
Circulating Tumor DNA Is a Variant of Liquid Biopsy with Predictive and Prognostic Clinical Value in Breast Cancer Patients

 , , ,
, , ,
Abstract
:1. Introduction
1.1. The Current State of the Problem of Breast Cancer around the World
1.2. Determination of Circulating Tumor DNA Is a New Approach
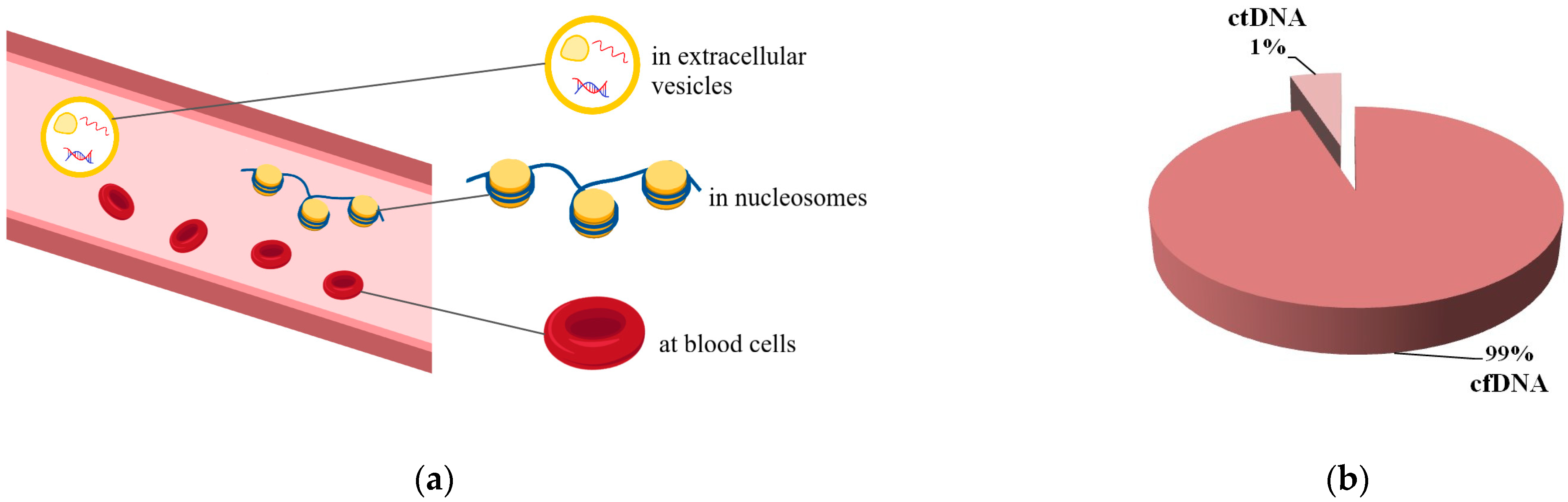
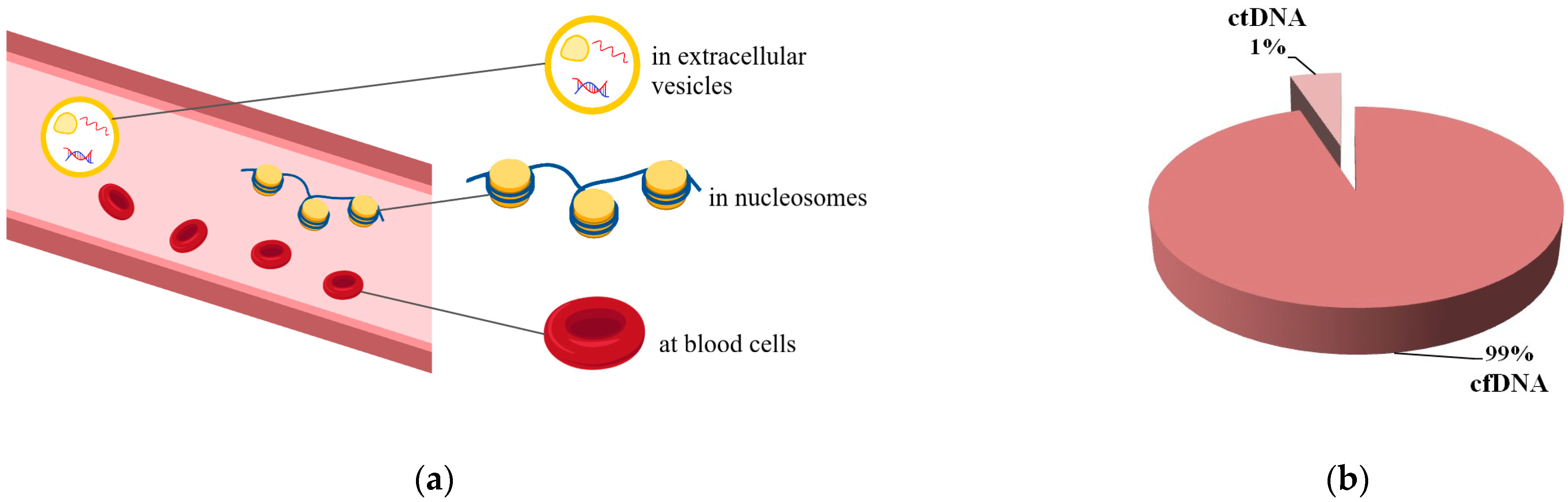
2. Circulating Tumor DNA: What Is It?
2.1. Circulating Tumor DNA Is a Variant of Liquid Biopsy
2.2. Circulating Tumor DNA and Its Main Features
2.3. Basic Methods for Studying ctDNA
2.3.1. PCR-Based Methods
2.3.2. NGS-Based Methods
2.3.3. Combined and New Approaches
2.4. Possible Applications of ctDNA in Oncology
3. Clinical Value of ctDNA in Breast Cancer Patients
3.1. Application of ctDNA for Early Detection/Screening of Breast Cancer
3.2. ctDNA as a Predictive and Prognostic Marker in Breast Cancer Treatment
3.2.1. Two Main Approaches to Determining ctDNA in Breast Cancer
3.2.2. Clinical Value of ctDNA Determination for Breast Cancer Treatment
Early Detection of Recurrence/Progression of Breast Cancer
ctDNA as a Surrogate Marker for Minimal Residual Disease after Primary Treatment for Breast Cancer
ctDNA Mutations as Prognostic and Predictive Factors for the Effectiveness of Therapy
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| BC | Breast Cancer |
| BEAMing | Beads, Emulsion, Amplification, Magnetics |
| CAPP-Seq | CAncer Personalized Profiling by deep Sequencing |
| cfDNA | Cell-Free DNA |
| cfNA | Cell-Free Nucleic Acids |
| CTC | Circulating tumor cells |
| ctDNA | Circulating tumor DNA |
| dPCR | Digital Polymerase Chain Reaction |
| ddPCR | digital droplet Polymerase Chain Reaction |
| DFS | Disease-Free Survival |
| HR | Hazard Ratio |
| MAF | Mutant Allele Fraction |
| MS-ddPCR | Methylation-Specific digital droplet PCR |
| MRD | Minimal Residual Disease |
| mTBI | Molecular Tumor Burden Index |
| NAC | Neoadjuvant Chemotherapy |
| NGS | Next Generation Sequencing |
| OS | Overall Survival |
| PCR | Polymerase Chain Reaction |
| pCR | Pathological Complete Response |
| PFS | Progression-Free Survival |
| SNV | Single Nucleotide Variants |
| WGS | Whole Genome Sequencing |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Jagsi, R.; King, T.A.; Burstein, H.J. Malignant Tumors of the Breast. In DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology, 12th ed.; DeVita, V.T., Lawrence, T.S., Rosenberg, S.A., Eds.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2023. [Google Scholar]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Jatoi, I.; Anderson, W.F.; Jeong, J.H.; Redmond, C.K. Breast cancer adjuvant therapy: Time to consider its time-dependent effects. J. Clin. Oncol. 2011, 29, 2301–2304. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of Precision Cancer Medicine: Evolution of the Treatment Paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Bennett, C.W.; Berchem, G.; Kim, Y.J.; El-Khoury, V. Cell-Free DNA and Next-Generation Sequencing in the Service of Personalized Medicine for Lung Cancer. Oncotarget 2016, 7, 71013–71035. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71. [Google Scholar] [CrossRef]
