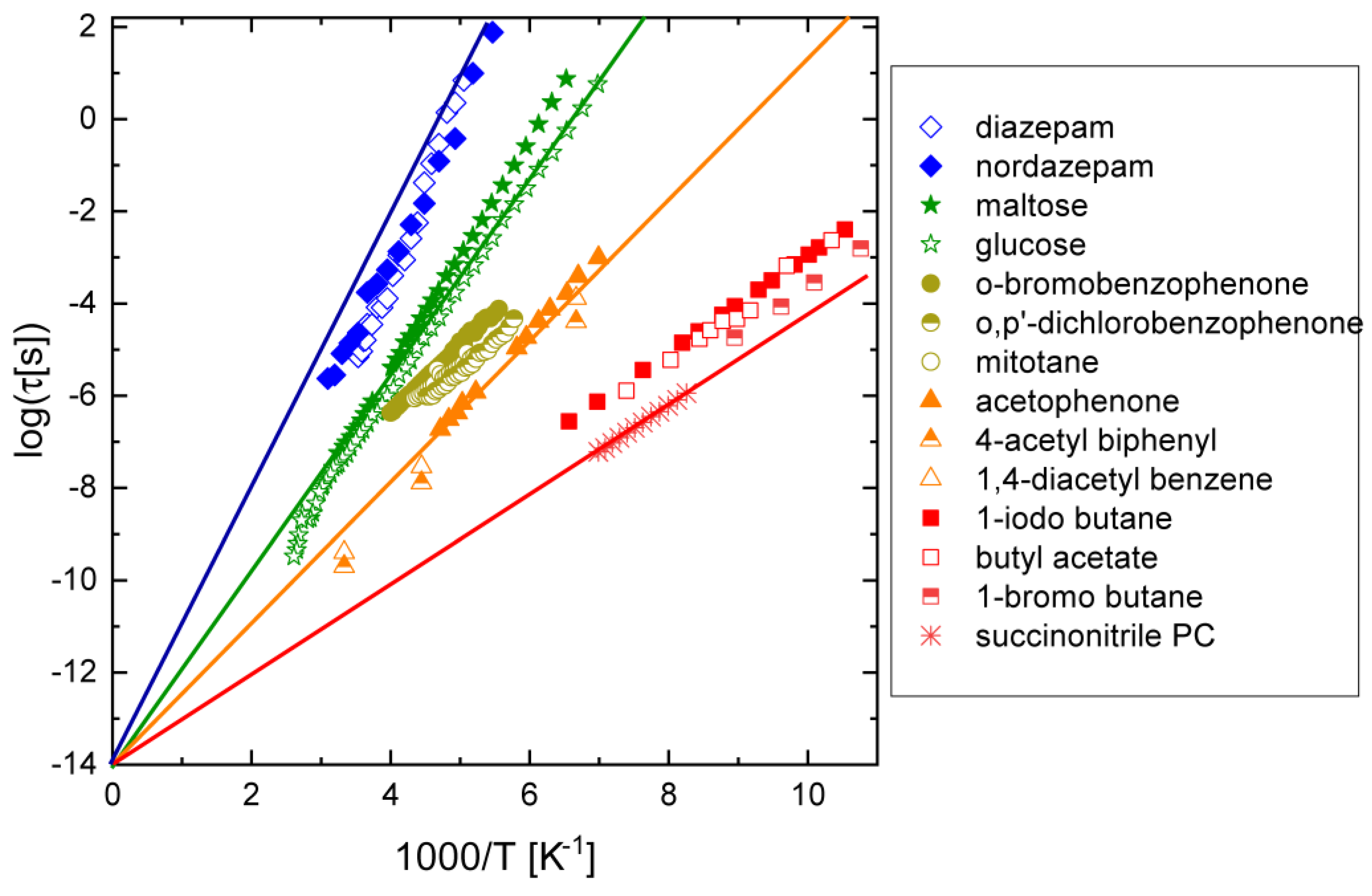
3.1. Haloalkanes, Acetylalkanes, and Simple Nitriles
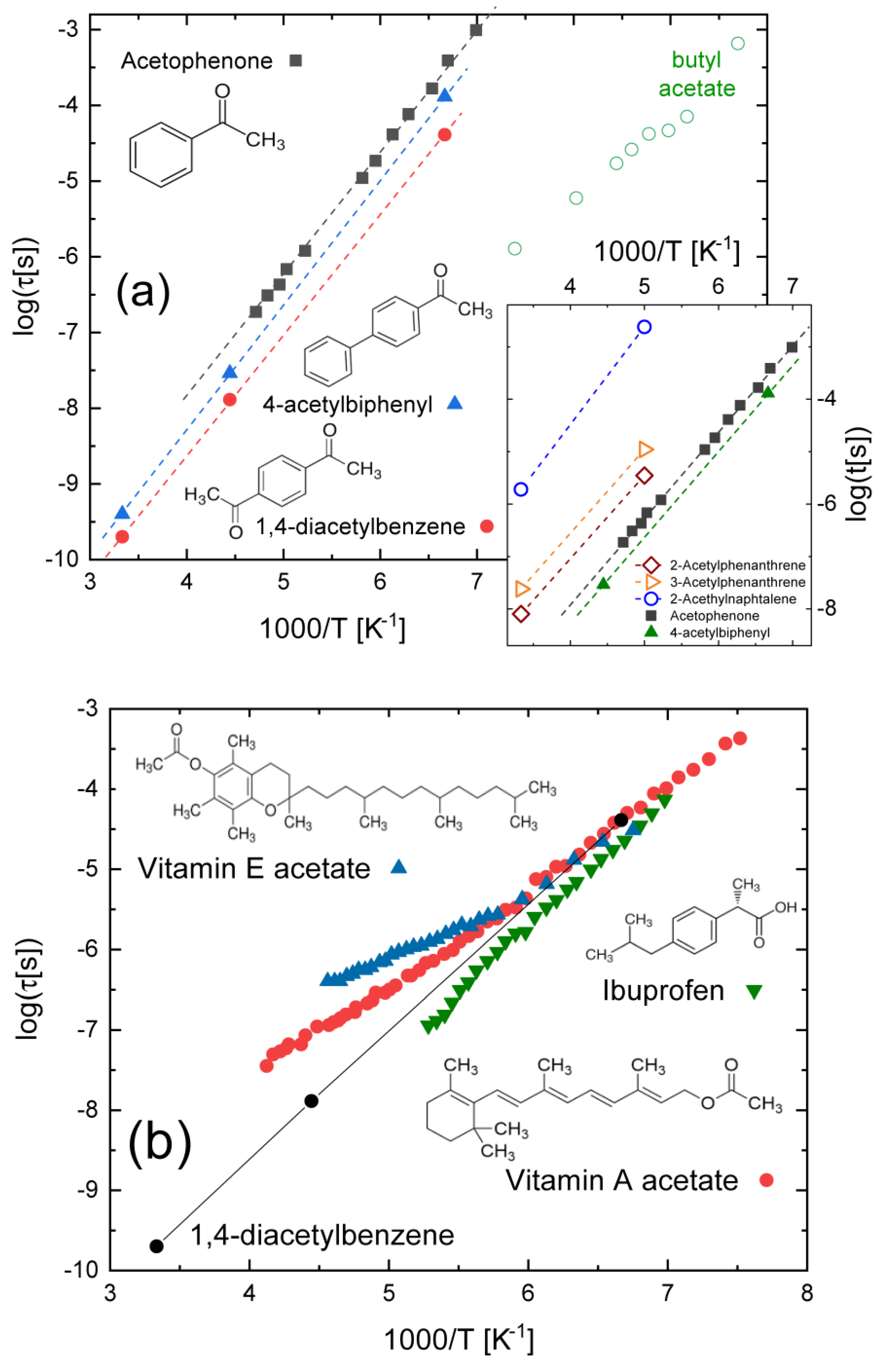
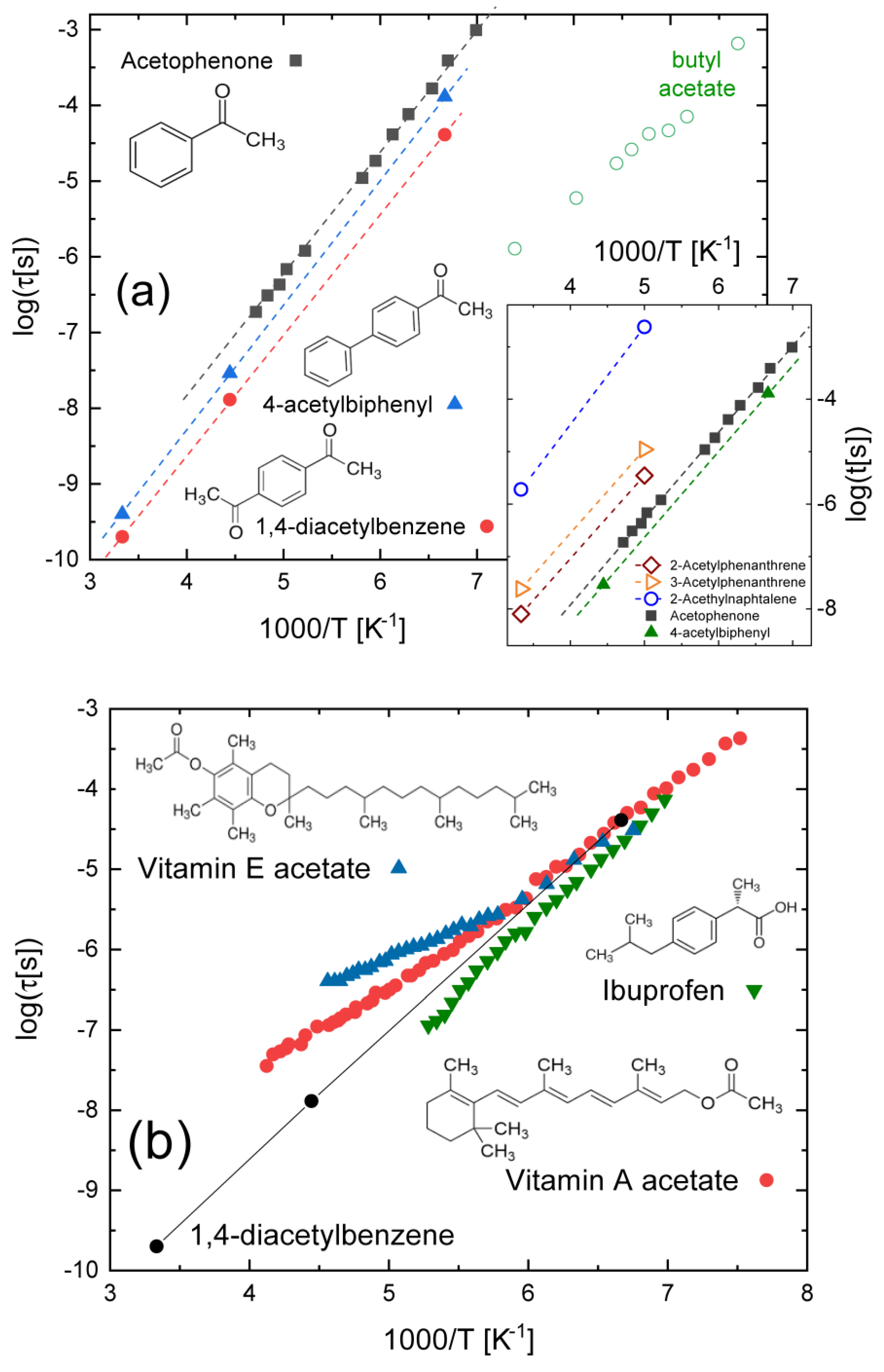
3.2. Compounds with Ketone–Benzene Cross-Conjugation
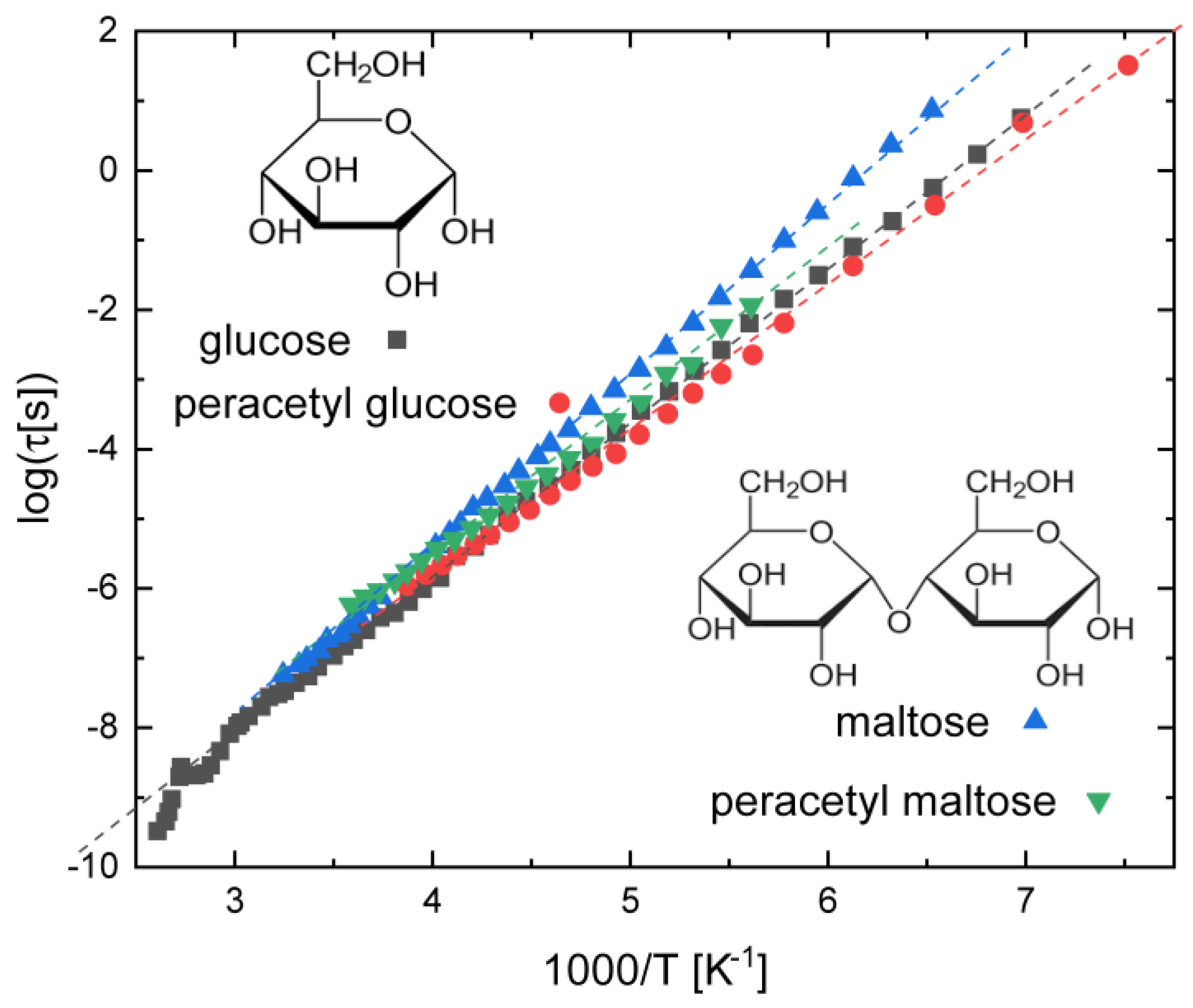
3.3. Saccharides
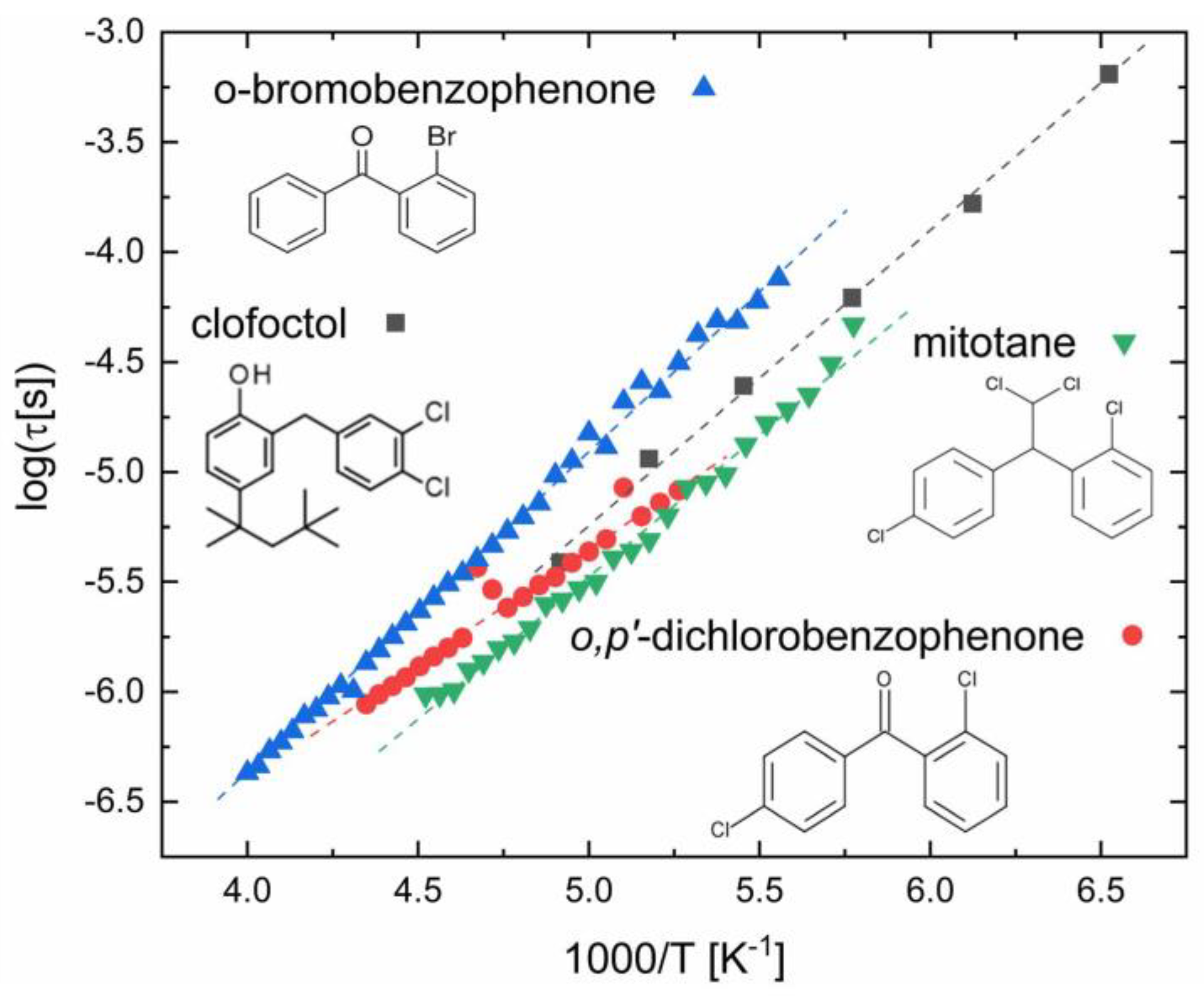
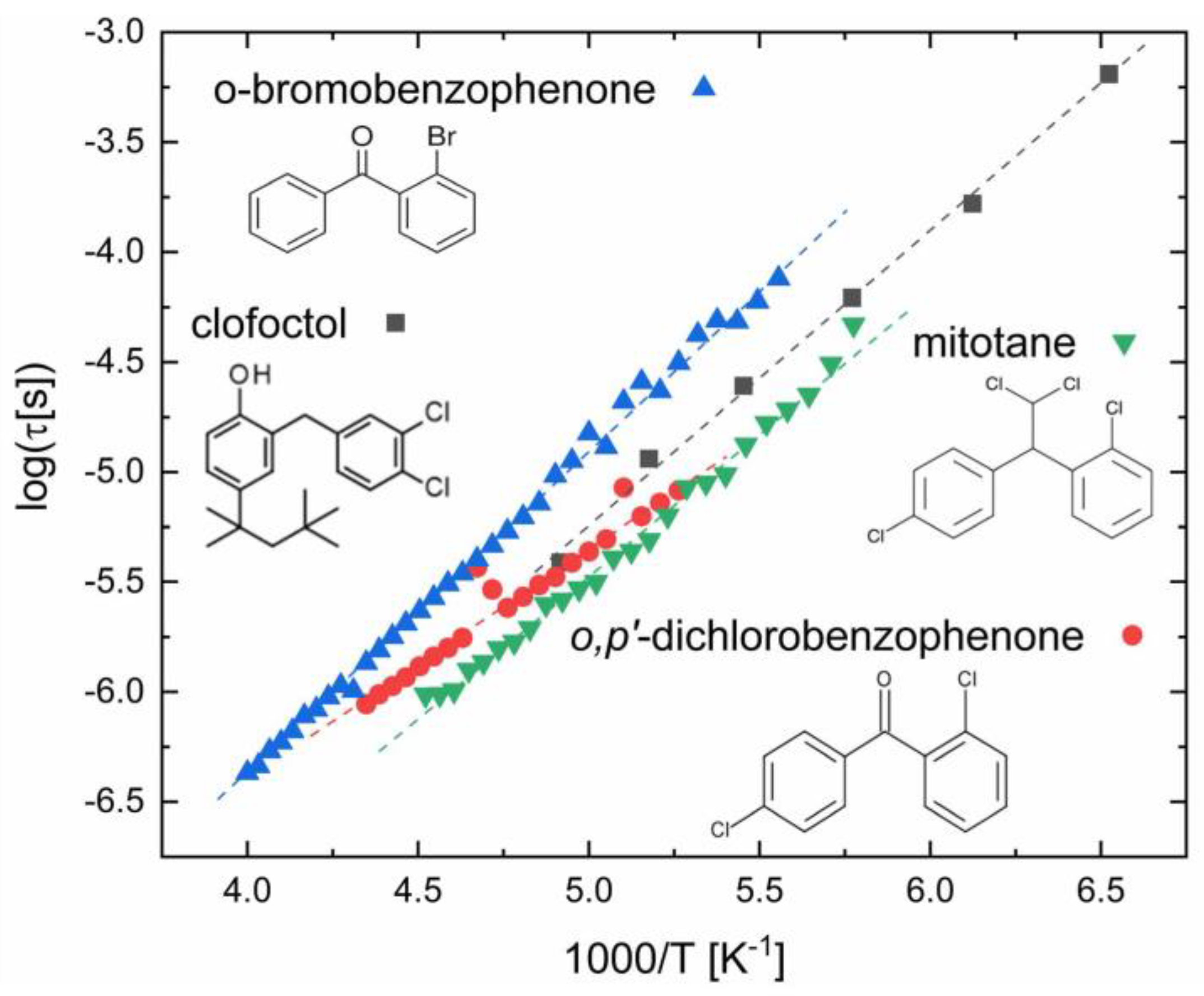
3.4. Compounds with Aromatic Halobenzene Rings
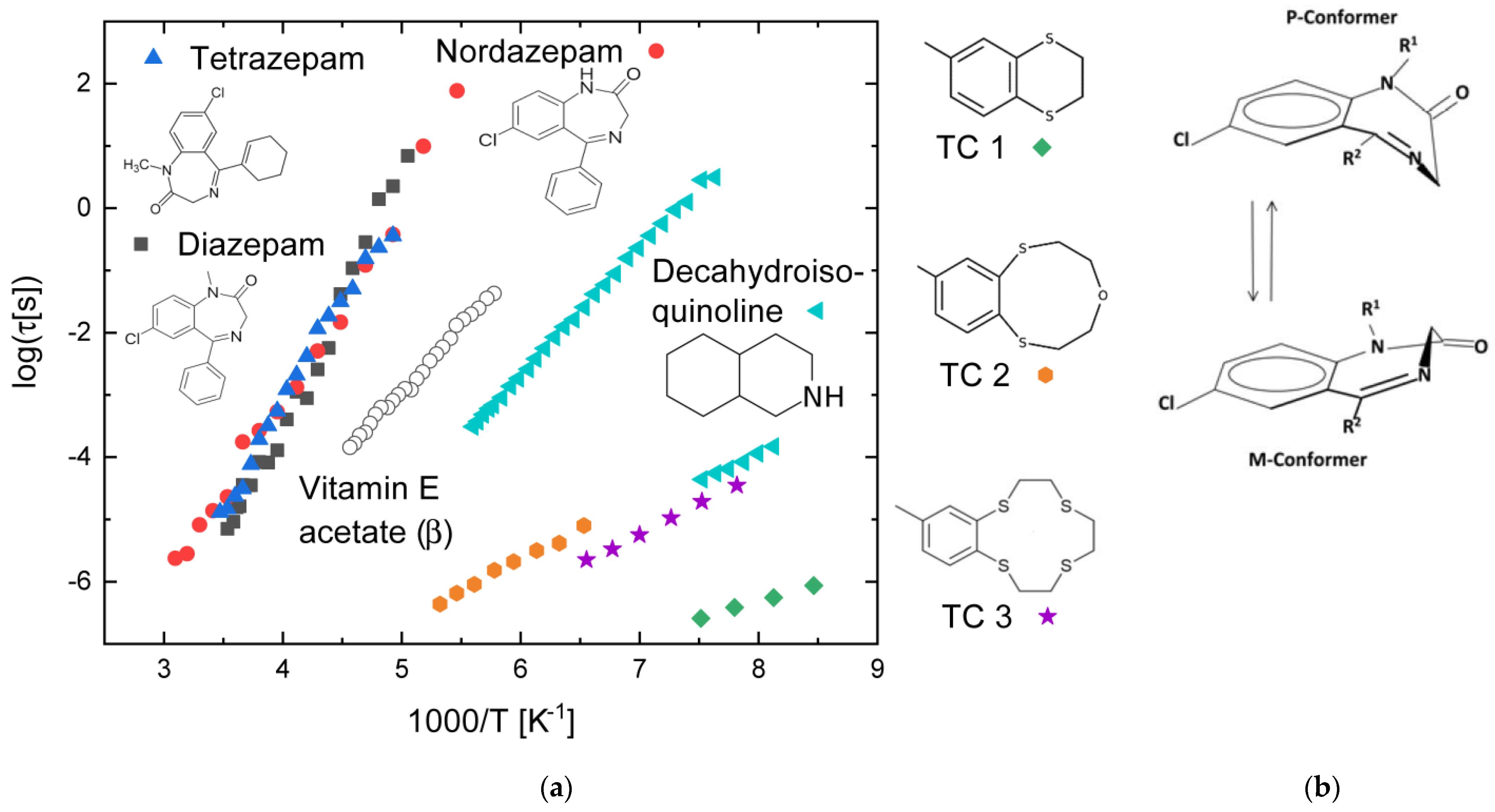
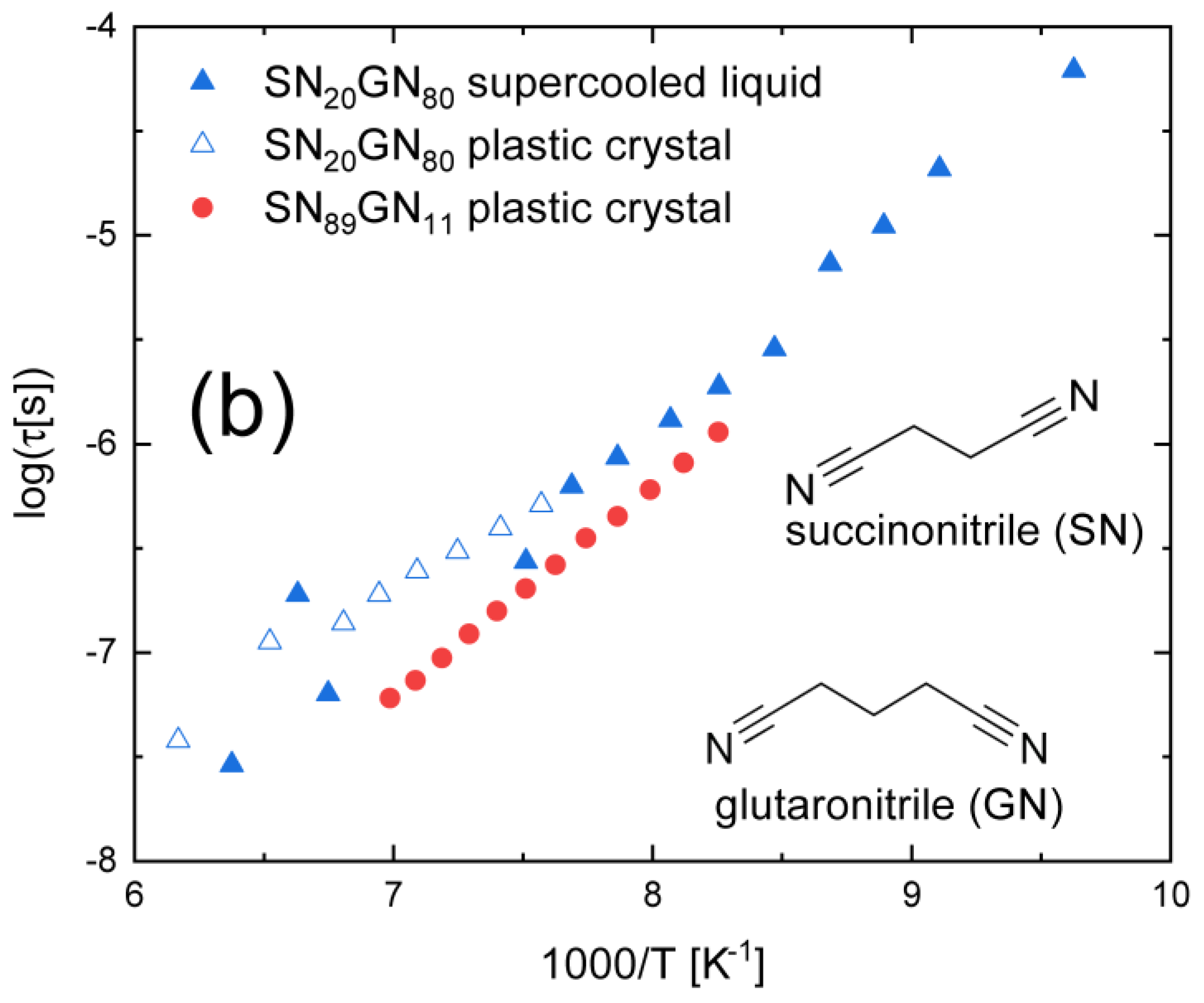
3.5. Compounds with Flexible Rings
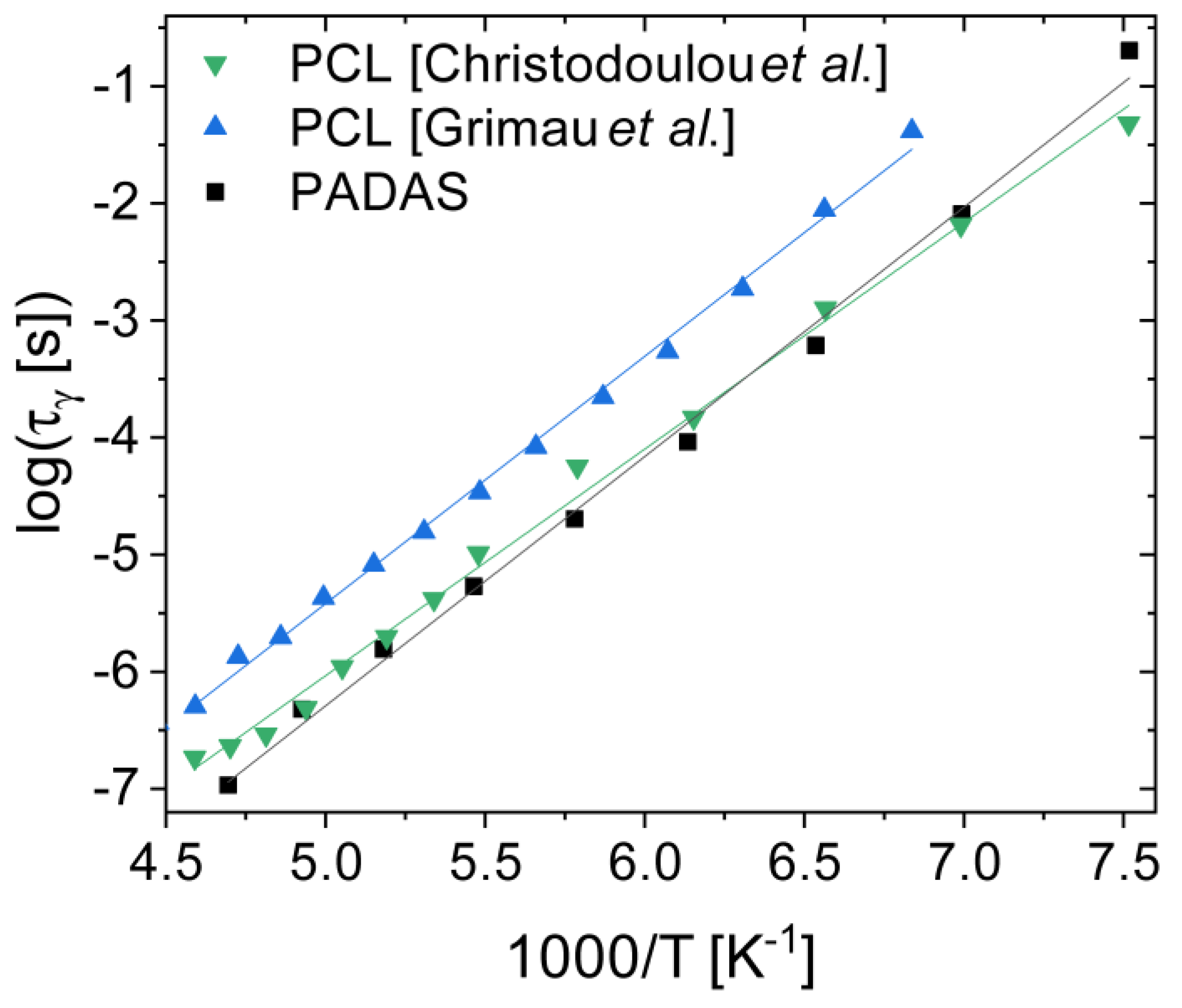
3.6. Polyesters and Polyesteramides
4. Discussion
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gutzow, I.S.; Schmelzer, J.W.P. The Vitreous State: Thermodynamics, Structure, Rheology, and Crystallization, 2nd ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA; Dordrecht, The Netherlands; London, UK, 2013. [Google Scholar]
- Böttcher, C.J.F.; Bordewijk, P. Theory of Electric Polarization, Vol. II. Dielectrics in Time-Dependent Fields; Elsevier: Amsterdam, The Netherlands; Oxford, UK; New York, NY, USA, 1978. [Google Scholar]
- Ngai, K.L. Universal properties of relaxation and diffusion in complex materials: Originating from fundamental physics with rich applications. Prog. Mater. Sci. 2023, 139, 101130. [Google Scholar] [CrossRef]
- Ngai, K.L. Relation between some secondary relaxations and the α relaxations in glass-forming materials according to the coupling model. J. Chem. Phys. 1998, 109, 6982–6994. [Google Scholar] [CrossRef]
- Ngai, K.L. Why the glass transition problem remains unsolved? J. Non-Cryst. Solids 2007, 353, 709–718. [Google Scholar] [CrossRef]
- Caporaletti, F.; Capaccioli, S.; Valenti, S.; Mikolasek, M.; Chumakov, A.I.; Monaco, G. A microscopic look at the Johari-Goldstein relaxation in a hydrogen-bonded glass-former. Sci. Rep. 2019, 9, 14319. [Google Scholar] [CrossRef] [PubMed]
- Ngai, K.L.; Paluch, M. Classification of secondary relaxation in glass-formers based on dynamic properties. J. Chem. Phys. 2004, 120, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Fong, F.K.; Smyth, C.P. Microwave Absorption and Molecular Structure in Liquids. L. The Dielectric Relaxation of Several Substituted Naphthalenes and 4-Acetyl-o-terphenyl. J. Am. Chem. Soc. 1963, 85, 548–550. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Crossley, J.; Hossain, M.S.; Saleh, M.A.; Walker, S. The nature of the β process for polar solutes in four glassy forming solids. J. Chem. Phys. 1984, 81, 448–451. [Google Scholar] [CrossRef]
