
Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions

 , , ,
, , ,
Abstract
:1. Introduction
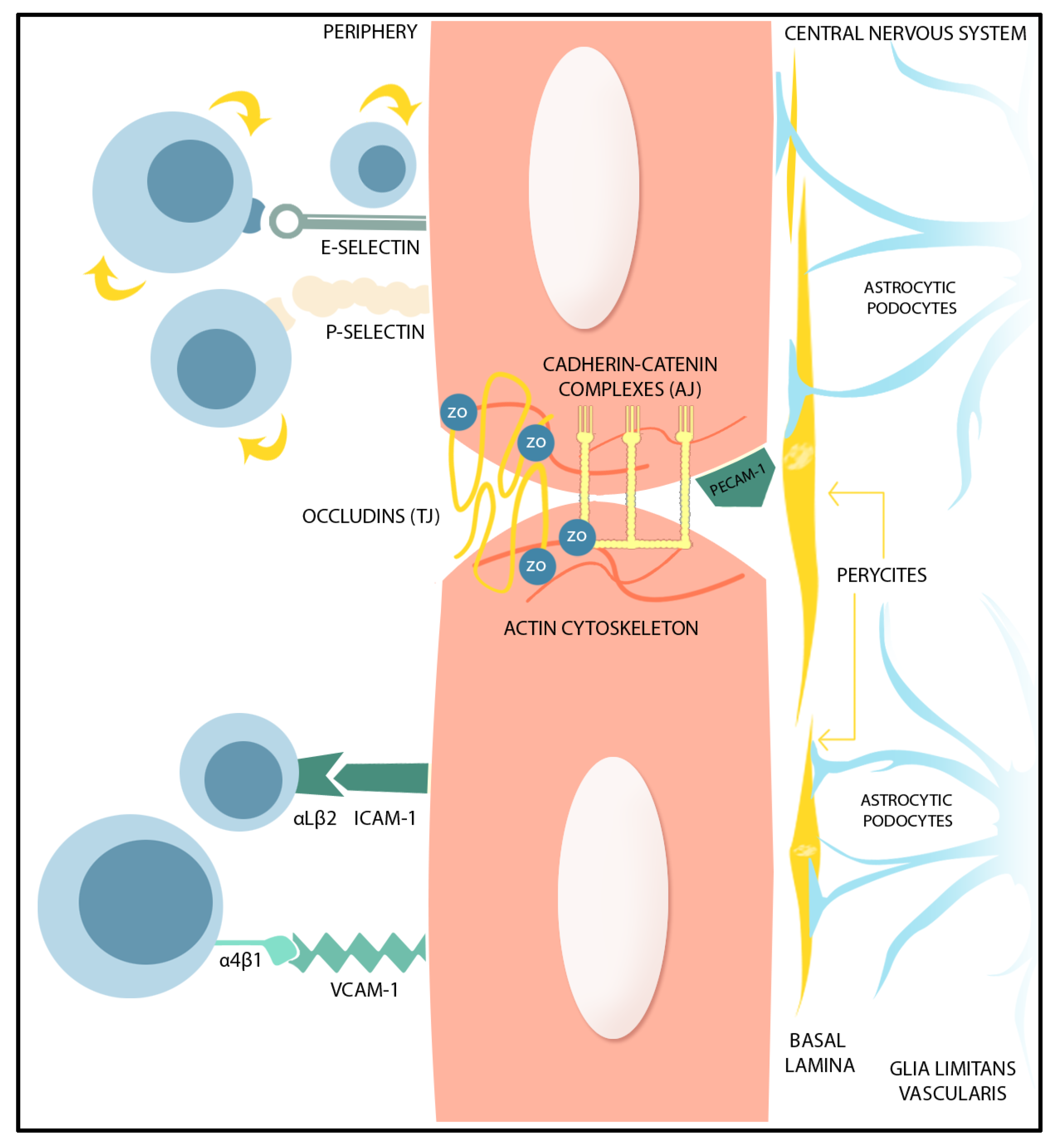
2. BBB Permeability-Regulating Proteins
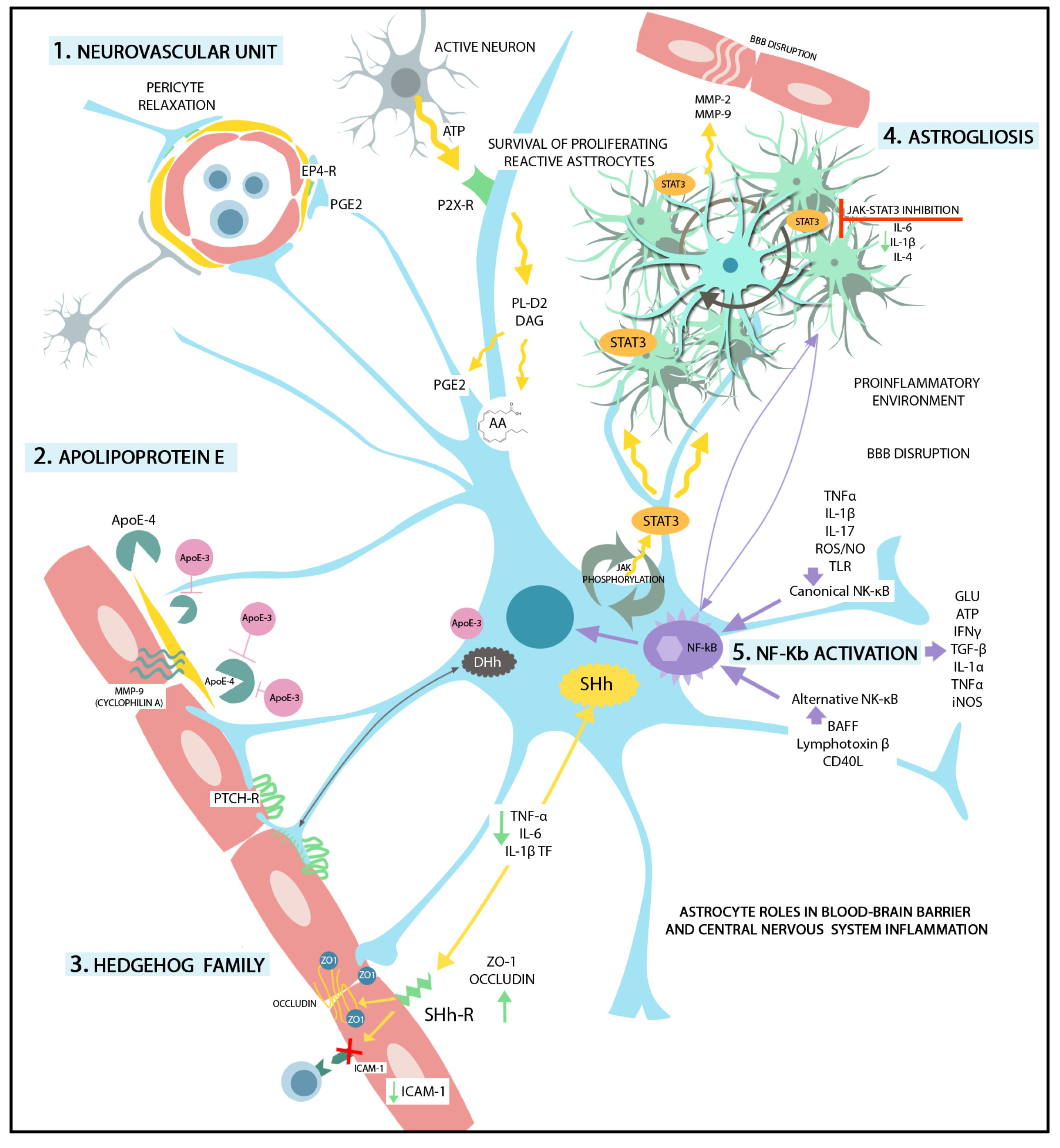
3. Endothelial Cells Interplay with Astrocytes within the Neurovascular Unit (NVU)
3.1. Astrocytic Factors Supporting BBB Integrity
3.2. Astrocyte-Derived Factors Increasing BBB Permeability
4. Reactive Astrogliosis in BBB Dysfunction
5. Astrocyte Functional Identity Is Driven by Transcriptional and Epigenetic Changes
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE-1 | Angiotensin converting enzyme-1 |
| AHR | Aryl hydrocarbon receptor |
| AJ | Adherens junction |
| AKT (PKB) | Protein kinase B |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ANG-1/2 | Angiopoetin-1/2 |
| ApoE | Apolipoprotein E |
| ATAC-seq | Transposase accessible chromatin sequencing |
| BAFF | B cell activating factor of the TNF family |
| BBB | Blood–brain barrier |
| BCR | B-cell receptor |
| BDNF | Brain-derived neurotrophic factor |
| BM | Basement membrane |
| CCL | Chemokine ligand |
| CD | Cluster of differentiation |
| CNS | Central nervous system |
| DhH | Desert Hedgehog |
| DNA | Deoxyribonucleic acid |
| ECs | Endothelial cells |
| ECM | Extracellular matrix |
| eNOS | Endothelial nitric oxide synthase |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ET | Endothelin |
| ETR | Endothelin receptor |
| FGF | Fibroblast Growth Factor |
| GDNF | Glial-derived Neurotrophic Factor |
| cGMP | Cyclic guanosine monophosphate |
| GTP | Guanosine-5′-triphosphate |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylases |
| HDM | Histone demethylase |
| HMT | Histone methyltransferase |
| ICAM-1 | Intercellular adhesion molecule -1 |
| IFN | Interferon |
| IGF-1 | Insulin-Like Growth Factor |
| IKK | IkappaB kinase |
| IL | Interleukin |
| IRAK | Interleukin-1 receptor-associated kinase |
| IRF-1 | Interferon regulatory factor-1 |
| JAK | Janus kinase |
| HIF | Hypoxia-inducible factor |
| HMGB1 | High mobility group box 1 protein |
| LaST | Large-area spatial transcriptomic |
| LIF | Leukemia inhibitory factor |
| LPS | Lipopolysaccharide |
| LRP1 | Low density lipoprotein receptor-related protein 1 |
| MAP | Mitogen-activated protein |
| MLC | Myosin light chain |
| MLCK | Myosin light chain kinase |
| MMP | Matrix metalloproteinase |
| mRNA | Messenger ribonucleic acid |
| miRNA | Micro ribonucleic acid |
| MRP | miRNAs after myeloid-related protein |
| NFAT | Nuclear factor of activated T-cells |
| NF-kB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric oxide |
| NVU | Neurovascular unit |
| PAK | Src–Rac1–p21-activated kinase |
| PHD2 | Prolyl hydroxylase domain protein 2 |
| PI3K | Phosphoinositide3 kinase |
| PK | Protein kinase |
| PL | Phospholipase |
| PTPN-2 | protein tyrosine phosphatase non-receptor type 2 |
| RA | Retinoic acid |
| RAGE | Advanced glycation end products |
| RNAs | Ribonucleic acids |
| ROS | Reactive oxygen species |
| ROCK | Rho-associated protein kinase |
| RRBS | Reduced representation bisulfite sequencing |
| scRNA-seq | Single cell RNA sequencing |
| snRNA-seq | Single nucleus RNA sequencing |
| STAT3 | Signal transducer and activator of transcription 3 |
| Smo | Signal transducer Smoothened |
| smFISH | Single-molecule fluorescent in situ hybridization |
| SOCS | Suppressor of cytokine signaling 1 |
| SHh | Sonic Hedgehog |
| TAK | TGFβ-activated kinase 1 |
| TCR | T-cell receptor |
| TGF-β | Transforming Growth Factor-β |
| TJ | Tight junction |
| TLR | Toll-like receptor |
| TNF | Tumor Necrosis Factor |
| VCAM-1 | Vascular cell adhesion molecule |
| VE | Vascular endothelial |
| VEGF | Vascular Endothelial Growth Factor |
| ZO | Zonula occludens |
References
- Balasa, R.; Barcutean, L.; Mosora, O.; Manu, D. Reviewing the Significance of Blood–Brain Barrier Disruption in Multiple Sclerosis Pathology and Treatment. Int. J. Mol. Sci. 2021, 22, 8370. [Google Scholar] [CrossRef] [PubMed]
- Burek, M.; König, A.; Lang, M.; Fiedler, J.; Oerter, S.; Roewer, N.; Bohnert, M.; Thal, S.C.; Blecharz-Lang, K.G.; Woitzik, J.; et al. Hypoxia-Induced MicroRNA-212/132 Alter Blood-Brain Barrier Integrity Through Inhibition of Tight Junction-Associated Proteins in Human and Mouse Brain Microvascular Endothelial Cells. Transl. Stroke Res. 2019, 10, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.W.; McGavern, D.B. Immune dynamics in the CNS and its barriers during homeostasis and disease. Immunol. Rev. 2022, 306, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Pivoriūnas, A.; Verkhratsky, A. Astrocyte–Endotheliocyte Axis in the Regulation of the Blood–Brain Barrier. Neurochem. Res. 2021, 46, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Brunner, J.; Ragupathy, S.; Borchard, G. Target specific tight junction modulators. Adv. Drug Deliv. Rev. 2021, 171, 266–288. [Google Scholar] [CrossRef] [PubMed]