- Abate, R.E.; Frezzetti, D.; Maiello, M.R.; Gallo, M.; Camerlingo, R.; De Luca, A.; De Cecio, R.; Morabito, A.; Normanno, N. Next Generation Sequencing-Based Profiling of Cell Free DNA in Patients With Advanced non-Small Cell Lung Cancer: Advantages and Pitfalls. Cancers 2020, 12, 3804. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xu, T.; Wang, S.; Chang, H.; Yu, T.; Zhu, Y.; Chen, J. Liquid Biopsy Applications in the Clinic. Mol. Diagn. Ther. 2020, 24, 125–132. [Google Scholar] [CrossRef]
- Romero, A.; Serna-Blasco, R.; Calvo, V.; Provencio, M. Use of Liquid Biopsy in the Care of Patients With Non-Small Cell Lung Cancer. Curr. Treat. Options Oncol. 2021, 22, 86. [Google Scholar] [CrossRef]
- Villatoro, S.; Mayo-de-las-Casas, C.; Jordana-Ariza, N.; Viteri-Ramírez, S.; Garzón-Ibañez, M.; Moya-Horno, I.; García-Peláez, B.; González-Cao, M.; Malapelle, U.; Balada-Bel, A.; et al. Prospective Detection of Mutations in Cerebrospinal Fluid, Pleural Effusion, and Ascites of Advanced Cancer Patients to Guide Treatment Decisions. Mol. Oncol. 2019, 13, 2633–2645. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabieres, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Appierto, V.; Di Cosimo, S.; Reduzzi, C.; Pala, V.; Cappelletti, V.; Daidone, M.G. How to study and overcome tumor heterogeneity with circulating biomarkers: The breast cancer case. Semin. Cancer Biol. 2017, 44, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Rubis, G.D.; Krishnan, S.R.; Bebawy, M. Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol. Sci. 2019, 40, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, H. Next-Generation Sequencing in Liquid Biopsy: Cancer Screening and Early Detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, H.; An, K.; Liu, Z.; Hai, L.; Li, R.; Zhou, Y.; Zhao, W.; Jia, Y.; Wu, N.; et al. Whole-genome bisulfite sequencing analysis of circulating tumour DNA for the detection and molecular classification of cancer. Clin. Transl. Med. 2022, 12, e1014. [Google Scholar] [CrossRef]
- Parsons, H.A.; Rhoades, J.; Reed, S.C.; Gydush, G.; Ram, P.; Exman, P.; Xiong, K.; Lo, C.C.; Li, T.; Fleharty, M.; et al. Sensitive Detection of Minimal Residual Disease in Patients Treated for Early-Stage Breast Cancer. Clin. Cancer Res. 2020, 26, 2556–2564. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Swigart, L.B.; Wu, H.T.; Hirst, G.L.; Yau, C.; Wolf, D.M.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant treated breast cancer reflects response and survival. Ann. Oncol. 2021, 32, 229–239. [Google Scholar] [CrossRef]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef]