- Umemoto, K.; Ouchi, K. Hindered internal rotation and intermolecular interactions. Proc. Indian Acad. Sci.-Chem. Sci. 1985, 94, 1–119. [Google Scholar] [CrossRef]
- Murthy, S.S.N.; Sobhanadri, J.; Gangasharan. The origin of β relaxation in organic glasses. J. Chem. Phys. 1994, 100, 4601–4606. [Google Scholar] [CrossRef]
- Götz, M.; Bauer, T.; Lunkenheimer, P.; Loidl, A. Supercooled-liquid and plastic-crystalline state in succinonitrile-glutaronitrile mixtures. J. Chem. Phys. 2014, 140, 094504. [Google Scholar] [CrossRef]
- Zachariah, M.; Romanini, M.; Tripathi, P.; Barrio, M.; Tamarit, J.L.; Macovez, R. Self-Diffusion, Phase Behavior, and Li+ Ion Conduction in Succinonitrile-Based Plastic Cocrystals. J. Phys. Chem. C 2015, 119, 27298–27306. [Google Scholar] [CrossRef]
- Kissi, E.O.; Grohganz, H.; Löbmann, K.; Ruggiero, M.T.; Zeitler, J.A.; Rades, T. Glass-Transition Temperature of the β-Relaxation as the Major Predictive Parameter for Recrystallization of Neat Amorphous Drugs. J. Phys. Chem. B 2018, 122, 2803–2808. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.E.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E., III; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef]
- Best, R.B.; Buchete, N.V.; Hummer, G. Are current molecular dynamics force fields too helical? Biophys. J. 2008, 95, L07–L09. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Lahey, S.-L.J.; Thien Phuc, T.N.; Rowley, C.N. Benchmarking Force Field and the ANI Neural Network Potentials for the Torsional Potential Energy Surface of Biaryl Drug Fragments. J. Chem. Inf. Model. 2020, 60, 6258–6268. [Google Scholar] [CrossRef]
- Kania, A.; Sarapata, K.; Gucwa, M.; Wójcik-Augustyn, A. Optimal Solution to the Torsional Coefficient Fitting Problem in Force Field Parametrization. J. Phys. Chem. A 2021, 125, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Romanini, M.; Barrio, M.; Macovez, R.; Capaccioli, S.; Tamarit, J.L. Mixtures of m-fluoroaniline with apolar aromatic molecules: Phase behaviour, suppression of H-bonded clusters, and local H-bond relaxation dynamics. J. Mol. Liquids 2019, 296, 111998. [Google Scholar] [CrossRef]