- Brunner, N.; Stein, L.; Cornelius, V.; Knittel, R.; Fallier-Becker, P.; Amasheh, S. Blood-Brain Barrier Protein Claudin-5 Expressed in Paired Xenopus laevis Oocytes Mediates Cell-Cell Interaction. Front. Physiol. 2020, 11, 857. [Google Scholar] [CrossRef]
- Markov, A.G.; Fedorova, A.A.; Kravtsova, V.V.; Bikmurzina, A.E.; Okorokova, L.S.; Matchkov, V.V.; Cornelius, V.; Amasheh, S.; Krivoi, I.I. Circulating Ouabain Modulates Expression of Claudins in Rat Intestine and Cerebral Blood Vessels. Int. J. Mol. Sci. 2020, 21, 5067. [Google Scholar] [CrossRef] [PubMed]
- Winkler, L.; Blasig, R.; Breitkreuz-Korff, O.; Berndt, P.; Dithmer, S.; Helms, H.C.; Puchkov, D.; Devraj, K.; Kaya, M.; Qin, Z.; et al. Tight junctions in the blood–brain barrier promote edema formation and infarct size in stroke—Ambivalent effects of sealing proteins. J. Cereb. Blood Flow. Metab. 2021, 41, 132–145. [Google Scholar] [CrossRef]
- Yang, Y.; Torbey, M.T. Angiogenesis and Blood-Brain Barrier Permeability in Vascular Remodeling after Stroke. Curr. Neuropharmacol. 2020, 18, 1250–1265. [Google Scholar] [CrossRef]
- Yeung, D.; Manias, J.L.; Stewart, D.J.; Nag, S. Decreased junctional adhesion molecule-A expression during blood–brain barrier breakdown. Acta Neuropathol. 2008, 115, 635–642. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Cristante, E.; McArthur, S.; Mauro, C.; Maggioli, E.; Romero, I.A.; Wylezinska-Arridge, M.; Couraud, P.O.; Lopez-Tremoleda, J.; Christian, H.C.; Weksler, B.B.; et al. Identification of an essential endogenous regulator of blood–brain barrier integrity, and its pathological and therapeutic implications. Proc. Natl. Acad. Sci. USA 2013, 110, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, Z.-L.; Norris, E.H.; Strickland, S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat. Commun. 2014, 5, 3413. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin Signaling. Science 1999, 285, 1028–1033. [Google Scholar] [CrossRef]
- McCarty, J.H. αvβ8 integrin adhesion and signaling pathways in development, physiology and disease. J. Cell Sci. 2020, 133, jcs239434. [Google Scholar] [CrossRef]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef]
- Steiner, E.; Enzmann, G.U.; Lyck, R.; Lin, S.; Rüegg, M.A.; Kröger, S.; Engelhardt, B. The heparan sulfate proteoglycan agrin contributes to barrier properties of mouse brain endothelial cells by stabilizing adherens junctions. Cell Tissue Res. 2014, 358, 465–479. [Google Scholar] [CrossRef]
- Marina, N.; Christie, I.N.; Korsak, A.; Doronin, M.; Brazhe, A.; Hosford, P.S.; Wells, J.A.; Sheikhbahaei, S.; Humoud, I.; Paton, J.F.R.; et al. Astrocytes monitor cerebral perfusion and control systemic circulation to maintain brain blood flow. Nat. Commun. 2020, 11, 131. [Google Scholar] [CrossRef]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef]
- Félétou, M. Calcium-activated potassium channels and endothelial dysfunction: Therapeutic options? Br. J. Pharmacol. 2009, 156, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β Induces Blood–Brain Barrier Disruption by Downregulating Sonic Hedgehog in Astrocytes. Kira, J. ichi, editor. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Allahyari, R.V.; Clark, K.L.; Shepard, K.A.; Garcia, A.D.R. Sonic hedgehog signaling is negatively regulated in reactive astrocytes after forebrain stab injury. Sci. Rep. 2019, 9, 565. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.A.; Fu, M.; Garcia, A.D.R. Sonic hedgehog signaling in astrocytes. Cell. Mol. Life Sci. 2021, 78, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Chechneva, O.V.; Mayrhofer, F.; Daugherty, D.J.; Krishnamurty, R.G.; Bannerman, P.; Pleasure, D.E.; Deng, W. A Smoothened receptor agonist is neuroprotective and promotes regeneration after ischemic brain injury. Cell Death Dis. 2014, 5, e1481. [Google Scholar] [CrossRef]
- Garcia, A.D.R. New Tricks for an Old (Hedge)Hog: Sonic Hedgehog Regulation of Astrocyte Function. Cells 2021, 10, 1353. [Google Scholar] [CrossRef]
- Zhu, S.-L.; Luo, M.-Q.; Peng, W.-X.; Li, Q.-X.; Feng, Z.-Y.; Li, Z.-X.; Wang, M.-X.; Feng, X.-X.; Liu, F.; Huang, J.-L. Sonic hedgehog signalling pathway regulates apoptosis through Smo protein in human umbilical vein endothelial cells. Rheumatology 2015, 54, 1093–1102. [Google Scholar] [CrossRef]
- Chapouly, C.; Guimbal, S.; Hollier, P.-L.; Renault, M.-A. Role of Hedgehog Signaling in Vasculature Development, Differentiation, and Maintenance. Int. J. Mol. Sci. 2019, 20, 3076. [Google Scholar] [CrossRef]