- Mandel, P.; Métais, P. Les acides nucléiques du plasma sanguin chez l’homme. Comptes Rendus Acad. Sci. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef]
- Esteller, M.; Sanchez-Cespedes, M.; Rosell, R.; Sidransky, D.; Baylin, S.B.; Herman, J.G. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59, 67–70. [Google Scholar] [PubMed]
- Silva, J.M.; Dominguez, G.; Garcia, J.M.; Gonzalez, R.; Villanueva, M.J.; Navarro, F.; Provencio, M.; San Martin, S.; España, P.; Bonilla, F. Presence of tumor DNA in plasma of breast cancer patients: Clinicopathological correlations. Cancer Res. 1999, 59, 3251–3256. [Google Scholar] [PubMed]
- Silva, J.M.; Dominguez, G.; Villanueva, M.J.; Gonzalez, R.; Garcia, J.M.; Corbacho, C.; Provencio, M.; España, P.; Bonilla, F. Aberrant DNA methylation of the p16INK4a gene in plasma DNA of breast cancer patients. Br. J. Cancer. 1999, 80, 1262–1264. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Gonzalez, R.; Dominguez, G.; Garcia, J.M.; Espana, P.; Bonilla, F. TP53 gene mutations in plasma DNA of cancer patients. Genes Chromosomes Cancer 1999, 24, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.; Pantel, K.; Muller, V.; Rack, B.; Kasimir-Bauer, S.; Janni, W.; Schwarzenbach, H. Apoptosis-related deregulation of proteolytic activities and high serum levels of circulating nucleosomes and DNA in blood correlate with breast cancer progression. BMC Cancer 2011, 11, 4. [Google Scholar] [CrossRef]
- Marsman, G.; Zeerleder, S.; Luken, B.M. Extracellular histones, cell-free DNA, or nucleosomes: Differences in immunostimula-tion. Cell Death Dis. 2016, 7, e2518. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, Structures, and Functions of Circulating DNA in Oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Fiala, C.; Diamandis, E.P. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018, 16, 166. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Danesi, R.; Lo, Y.M.D.; Oellerich, M.; Beck, J.; Galbiati, S.; Re, M.D.; Lianidou, E.; Neumaier, M.; van Schaik, R.H.N. What do we need to obtain high quality circulating tumor DNA (ctDNA) for routine diagnostic test in oncology?—Considerations on pre-analytical aspects by the IFCC workgroup cfDNA. Clin. Chim. Acta 2021, 520, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, A.M.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N. Pathophysiology of ctDNA Release into the Circulation and Its Characteristics: What Is Important for Clinical Applications. Recent Results Cancer Res. 2020, 215, 163–180. [Google Scholar] [CrossRef]