- Böhmer, R.; Gainaru, C.; Richert, R. Structure and dynamics of monohydroxy alcohols—Milestones towards their microscopic understanding, 100 years after Debye. Phys. Rep. 2014, 545, 125–195. [Google Scholar] [CrossRef]
- Bama, J.-A.; Dudognon, E.; Affouard, F. Impact of Low Concentration of Strongly Hydrogen-Bonded Water Molecules on the Dynamics of Amorphous Terfenadine: Insights from Molecular Dynamics Simulations and Dielectric Relaxation Spectroscopy. J. Phys. Chem. B 2021, 125, 11292–11307. [Google Scholar] [CrossRef]
- Hansen, C.; Stickel, F.; Berger, T.; Richert, R.; Fischer, E.W. Dynamics of glass-forming liquids. III. Comparing the dielectric α- and β-relaxation of 1-propanol and o-terphenyl. J. Chem. Phys. 1997, 107, 1086–1093. [Google Scholar] [CrossRef]
- Ngai, K.L.; Wang, L.-M. Relations between the Structural α-Relaxation and the Johari–Goldstein β-Relaxation in Two Mono-hydroxyl Alcohols: 1-Propanol and 5-Methyl-2-hexanol. J. Phys. Chem. B 2019, 123, 714–719. [Google Scholar] [CrossRef]
- Johari, G.P.; Kalinovskaya, O.E.; Vij, J.K. Effects of induced steric hindrance on the dielectric behavior and H bonding in the supercooled liquid and vitreous alcohol. J. Chem. Phys. 2001, 114, 4634–4642. [Google Scholar] [CrossRef]
- Vij, J.K. Primary and secondary relaxations in aliphatic alcohols: Complex dielectric permittivity spectra of 5-methyl 2-hexanol. J. Mol. Liq. 1996, 69, 19–29. [Google Scholar]
- Kalinovskaya, O.E.; Vij, J.K. The exponential dielectric relaxation dynamics in a secondary alcohol’s supercooled liquid and glassy states. J. Chem. Phys. 2000, 112, 3262–3266. [Google Scholar] [CrossRef]
- Nowok, A.; Dulski, M.; Grelska, J.; Szeremeta, A.Z.; Jurkiewicz, K.; Grzybowska, K.; Musial, M.; Pawlus, S. Phenyl Ring: A Steric Hindrance or a Source of Different Hydrogen Bonding Patterns in Self-Organizing Systems? J. Phys. Chem. Lett. 2021, 12, 2142–2147. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Schönhals, A.; Kremer, F. Broadband Dielectric Measurement Techniques (10−6 Hz to 1012 Hz). In Broadband Dielectric Spectroscopy; Kremer, F., Schönhals, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics—I. Alternating Current Characteristics. J. Chem. Phys. 1941, 9, 341–352. [Google Scholar] [CrossRef]