- Xiao, Y.; Sun, Y.; Liu, W.; Zeng, F.; Shi, J.; Li, J.; Chen, H.T.C.; Xu, Y.; Tan, Z.; Gong, F.; et al. HMGB1 Promotes the Release of Sonic Hedgehog from Astrocytes. Front. Immunol. 2021, 12, 584097. [Google Scholar] [CrossRef] [PubMed]
- Mora, P.; Hollier, P.-L.; Guimbal, S.; Abelanet, A.; Diop, A.; Cornuault, L.; Couffinhal, T.; Horng, S.; Gadeau, A.P.; Renault, M.A.; et al. Blood–brain barrier genetic disruption leads to protective barrier formation at the Glia Limitans. PLoS Biol. 2020, 18, e3000946. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Sako, K.; Minami, T.; Noda, K.; Kim, H.Z.; Kodama, T.; Shibuya, M.; Takakura, N.; Koh, G.Y.; Mochizuki, N. Differential function of Tie2 at cell–cell contacts and cell–substratum contacts regulated by angiopoietin-1. Nat. Cell Biol. 2008, 10, 513–526. [Google Scholar] [CrossRef]
- Meng, Z.; Li, M.; He, Q.; Jiang, S.; Zhang, X.; Xiao, J.; Bai, Y. Ectopic expression of human angiopoietin-1 promotes functional recovery and neurogenesis after focal cerebral ischemia. Neuroscience 2014, 267, 135–146. [Google Scholar] [CrossRef]
- Brindle, N.P.; Saharinen, P.; Alitalo, K. Signaling and Functions of Angiopoietin-1 in Vascular Protection. Circ. Res. 2006, 98, 1014–1023. [Google Scholar] [CrossRef]
- Gavard, J.; Patel, V.; Gutkind, J.S. Angiopoietin-1 Prevents VEGF-Induced Endothelial Permeability by Sequestering Src through mDia. Dev. Cell 2008, 14, 25–36. [Google Scholar] [CrossRef]
- Xia, Y.-P.; He, Q.-W.; Li, Y.-N.; Chen, S.-C.; Huang, M.; Wang, Y.; Gao, Y.; Huang, Y.; Wang, M.-D.; Mao, L.; et al. Recombinant Human Sonic Hedgehog Protein Regulates the Expression of ZO-1 and Occludin by Activating Angiopoietin-1 in Stroke Damage. Zhou R, editor. PLoS ONE 2013, 8, e68891. [Google Scholar] [CrossRef]
- Siddiqui, M.R.; Mayanil, C.S.; Kim, K.S.; Tomita, T. Angiopoietin-1 Regulates Brain Endothelial Permeability through PTPN-2 Mediated Tyrosine Dephosphorylation of Occludin. Johnson R, editor. PLoS ONE 2015, 10, e0130857. [Google Scholar] [CrossRef]
- Shen, F.; Walker, E.J.; Jiang, L.; Degos, V.; Li, J.; Sun, B.; Heriyanto, F.; Young, W.L.; Su, H. Coexpression of Angiopoietin-1 with VEGF Increases the Structural Integrity of the Blood–Brain Barrier and Reduces Atrophy Volume. J. Cereb. Blood Flow Metab. 2011, 31, 2343–2351. [Google Scholar] [CrossRef]
- Nourhaghighi, N.; Teichert-Kuliszewska, K.; Davis, J.; Stewart, D.J.; Nag, S. Altered Expression of Angiopoietins During Blood-Brain Barrier Breakdown and Angiogenesis. Lab. Investig. 2003, 83, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Wosik, K.; Cayrol, R.; Dodelet-Devillers, A.; Berthelet, F.; Bernard, M.; Moumdjian, R.; Bouthillier, A.; Reudelhuber, T.L.; Prat, A. Angiotensin II Controls Occludin Function and Is Required for Blood–Brain Barrier Maintenance: Relevance to Multiple Sclerosis. J. Neurosci. 2007, 27, 9032–9042. [Google Scholar] [CrossRef] [PubMed]
- Mizee, M.R.; Nijland, P.G.; van der Pol, S.M.A.; Drexhage, J.A.R.; Hof, B.v.H.; Mebius, R.; van der Valk, P.; van Horssen, J.; Reijerkerk, A.; de Vries, H.E. Astrocyte-derived retinoic acid: A novel regulator of blood–brain barrier function in multiple sclerosis. Acta Neuropathol. 2014, 128, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Wang, Y.; Wang, X.J.; Wang, X.T.; Zhao, Y.; Wang, L.M.; Chen, Z.Y. Retinoic acid ameliorates blood–brain barrier disruption following ischemic stroke in rats. Pharmacol. Res. 2015, 99, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Gille, J.; Paxton, L.L.; Lawley, T.J.; Caughman, S.W.; Swerlick, R.A. Retinoic acid inhibits the regulated expression of vascular cell adhesion molecule-1 by cultured dermal microvascular endothelial cells. J. Clin. Investig. 1997, 99, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Al-Ahmad, A.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 2014, 4, 4160. [Google Scholar] [CrossRef]
- Bonney, S.; Dennison, B.J.C.; Wendlandt, M.; Siegenthaler, J.A. Retinoic Acid Regulates Endothelial β-catenin Expression and Pericyte Numbers in the Developing Brain Vasculature. Front. Cell. Neurosci. 2018, 12, 476. [Google Scholar] [CrossRef]
- Guérit, S.; Fidan, E.; Macas, J.; Czupalla, C.J.; Figueiredo, R.; Vijikumar, A.; Yalcin, B.H.; Thom, S.; Winter, P.; Gerhardt, H.; et al. Astrocyte-derived Wnt growth factors are required for endothelial blood-brain barrier maintenance. Prog. Neurobiol. 2021, 199, 101937. [Google Scholar] [CrossRef]
- Manukjan, N.; Ahmed, Z.; Fulton, D.; Blankesteijn, W.M.; Foulquier, S. A Systematic Review of WNT Signaling in Endothelial Cell Oligodendrocyte Interactions: Potential Relevance to Cerebral Small Vessel Disease. Cells 2020, 9, 1545. [Google Scholar] [CrossRef]