- Thijssen, M.A.; Swinkels, D.W.; Ruers, T.J.; de Kok, J.B. Difference between free circulating plasma and serum DNA in patients with colorectal liver metastases. Anticancer Res. 2002, 22, 421–425. [Google Scholar]
- Lui, Y.Y.; Chik, K.W.; Chiu, R.W.; Ho, C.Y.; Lam, C.W.; Lo, Y.M.D. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [PubMed]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Risberg, B.; Tsui, D.W.; Biggs, H.; de Almagro, A.R.-V.M.; Dawson, S.J.; Hodgkin, C.; Jones, L.; Parkinson, C.; Piskorz, A.; Marass, F.; et al. Effects of Collection and Processing Procedures on Plasma Circulating Cell-Free DNA from Cancer Patients. J. Mol. Diagn. 2018, 20, 883–892. [Google Scholar] [CrossRef]
- Panabières, C.A.; Pantel, K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef]
- Fackler, M.J.; Tulac, S.; Venkatesan, N.; Aslam, A.J.; de Guzman, T.N.; Mercado-Rodriguez, C.; Cope, L.M.; Downs, B.M.; Vali, A.H.; Ding, W.; et al. Development of an automated liquid biopsy assay for methylated markers in advanced breast cancer. Cancer Res. Commun. 2022, 2, 391–401. [Google Scholar] [CrossRef]
- Moss, J.; Zick, A.; Grinshpun, A.; Carmon, E.; Maoz, M.; Ochana, B.L.; Abraham, O.; Arieli, O.; Germansky, L.; Meir, K.; et al. Circulating breast-derived DNA allows universal detection and monitoring of localized breast cancer. Ann. Oncol. 2020, 31, 395–403. [Google Scholar] [CrossRef]
- Pierga, J.-Y.; Silveira, A.; Girard, E.; Lorgis, V.; Tanguy, M.-L.; Albaud, B.; Tredan, O.; Dubot, C.; Hego, C.; Jacot, W.; et al. Abstract 3390: Predictive and prognostic value of circulating tumor DNA (ctDNA) compared to circulating tumor cells (CTC) in a prospective cohort of metastatic breast cancer patients: The UCBG COMET trial. Cancer Res. 2020, 80, 3390. [Google Scholar] [CrossRef]
- Kong, S.L.; Liu, X.; Tan, S.J.; Tai, J.A.; Phua, L.Y.; Poh, H.M.; Yeo, T.; Chua, Y.W.; Haw, Y.X.; Ling, W.H.; et al. Complementary Sequential Circulating Tumor Cell (CTC) and Cell-Free Tumor DNA (ctDNA) Profiling Reveals Metastatic Heterogeneity and Genomic Changes in Lung Cancer and Breast Cancer. Front. Oncol. 2021, 16, 698551. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wen, L.; Li, Z.; Xia, C. Analysis of Diagnostic Value of CTC and CTDNA in Early Lung Cancer. Cell Mol. Biol. 2023, 69, 57–62. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Atwal, S.; Liu, W.; Raja, R.; Fine, B.M.; Hughes, B.G.; Hicks, R.J.; Hampton, G.M.; Amler, L.C.; Pirzkall, A.; et al. Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: Association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib. Clin. Cancer Res. 2012, 18, 2391–2401. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Evans, I.; Jones, A.; Ghazali, S.; Reisel, D.; Ryan, A.; Gentry-Maharaj, A.; Zikan, M.; Cibula, D.; Eichner, J.; et al. Methylation patterns in serum DNA for early identification of disseminated breast cancer. Genome Med. 2017, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Kamel, A.M.; Teama, S.; Fawzy, A.; El Deftar, M. Plasma DNA integrity index as a potential molecular diagnostic marker for breast cancer. Tumour Biol. 2016, 37, 7565–7572. [Google Scholar] [CrossRef] [PubMed]