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics—II. Direct Current Characteristics. J. Chem. Phys. 1942, 10, 98–105. [Google Scholar] [CrossRef]
- Romanini, M.; Macovez, R.; Barrio, M.; Tamarit, J.L. Inter-enantiomer conversion dynamics and Johari–Goldstein relaxation of benzophenones. Sci. Rep. 2021, 11, 20248. [Google Scholar] [CrossRef]
- Glasstone, S.; Laidler, K.J.; Eyring, H. The Theory of Rate Processes; McGraw-Hill: New York, NY, USA, 1941. [Google Scholar]
- Kauzmann, W. Dielectric Relaxation as a Chemical Rate Process. Rev. Mod. Phys. 1942, 14, 12. [Google Scholar] [CrossRef]
- Crossley, J.; Smyth, C.P. Microwave absorption and molecular structure in liquids. LXXIII. A dielectric study of solute-solvent interactions. J. Am. Chem. Soc. 1969, 91, 2482–2487. [Google Scholar] [CrossRef]
- Madan, M.P. Microwave Absorption and Dielectric Relaxation of 1,2-Dihalogenoethanes in Dilute Solutions. J. Chem. Soc. Faraday Trans. 2 1975, 71, 67–70. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Walker, S. Two Resolved Dielectric Absorption Peaks of the 1-Bromoalkanes. J. Chem. Soc. Faraday Trans. 2 1985, 81, 479–489. [Google Scholar] [CrossRef]
- Shahin, M.; Murthy, S.S.N. Sub-Tg relaxations due to dipolar solutes in nonpolar glass-forming solvents. J. Chem. Phys. 2005, 122, 014507. [Google Scholar] [CrossRef]
- Nunes, T.G.; Viciosa, M.T.; Correia, N.T.; Danède, F.; Nunes, R.G.; Diogo, H.P. A Stable Amorphous Statin: Solid-State NMR and Dielectric Studies on Dynamic Heterogeneity of Simvastatin. Mol. Pharm. 2014, 11, 727–737. [Google Scholar] [CrossRef]
- Grzybowska, K.; Paluch, M.; Grzybowski, A.; Wojnarowska, Z.; Hawelek, L.; Kolodziejczyk, K.; Ngai, K.L. Molecular Dynamics and Physical Stability of Amorphous Anti-Inflammatory Drug: Celecoxib. J. Phys. Chem. B 2010, 114, 12792–12801. [Google Scholar] [CrossRef]
- Bauer, T.; Köhler, M.; Lunkenheimer, P.; Loidl, A.; Angell, C.A. Relaxation dynamics and ionic conductivity in a fragile plastic crystal. J. Chem. Phys. 2010, 133, 144509. [Google Scholar] [CrossRef]
- Zachariah, M.; Romanini, M.; Tripathi, P.; Tamarit, J.L.; Macovez, R. Molecular diffusion and dc conductivity perfectly correlated with molecular rotational dynamics in a plastic crystalline electrolyte. Phys. Chem. Chem. Phys. 2015, 17, 16053–16057. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.C.K.; Walker, S. Acetyl group relaxation in four aromatic ketones. Can. J. Chem. 1977, 55, 583–587. [Google Scholar] [CrossRef]