- Bake, S.; Okoreeh, A.K.; Alaniz, R.C.; Sohrabji, F. Insulin-Like Growth Factor (IGF)-I Modulates Endothelial Blood-Brain Barrier Function in Ischemic Middle-Aged Female Rats. Endocrinology 2016, 157, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Igarashiac, Y.; Utsumi, H.; Chibaa, H.; Yamada-Sasamori, Y.; Tobiokaa, H.; Kamimura, Y.; Furuuchi, K.; Kokai, Y.; Nakagawa, T.; Mori, M.; et al. Glial Cell Line-Derived Neurotrophic Factor Induces Barrier Function of Endothelial Cells Forming the Blood–Brain Barrier. Biochem. Biophys. Res. Commun. 1999, 261, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Hoi, M.P.M. Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease. Neural Regen. Res. 2023, 18, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, L.; Liu, P.; Ji, X.; Qi, X.; Wang, Z.; Chi, T.; Zou, L. Sigma-1 receptor activation alleviates blood-brain barrier disruption post cerebral ischemia stroke by stimulating the GDNF-GFRα1-RET pathway. Exp. Neurol. 2022, 347, 113867. [Google Scholar] [CrossRef]
- Kang, W.; Balordi, F.; Su, N.; Chen, L.; Fishell, G.; Hébert, J.M. Astrocyte activation is suppressed in both normal and injured brain by FGF signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E2987–E2995. Available online: https://pnas.org/doi/full/10.1073/pnas.1320401111 (accessed on 26 March 2023). [CrossRef]
- Klimaschewski, L.; Claus, P. Fibroblast Growth Factor Signalling in the Diseased Nervous System. Mol. Neurobiol. 2021, 58, 3884–3902. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Z.; Cheng, Y.; Kardami, E.; Loh, Y.P. Low and High Molecular Weight FGF-2 Have Differential Effects on Astrocyte Proliferation, but Are Both Protective Against Aβ-Induced Cytotoxicity. Front. Mol. Neurosci. 2020, 12, 328. [Google Scholar] [CrossRef]
- Kang, K.; Lee, S.-W.; Han, J.E.; Choi, J.W.; Song, M.-R. The complex morphology of reactive astrocytes controlled by fibroblast growth factor signaling: Reactive Astrocyte Morphology Involves FGF. Glia 2014, 62, 1328–1344. [Google Scholar] [CrossRef]
- Reuss, B.; Dono, R.; Unsicker, K. Functions of Fibroblast Growth Factor (FGF)-2 and FGF-5 in Astroglial Differentiation and Blood-Brain Barrier Permeability: Evidence from Mouse Mutants. J. Neurosci. 2003, 23, 6404–6412. [Google Scholar] [CrossRef]
- Wang, Z.-G.; Cheng, Y.; Yu, X.-C.; Ye, L.-B.; Xia, Q.-H.; Johnson, N.R.; Wei, X.; Chen, D.-Q.; Cao, G.; Fu, X.-B.; et al. bFGF Protects Against Blood-Brain Barrier Damage Through Junction Protein Regulation via PI3K-Akt-Rac1 Pathway Following Traumatic Brain Injury. Mol. Neurobiol. 2016, 53, 7298–7311. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [PubMed]
- Hussain, B.; Fang, C.; Chang, J. Blood–Brain Barrier Breakdown: An Emerging Biomarker of Cognitive Impairment in Normal Aging and Dementia. Front. Neurosci. 2021, 15, 688090. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Nikolakopoulou, A.M.; Wang, Y.; Ma, Q.; Sagare, A.P.; Montagne, A.; Huuskonen, M.T.; Rege, S.V.; Kisler, K.; Dai, Z.; Körbelin, J.; et al. Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J. Exp. Med. 2021, 218, e20202207. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Chang, M.S.; Koh, S.H.; Choi, Y.K. Repair Mechanisms of the Neurovascular Unit after Ischemic Stroke with a Focus on VEGF. Int. J. Mol. Sci. 2021, 22, 8543. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- Baumann, J.; Tsao, C.-C.; Huang, S.-F.; Gassmann, M.; Ogunshola, O.O. Astrocyte-specific hypoxia-inducible factor 1 (HIF-1) does not disrupt the endothelial barrier during hypoxia in vitro. Fluids Barriers CNS 2021, 18, 13. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.V.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Patiño-López, G.; Gómez-Lira, G.; Gutiérrez, R.; Becerril-Pérez, F.; Rosales-Córdova, A.; León-Contreras, J.C.; Hernández-Pando, R.; León-Rivera, I.; Soto-Cruz, I.; et al. Activation of STAT3 Regulates Reactive Astrogliosis and Neuronal Death Induced by AβO Neurotoxicity. Int. J. Mol. Sci. 2020, 21, 7458. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, X.; Miao, C.; Chen, J. Propofol attenuated TNF-α-modulated occludin expression by inhibiting Hif-1α/ VEGF/ VEGFR-2/ ERK signaling pathway in hCMEC/D3 cells. BMC Anesthesiol. 2019, 19, 127. [Google Scholar] [CrossRef]
- Sharp, C.D.; Hines, I.; Houghton, J.; Warren, A.; Jackson, T.H.; Jawahar, A.; Nanda, A.; Elrod, J.W.; Long, A.; Chi, A.; et al. Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am. J. Physiol. Circ. Physiol. 2003, 285, H2592–H2598. [Google Scholar] [CrossRef]