- Crossley, J.; Mazid, M.A.; McLellan, C.K.; Mountain, P.F.; Walker, S. Dielectric relaxation in 2-acetylnaphthalene and related compounds. Can. J. Chem. 1978, 56, 567–573. [Google Scholar] [CrossRef]
- Khwaja, H.A.; Mazid, M.A.; Walker, S. Enhanced Energy Barriers in Group Relaxation of some Heterocyclic Aldehydes and Ketones. Z. Phys. Chem. 1981, 128, 147–161. [Google Scholar] [CrossRef]
- Wojnarowska, Z.; Paluch, M.; Wlodarczyk, P.; Hawelek, L.; Wrzalik, R.; Zioło, J.; Wygledowska-Kania, M.; Bergler-Czop, B.; Brzezinska-Wcislo, L.; Bujak, P. Nanoscale domains with nematic order in supercooled vitamin-A acetate: Molecular dynamics studies. Phys. Rev. E 2011, 83, 051502. [Google Scholar] [CrossRef]
- Kaminski, K.; Maslanka, S.; Ziolo, J.; Paluch, M.; McGrath, K.J.; Roland, C.M. Dielectric relaxation of α-tocopherol acetate (vitamin E). Phys. Rev. E 2007, 75, 011903. [Google Scholar] [CrossRef]
- Bras, A.R.; Noronha, J.P.; Antunes, A.M.M.; Cardoso, M.M.; Schönhals, A.; Affouard, F.; Dionisio, M.; Correia, N.T. Molecular Motions in Amorphous Ibuprofen as Studied by Broadband Dielectric Spectroscopy. J. Phys. Chem. B 2008, 112, 11087–11099. [Google Scholar] [CrossRef]
- Kaminski, K.; Kaminska, E.; Ngai, K.L.; Paluch, M.; Wlodarczyk, P.; Kasprzycka, A.; Szeja, W. Identifying the Origins of Two Secondary Relaxations in Polysaccharides. J. Phys. Chem. B 2009, 113, 10088–10096. [Google Scholar] [CrossRef]
- Kaminski, K.; Wlodarczyk, P.; Adrjanowicz, K.; Kaminska, E.; Wojnarowska, Z.; Paluch, M. Origin of the Commonly Observed Secondary Relaxation Process in Saccharides. J. Phys. Chem. B 2010, 114, 11272–11281. [Google Scholar] [CrossRef]
- Kaminski, K.; Wlodarczyk, P.; Hawelek, L.; Adrjanowicz, K.; Wojnarowska, Z.; Paluch, M.; Kaminska, E. Comparative dielectric studies on two hydrogen-bonded and van der Waals liquids. Phys. Rev. E 2011, 83, 061506. [Google Scholar] [CrossRef]
- Kaminski, K.; Adrjanowicz, K.; Wojnarowska, Z.; Dulski, M.; Wrzalik, R.; Paluch, M.; Kaminska, E.; Kasprzycka, A. Do Intermolecular Interactions Control Crystallization Abilities of Glass-Forming Liquids? J. Phys. Chem. B 2011, 115, 11537–11547. [Google Scholar] [CrossRef] [PubMed]
- Havriliak, S.; Negami, S. A complex plane representation of dielectric and mechanical relaxation processes in some polymers. Polymer 1967, 8, 161–210. [Google Scholar] [CrossRef]
- Romanini, M.; Valmaseda, A.; Macovez, R. Glass transition, crystallization kinetics, and inter-conformer relaxation dynamics of amorphous mitotane and related compounds. Int. J. Pharm. 2022, 629, 122390. [Google Scholar] [CrossRef] [PubMed]
- Sahra, M.; Thayyila, M.S.; Bansal, A.K.; Ngai, K.L.; Sulaiman, M.K.; Shete, G.; Safna Hussan, K.P. Dielectric spectroscopic studies of three important active pharmaceutical ingredients—Clofoctol, droperidol and probucol. J. Non-Cryst. Solids 2019, 505, 28–36. [Google Scholar] [CrossRef]
- Baraldi, I.; Gallinella, E.; Momicchioli, F. Conformations and internal rotation properties of molecules containing one geminal diphenylgroup: Diphenylethylene, diphenylketimine, benzophenone, diphenylether and diphenylmethane. J. Chim. Phys. 1986, 83, 653–664. [Google Scholar] [CrossRef]
- Mazid, M.A.; Walker, S.; Weir, N.A. Intramolecular relaxation of phenyl-alkyl ketones. Adv. Mol. Relax. Interact. Process. 1981, 19, 249–259. [Google Scholar] [CrossRef]
- Tripathi, P.; Romanini, M.; Tamarit, J.L.; Macovez, R. Collective relaxation dynamics and crystallization kinetics of the amorphous Biclotymol antiseptic. Int. J. Pharm. 2015, 495, 420–427. [Google Scholar] [CrossRef]
- Romanini, M.; Lorente, M.; Schammé, B.; Delbreilh, L.; Dupray, V.; Coquerel, G.; Tamarit, J.L.; Macovez, R. Enhancement of the Physical and Chemical Stability of Amorphous Drug−Polymer Mixtures via Cryogenic Comilling. Macromolecules 2018, 51, 9382–9392. [Google Scholar] [CrossRef]
- Valenti, S.; Barrio, M.; Negrier, P.; Romanini, M.; Macovez, R.; Tamarit, J.L. Comparative Physical Study of Three Pharmaceutically Active Benzodiazepine Derivatives: Crystalline versus Amorphous State and Crystallization Tendency. Mol. Pharm. 2021, 18, 1819–1832, Erratum in Mol. Pharm. 2021, 18, 3926–3927. [Google Scholar] [CrossRef]
- Mielcarek, J.; Nowak, D.; Pajzderska, A.; Peplinska, B.; Wasicki, J. A hybrid method for estimation of molecular dynamics of diazepam—Density functional theory combined with NMR and FT-IR spectroscopy. Int. J. Pharm. 2011, 404, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Linscheid, P.; Lehn, J.M. Etudes cinétiques et conformationelles par résonance magnétique nucléaire: Inversion de cycle dans desbenzodiazépinones. Bull. Soc. Chim. Fr. 1967, 992–997. [Google Scholar]