- Liu, X.; Su, P.; Meng, S.; Aschner, M.; Cao, Y.; Luo, W.; Zheng, L.; Liu, M. Role of matrix metalloproteinase-2/9 (MMP2/9) in lead-induced changes in an in vitro blood-brain barrier model. Int. J. Biol. Sci. 2017, 13, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.-J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.F.; Turk, J.; et al. Matrix Metalloproteinases Expressed by Astrocytes Mediate Extracellular Amyloid-β Peptide Catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Xu, L.; Wang, X.; Sun, X. MMP-9 expression and activity is concurrent with endothelial cell apoptosis in the basilar artery after subarachnoid hemorrhaging in rats. Neurol. Sci. 2015, 36, 1241–1245. [Google Scholar] [CrossRef]
- Lee, S.-R.; Lo, E.H. Induction of Caspase-Mediated Cell Death by Matrix Metalloproteinases in Cerebral Endothelial Cells after Hypoxia—Reoxygenation. J. Cereb. Blood Flow Metab. 2004, 24, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.; Soldner, E.L.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix Metalloproteinase-Mediated Blood-Brain Barrier Dysfunction in Epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- D’Orléans-Juste, P.; Ndunge, O.B.A.; Desbiens, L.; Tanowitz, H.B.; Desruisseaux, M.S. Endothelins in inflammatory neurological diseases. Pharmacol. Ther. 2019, 194, 145–160. [Google Scholar] [CrossRef]
- Wang, H.H.; Hsieh, H.L.; Wu, C.Y.; Yang, C.M. Endothelin-1 enhances cell migration via matrix metalloproteinase-9 up-regulation in brain astrocytes. J. Neurochem. 2010, 113, 1133–1149. Available online: https://onlinelibrary.wiley.com/doi/10.1111/j.1471-4159.2010.06680.x (accessed on 23 March 2023). [CrossRef]
- Koyama, Y.; Michinaga, S. Regulations of Astrocytic Functions by Endothelins: Roles in the Pathophysiological Responses of Damaged Brains. J. Pharmacol. Sci. 2012, 118, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Meng, X.; Wang, L.; Ren, S.; Matei, N.; Wu, G. Mitigating the effects of Endothelin-1 following a minimally invasive surgery reduces the blood-brain barrier permeability in a rabbit model of intracerebral hemorrhage. Brain Hemorrhages 2022, 3, 177–183. [Google Scholar] [CrossRef]
- Czigler, A.; Toth, L.; Szarka, N.; Szilágyi, K.; Kellermayer, Z.; Harci, A.; Vecsernyes, M.; Ungvari, Z.; Szolics, A.; Koller, A.; et al. Prostaglandin E2, a postulated mediator of neurovascular coupling, at low concentrations dilates whereas at higher concentrations constricts human cerebral parenchymal arterioles. Prostaglandins Other Lipid Mediat. 2020, 146, 106389. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Hsieh, H.L.; Chi, P.L.; Yang, C.C.; Hsiao, L.D.; Yang, C.M. Upregulation of COX-2/PGE2 by ET-1 mediated through Ca2+-dependent signals in mouse brain microvascular endothelial cells. Mol. Neurobiol. 2014, 49, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Persaud, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic Inhibition of Astroglial NF-κB Improves Functional Outcome in Experimental Autoimmune Encephalomyelitis by Suppressing Chronic Central Nervous System Inflammation. J. Immunol. 2009, 182, 2628–2640. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Wang, Q.; Mergia, E.; Koesling, D.; Mittmann, T. Nitric Oxide/Cyclic Guanosine Monophosphate Signaling via Guanylyl Cyclase Isoform 1 Mediates Early Changes in Synaptic Transmission and Brain Edema Formation after Traumatic Brain Injury. J. Neurotrauma 2021, 38, 1689–1701. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, Z.-Y.; Huang, T.-T.; Zhou, Y.-X.; Wang, X.; Yang, L.-Q.; Chen, Z.-A.; Yu, W.-F.; Li, P.-Y. The peripheral immune response after stroke-A double edge sword for blood-brain barrier integrity. CNS Neurosci. Ther. 2018, 24, 1115–1128. [Google Scholar] [CrossRef]
- Bonney, S.; Seitz, S.; Ryan, C.A.; Jones, K.L.; Clarke, P.; Tyler, K.L.; Siegenthaler, J.A. Gamma Interferon Alters Junctional Integrity via Rho Kinase, Resulting in Blood-Brain Barrier Leakage in Experimental Viral Encephalitis. Mbio 2019, 10, e01675-19. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hu, Y.; Li, H.; Huang, X.; Zheng, H.; Hu, Y.; Wang, J.; Jiang, X.; Li, J.; Yang, Z.; et al. miR-1303 regulates BBB permeability and promotes CNS lesions following CA16 infections by directly targeting MMP9. Emerg. Microbes Infect. 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; A Trauger, S.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Barroso, A.; Mahler, J.V.; Fonseca-Castro, P.H.; Quintana, F.J. The aryl hydrocarbon receptor and the gut–brain axis. Cell. Mol. Immunol. 2021, 18, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Aveleira, C.A.; Lin, C.-M.; Abcouwer, S.F.; Ambrósio, A.F.; Antonetti, D.A. TNF-α Signals Through PKCζ/NF-κB to Alter the Tight Junction Complex and Increase Retinal Endothelial Cell Permeability. Diabetes 2010, 59, 2872–2882. [Google Scholar] [CrossRef]