- Paizs, B.; Simonyi, M. Ring inversion barrier of diazepam and derivatives: An ab initio study. Chirality 1999, 11, 651–658. [Google Scholar] [CrossRef]
- Hellwig, H.; Nowok, A.; Malecki, J.G.; Kus, P.; Jedrzejowska, A.; Grzybowska, K.; Pawlus, S. Conformational analysis and molecular dynamics of glass-forming aromatic thiacrown ethers. Phys. Chem. Chem. Phys. 2020, 22, 17948–17959. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, H.; Nowok, A.; Peksa, P.; Dulski, M.; Musioł, R.; Pawlus, S.; Kuś, P. Molecular Dynamics and Near-Tg Phenomena of Cyclic Thioethers. Int. J. Mol. Sci. 2023; in press. [Google Scholar]
- Paluch, M.; Pawlus, S.; Hensel-Bielowka, S.; Kaminska, E.; Prevosto, D.; Capaccioli, S.; Rolla, P.A.; Ngai, K.L. Two secondary modes in decahydroisoquinoline: Which one is the true Johari Goldstein process? J. Chem. Phys. 2005, 122, 234506. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, P.; Czarnota, B.; Paluch, M.; Pawlus, S.; Ziolo, J. Microscopic origin of secondary modes observed in decahydroisoquinoline. J. Mol. Struct. 2010, 975, 200–204. [Google Scholar] [CrossRef]
- Grimau, M.; Laredo, E.; Pérez, Y.M.; Bello, A. Study of dielectric relaxation modes in poly (ε-caprolactone): Molecular weight, water sorption, and merging effects. J. Chem. Phys. 2001, 114, 6417–6425. [Google Scholar] [CrossRef]
- Christodoulou, E.; Klonos, P.A.; Tsachouridis, K.; Zamboulis, A.; Kyritsis, A.; Bikiaris, D.N. Synthesis, crystallization, and molecular mobility in poly (ε-caprolactone) copolyesters of different architectures for biomedical applications studied by calorimetry and dielectric spectroscopy. Soft Matter 2020, 16, 8187–8201. [Google Scholar] [CrossRef]
- Valenti, S.; Arioli, M.; Jamett, A.; Tamarit, J.L.; Puiggalí, J.; Macovez, R. Amorphous solid dispersions of curcumin in a poly(ester amide): Antiplasticizing effect on the glass transition and macromolecular relaxation dynamics, and controlled release. Int. J. Pharm. 2023, 644, 123333. [Google Scholar] [CrossRef]
- Kortaberria, G.; Jimeno, A.; Arruti, P.; de la Caba, K.; Remiro, P.; Eceiza, A.; Mondragon, I. Molecular Dynamics of PGA Bioabsorbable Polymer During Isothermal Cold Crystallization. Macromol. Symp. 2006, 239, 152–158. [Google Scholar] [CrossRef]
- Valenti, S.; del Valle, L.J.; Romanini, M.; Mitjana, M.; Puiggalí, J.; Tamarit, J.L.; Macovez, R. Drug-Biopolymer Dispersions: Morphology- and Temperature-Dependent (Anti)Plasticizer Effect of the Drug and Component-Specific Johari–Goldstein Relaxations. Int. J. Mol. Sci. 2022, 23, 2456. [Google Scholar] [CrossRef]
- Feixas, F.; Lindert, S.; Sinko, W.; McCammon, J.A. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys. Chem. 2014, 186, 31–45. [Google Scholar] [CrossRef]
- Light, S.H.; Cahoon, L.A.; Mahasenan, K.V.; Lee, M.; Boggess, B.; Halavaty, A.S.; Mobashery, S.; Freitag, N.E.; Anderson, W.F. Transferase Versus Hydrolase: The Role of Conformational Flexibility in Reaction Specificity. Structure 2017, 25, 295–304. [Google Scholar] [CrossRef]
- Ben Bdira, F.; Waudby, C.A.; Volkov, A.N.; Schröder, S.P.; Ab, E.; Codée, J.D.C.; Overkleeft, H.S.; Aerts, J.M.F.G.; van Ingen, H.; Ubbink, M. Dynamics of LigandBinding to a Rigid Glycosidase. Angew. Chem. Int. Ed. 2020, 59, 20508–20514. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanini, M.; Macovez, R.; Valenti, S.; Noor, W.; Tamarit, J.L. Dielectric Spectroscopy Studies of Conformational Relaxation Dynamics in Molecular Glass-Forming Liquids. Int. J. Mol. Sci. 2023, 24, 17189. https://doi.org/10.3390/ijms242417189
Romanini M, Macovez R, Valenti S, Noor W, Tamarit JL. Dielectric Spectroscopy Studies of Conformational Relaxation Dynamics in Molecular Glass-Forming Liquids. International Journal of Molecular Sciences. 2023; 24(24):17189. https://doi.org/10.3390/ijms242417189
Chicago/Turabian StyleRomanini, Michela, Roberto Macovez, Sofia Valenti, Wahi Noor, and Josep Lluís Tamarit. 2023. "Dielectric Spectroscopy Studies of Conformational Relaxation Dynamics in Molecular Glass-Forming Liquids" International Journal of Molecular Sciences 24, no. 24: 17189. https://doi.org/10.3390/ijms242417189
APA StyleRomanini, M., Macovez, R., Valenti, S., Noor, W., & Tamarit, J. L. (2023). Dielectric Spectroscopy Studies of Conformational Relaxation Dynamics in Molecular Glass-Forming Liquids. International Journal of Molecular Sciences, 24(24), 17189. https://doi.org/10.3390/ijms242417189