- Ridder, D.A.; Wenzel, J.; Müller, K.; Töllner, K.; Tong, X.-K.; Assmann, J.C.; Stroobants, S.; Weber, T.; Niturad, C.; Fischer, L.; et al. Brain endothelial TAK1 and NEMO safeguard the neurovascular unit. J. Exp. Med. 2015, 212, 1529–1549. [Google Scholar] [CrossRef]
- Rakers, C.; Schleif, M.; Blank, N.; Matušková, H.; Ulas, T.; Händler, K.; Torres, S.V.; Schumacher, T.; Tai, K.; Schultze, J.L.; et al. Stroke target identification guided by astrocyte transcriptome analysis. Glia 2019, 67, 619–633. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Wang, Z.; Zhang, X.; Yao, L.; Wang, F.; Liu, S.; Yin, J.; Ling, E.A.; Wang, L.; et al. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience 2012, 202, 58–68. [Google Scholar] [CrossRef]
- Liu, X.; Tian, Y.; Lu, N.; Gin, T.; Cheng, C.H.K.; Chan, M.T.V. Stat3 Inhibition Attenuates Mechanical Allodynia through Transcriptional Regulation of Chemokine Expression in Spinal Astrocytes. PLoS ONE 2013, 8, e75804. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Leng, K.; Park, J.; Sorets, A.G.; Kim, S.; Shostak, A.; Embalabala, R.J.; Mlouk., K.; Katdare, K.A.; Rose, I.V.L.; et al. Reactive astrocytes transduce inflammation in a blood-brain barrier model through a TNF-STAT3 signaling axis and secretion of alpha 1-antichymotrypsin. Nat. Commun. 2022, 13, 6581. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Freed, D.M.; Schlessinger, J.; Kiyatkin, A. The Dark Side of Cell Signaling: Positive Roles for Negative Regulators. Cell 2016, 164, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.A.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins Are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef]
- Tong, M.; Jiang, Y. FK506-Binding Proteins and Their Diverse Functions. Curr. Mol. Pharmacol. 2015, 9, 48–65. [Google Scholar] [CrossRef]
- Park, Y.-J.; Yoo, S.-A.; Kim, M.; Kim, W.-U. The Role of Calcium–Calcineurin–NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef]
- Sompol, P.; Norris, C.M. Ca2+, Astrocyte Activation and Calcineurin/NFAT Signaling in Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10, 199. [Google Scholar] [CrossRef]
- Chang, C.; Li, J.; Chen, W.; Ou, Y.; Lai, C.; Hu, Y.; Wu, C.C.; Chang, C.J.; Chen, C.J. Disruption of in vitro endothelial barrier integrity by J apanese encephalitis virus-Infected astrocytes. Glia 2015, 63, 1915–1932. [Google Scholar] [CrossRef]
- Deng, Z.; Zhou, L.; Wang, Y.; Liao, S.; Huang, Y.; Shan, Y.; Tan, S.; Zeng, Q.; Peng, L.; Huang, H.; et al. Astrocyte-derived VEGF increases cerebral microvascular permeability under high salt conditions. Aging 2020, 12, 11781–11793. [Google Scholar] [CrossRef]
- Hudson, L.C.; Bragg, D.C.; Tompkins, M.B.; Meeker, R.B. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res. 2005, 1058, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Ghorpade, A.; Rasmussen, J.; Limoges, J.; Liu, X.J.; Stins, M.; Fiala, M.; Way, D.; Kim, K.S.; Witte, M.H.; et al. Microglial and Astrocyte Chemokines Regulate Monocyte Migration through the Blood-Brain Barrier in Human Immunodeficiency Virus-1 Encephalitis. Am. J. Pathol. 1999, 155, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Ãvila, M.; Gonzalez, J.; El-Bachá, R.S.; Báez, E.; Garcia-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 211. Available online: http://journal.frontiersin.org/article/10.3389/fncel.2014.00211 (accessed on 27 October 2023). [CrossRef] [PubMed]
- Liu, C.-Y.; Yang, Y.; Ju, W.-N.; Wang, X.; Zhang, H.-L. Emerging Roles of Astrocytes in Neuro-Vascular Unit and the Tripartite Synapse with Emphasis on Reactive Gliosis in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Rothhammer, V. Protective Functions of Reactive Astrocytes Following Central Nervous System Insult. Front. Immunol. 2020, 11, 573256. [Google Scholar] [CrossRef] [PubMed]
- Myer, D.J. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 2006, 129, 2761–2772. [Google Scholar] [CrossRef]
- Bayraktar, O.A.; Bartels, T.; Holmqvist, S.; Kleshchevnikov, V.; Martirosyan, A.; Polioudakis, D.; Ben Haim, L.; Young, A.M.H.; Batiuk, M.Y.; Prakash, K.; et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 2020, 23, 500–509. [Google Scholar] [CrossRef]
- Lozzi, B.; Huang, T.-W.; Sardar, D.; Huang, A.Y.-S.; Deneen, B. Regionally Distinct Astrocytes Display Unique Transcription Factor Profiles in the Adult Brain. Front. Neurosci. 2020, 14, 61. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Quintana, F.J. Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a029009. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef]
- Moulson, A.J.; Squair, J.W.; Franklin, R.J.M.; Tetzlaff, W.; Assinck, P. Diversity of Reactive Astrogliosis in CNS Pathology: Heterogeneity or Plasticity? Front. Cell. Neurosci. 2021, 15, 703810. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.A.S.; Grandbarbe, L.; Buckley, N.J.; Niclou, S.P.; Michelucci, A. Transcriptional and epigenetic mechanisms underlying astrocyte identity. Prog. Neurobiol. 2019, 174, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, A.; Vidos, C.; Busso, M.M.; Cieri, M.B.; Ramos, A.J. Pathological Neuroinflammatory Conversion of Reactive Astrocytes Is Induced by Microglia and Involves Chromatin Remodeling. Front. Pharmacol. 2021, 12, 689346. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef]
- Dhar, G.A.; Saha, S.; Mitra, P.; Chaudhuri, R.N. DNA methylation and regulation of gene expression: Guardian of our health. The Nucleus 2021, 64, 259–270. [Google Scholar] [CrossRef]
- Welle, A.; Kasakow, C.V.; Jungmann, A.M.; Gobbo, D.; Stopper, L.; Nordström, K.; Salhab, A.; Gasparoni, G.; Scheller, A.; Kirchhoff, F.; et al. Epigenetic control of region-specific transcriptional programs in mouse cerebellar and cortical astrocytes. Glia 2021, 69, 2160–2177. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.-D.; Xia, Y.-P.; Gao, Y.; Zhu, Y.-Y.; Chen, S.-C.; Mao, L.; He, Q.W.; Yue, Z.Y.; Hu, B. MicroRNA-130a regulates cerebral ischemia–induced blood–brain barrier permeability by targeting Homeobox A5. FASEB J. 2018, 32, 935–944. [Google Scholar] [CrossRef]
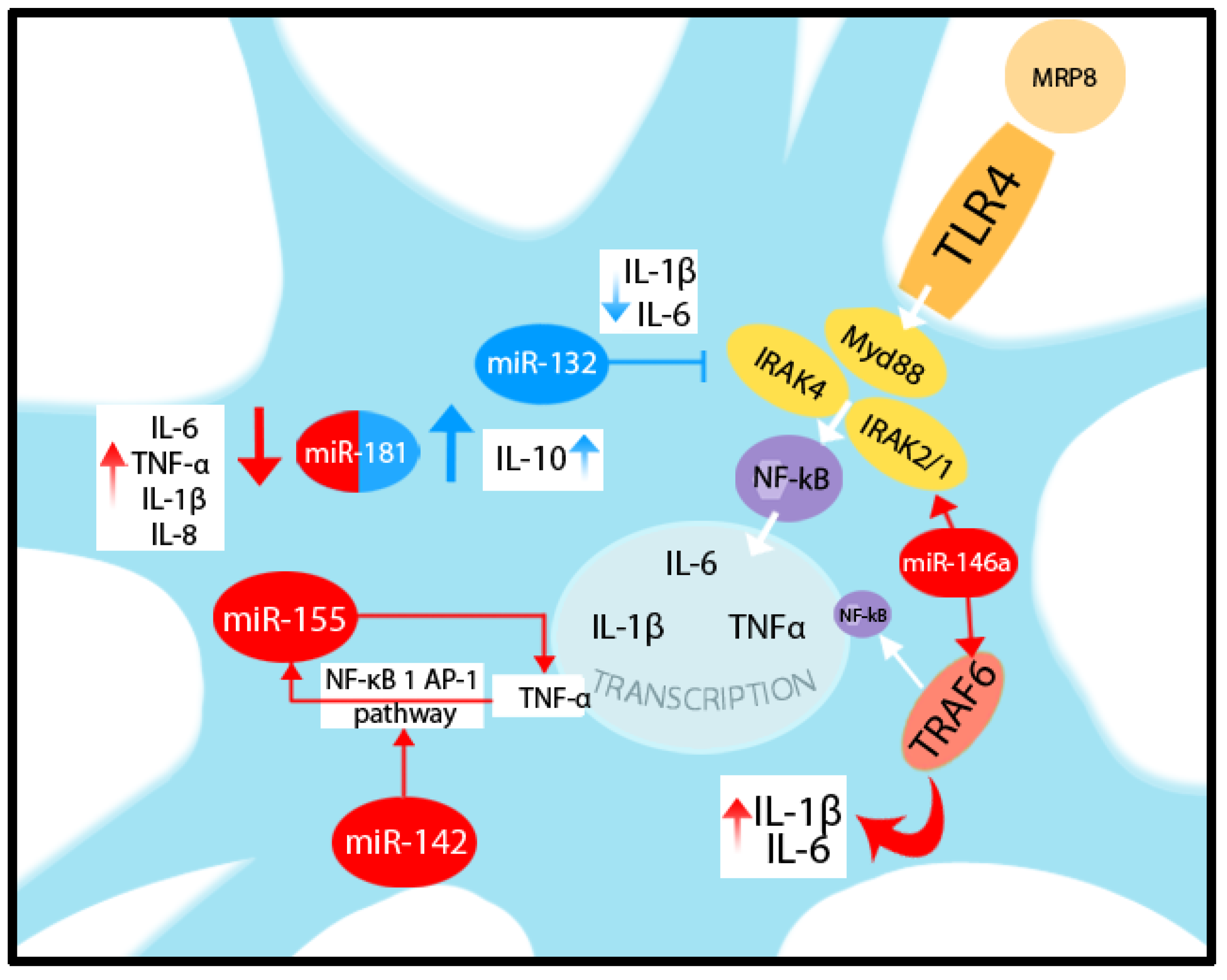
- Kong, H.; Yin, F.; He, F.; Omran, A.; Li, L.; Wu, T.; Wang, Y.; Peng, J. The Effect of miR-132, miR-146a, and miR-155 on MRP8/TLR4-Induced Astrocyte-Related Inflammation. J. Mol. Neurosci. 2015, 57, 28–37. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Guedes, J.R.; de Almeida, L.P.; de Lima, M.C.P. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production: miR-155 role during microglia activation. Immunology 2012, 135, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, L. Inflamma-MicroRNAs in Alzheimer’s Disease: From Disease Pathogenesis to Therapeutic Potentials. Front. Cell. Neurosci. 2021, 15, 785433. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, A.; Puhakka, N.; Das Gupta, S.; Vuokila, N.; Broekaart, D.W.M.; Anink, J.J.; Heiskanen, M.; Karttunen, J.; van Scheppingen, J.; Huitinga, I.; et al. Increased expression of miR142 and miR155 in glial and immune cells after traumatic brain injury may contribute to neuroinflammation via astrocyte activation. Brain Pathol. 2020, 30, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H.; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes: miR-181 and Astrocyte Inflammatory Responses. Glia 2013, 61, 1018–1028. [Google Scholar] [CrossRef]
- Wu, L.; Ye, Z.; Pan, Y.; Li, X.; Fu, X.; Zhang, B.; Li, Y.; Lin, W.; Li, X.; Gao, Q. Vascular endothelial growth factor aggravates cerebral ischemia and reperfusion-induced blood-brain-barrier disruption through regulating LOC102640519/HOXC13/ZO-1 signaling. Exp. Cell Res. 2018, 369, 275–283. [Google Scholar] [CrossRef]
- Chen, C.; Chao, Y.; Lin, H.; Chen, C.; Chen, C.; Yang, J.; Chan, J.Y.H.; Juo, S.-H.H. miR-195 reduces age-related blood–brain barrier leakage caused by thrombospondin-1-mediated selective autophagy. Aging Cell 2020, 19, e13236. Available online: https://onlinelibrary.wiley.com/doi/10.1111/acel.13236 (accessed on 5 April 2023). [CrossRef]
- Zhao, S.; Sheng, S.; Wang, Y.; Ding, L.; Xu, X.; Xia, X.; Zheng, J.C. Astrocyte-derived extracellular vesicles: A double-edged sword in central nervous system disorders. Neurosci. Biobehav. Rev. 2021, 125, 148–159. [Google Scholar] [CrossRef]
- Jovičić, A.; Gitler, A.D. Distinct repertoires of microRNAs present in mouse astrocytes compared to astrocyte-secreted exosomes. PLoS ONE 2017, 12, e0171418. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors Promoting BBB Integrity | Mechanism of Action in Brain ECs | References |
|---|---|---|
| Sonic Hedgehog | Activates PTCH1 and SMO followed by GLI1 nuclear translocation. Increases expression of junctional proteins claudin-3, -5, occludin, junction adhesion molecule-A, VE-cadherin, p120, and laminin expression. Decreases the level of microglial TNF-α, IL-6, and IL-1β, CCL2, and of ICAM-1 in ECs. | [23,24,27,28,30,85] |
| Angiopoetin-1 | Binds on angiopoietin-1 receptor Tie2, activation of PI3K /AKT-myocyte enhancer factor-2 (MEF2)-Krüppel-like factor 2 (KLF2) pathway stabilizes TJ and AJ. Counteracts VEGF-induced endothelial permeability by inhibiting phosphorylation of VE-cadherin. Up-regulates occludin and ZO-1 expression and prevents occludin phosphorylation, favoring occludin interaction with ZO-1. Suppresses VEGF-induced expression of ICAM-1 and VCAM-1. | [33,34,35,36,37,38,39] |
| Retinoic acid | Interacts with RAR in ECs, interferes with ShH and Wnt pathways. Increases expression of ZO-1, occludin, claudin-5, and VE-cadherin. Activates nuclear factor erythroid 2-related factor 2 (NRF2) pathway, which results in ICAM-1 and VCAM-1 decreased expression in BMECs. | [43,44,45,46,47,48,81] |
| Wnt Growth Factors | WNT/β-catenin pathway activation and nuclear translocation of β-catenin increase endothelial expression of claudin-3, claudin-5, and occludin reducing BBB permeability. Decrease caveolin-1 expression and decreased transcellular vesicular traffic in brain ECs. | [49,50,86] |
| Glial-derived Neurotrophic Factor | Binds on GDNF family receptor α1 and RET receptor kinase, activates sirtuin 1/ eNOS, PI3K/Akt, cAMP/PKA pathway and inactivates p38 MAPK, preventing TJ protein degradation; increases claudin-5, occludin and ZO-1 expression. | [52,53,54] |
| Fibroblast Growth Factor | Binds to FGFR1 and activates S1PR1, ERK, and PI3K/AKT/Rac-1 pathways preventing TJ and AJ proteins degradation. Increases astrocytic proliferation, influences astrocyte morphology. | [55,56,57,58,59,60,61] |
| Apolipoprotein E3 | APOE4 binds on LRP1 in pericytes and induces MMP-9 secretion from pericyte via CypA/NFκB pathway. ApoE3 counteracts BBB disruption through MMP-9 activation by ApoE4. | [62,63,64] |
| Factors promoting BBB permeability | ||
| Vascular Endothelial Growth Factor (VEGF) | Activation of nuclear factor κB (NF-κB) by TLRs and RAGE or activation of JAK2/STAT3/ HIF1α pathway are involved in VEGF production in astrocytes. Activation of HIF1α/VEGF/VEGFR2/ERK pathway is involved in the TNF-α-induced down-regulation of TJ proteins. IL-1β induces HIF1α-mediated VEGF expression. VEGF signaling increases BBB permeability through PI3K/Akt/eNOS, PLCγ/ PKC/ ERK, p38, and Src pathways, resulting in TJ-related protein down-regulation (occludin and claudin-5) and eNOS up-regulation. VEGF induces expressions of ICAM-1, VCAM-1, and MMP-9. eNOS activation and NO produce TJ and AJ complex disruption. Activation of Src and focal adhesion kinase result in phosphorylation and internalization of the VE-Cadherin and AJ breakdown. | [40,65,66,67,68,69,70] |
| Nitric oxide (NO) Reactive oxygen species (ROS) | NO production in reactive astrocytes after iNOS up-regulation is followed by activation of GMP/PKG and endothelial TJ protein down-regulation. ROS lead to arachidonic acids-mediated MMP activation and increased cytokine production. ROS induce down-regulation and degradation of TJ-related proteins through activation of the ROCK /MLC /MLCK pathway. | [61,80,86,87,88,89] |
| Matrix metalloproteinases (MMPs) | MMPs degrade the extracellular matrix proteins (collagen, fibronectin, and laminin), and TJ-related proteins. Astrocytic MMP-2 and MMP-9 stimulate NF-κB activation, leading to chemokine expression. | [61,72,73,74,75,76,77,90] |
| Glutamate | Decreased glutamate reuptake in reactive astrocytes results in excessive extracellular glutamate and increased MMP-2 and MMP-9 expression. | [71,77,78,86] |
| Endothelins (ETs) and other vasoactive mediators | Endothelin-1 (ET-1), arachidonic acids, and arachidonic acids metabolite PGE2 are up-regulated in reactive astrocyte via PLA2 activation. PGE2 modulates BBB integrity and induces endothelial migration via cAMP/PKA pathway activation. Increased expression of ET-1 in reactive astrocytes impairs BBB integrity through endothelial MMP-2, -9, and VEGF up-regulation. ET-1 triggers astrocytic AQP4 down-regulation, and affects ECs contact with astrocytic end-feet. ET-1 binding on endothelial ET-A and ET-B results in NF-κB activation, vascular inflammation, increased PGE2 production via COX2 activation, and immune cell migration due to up-regulated ICAM-1, and VCAM-1, and E-selectin. | [79,80,81,82,83,84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. https://doi.org/10.3390/ijms242417146
Manu DR, Slevin M, Barcutean L, Forro T, Boghitoiu T, Balasa R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. International Journal of Molecular Sciences. 2023; 24(24):17146. https://doi.org/10.3390/ijms242417146
Chicago/Turabian StyleManu, Doina Ramona, Mark Slevin, Laura Barcutean, Timea Forro, Tudor Boghitoiu, and Rodica Balasa. 2023. "Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions" International Journal of Molecular Sciences 24, no. 24: 17146. https://doi.org/10.3390/ijms242417146
APA StyleManu, D. R., Slevin, M., Barcutean, L., Forro, T., Boghitoiu, T., & Balasa, R. (2023). Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. International Journal of Molecular Sciences, 24(24), 17146. https://doi.org/10.3390/ijms242417146