- Elhelaly, R.; Effat, N.; Hegazy, M.A.E.; Abdelwahab, K.; Hamdy, O.; Abo Hashem, E.M.; Elzehery, R.R. Circulating Cell Free DNA and DNA Integrity Index as Discriminating Tools between Breast Cancer and Benign Breast Disease. Asian Pac. J. Cancer Prev. 2022, 23, 545–552. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, W.; Su, L.; Sang, J.; Wang, S.; Yao, Y. Plasma cell-free DNA integrity: A potential biomarker to monitor the response of breast cancer to neoadjuvant chemotherapy. Transl. Cancer Res. 2019, 8, 1531–1539. [Google Scholar] [CrossRef]
- Pons-Belda, O.D.; Fernandez-Uriarte, A.; Diamandis, E.P. Can Circulating Tumor DNA Support a Successful Screening Test for Early Cancer Detection? The Grail Paradigm. Diagnostics 2021, 11, 2171. [Google Scholar] [CrossRef]
- Alese, O.B.; Cook, N.; Ortega-Franco, A.; Ulanja, M.B.; Tan, L.; Tie, J. Circulating Tumor DNA: An Emerging Tool in Gastro-intestinal Cancers. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Riva, F.; Bidard, F.-C.; Houy, A.; Saliou, A.; Madic, J.; Rampanou, A.; Hego, C.; Milder, M.; Cottu, P.; Sablin, M.P.; et al. Patient-Specific Circulating Tumor DNA Detection during Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer. Clin. Chem. 2017, 63, 691–699. [Google Scholar] [CrossRef]
- Bidard, F.-C.; Hardy-Bessard, A.-C.; Dalenc, F.; Bachelot, T.; Pierga, J.Y.; de la Motte Rouge, T.; Sabatier, R.; Dubot, C.; Frenel, J.S.; Ferrero, J.M.; et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2022, 23, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Toi, M.; Neven, P.; Sohn, J.; Grischke, E.M.; Llombart-Cussac, A.; Soliman, H.; Wang, H.; Wijayawardana, S.; Jansen, V.M.; et al. Clinical Significance of PIK3CA and ESR1 Mutations in Circulating Tumor DNA: Analysis from the MONARCH 2 Study of Abemaciclib plus Fulvestrant. Clin. Cancer Res. 2022, 28, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, R.; Vicier, C.; Garnier, S.; Guille, A.; Carbuccia, N.; Isambert, N.; Dalenc, F.; Robert, M.; Levy, C.; Pakradouni, J.; et al. Circulating tumor DNA predicts efficacy of a dual AKT/p70S6K inhibitor (LY2780301) plus paclitaxel in metastatic breast cancer: Plasma analysis of the TAKTIC phase IB/II study. Mol. Oncol. 2022, 16, 2057–2070. [Google Scholar] [CrossRef]
- Holdhoff, M.; Schmidt, K.; Diehl, F.; Aggrawal, N.; Angenendt, P.; Romans, K.; Edelstein, D.L.; Torbenson, M.; Kinzler, K.W.; Vogelstein, B.; et al. Detection of tumor DNA at the margins of colorectal cancer liver metastasis. Clin. Cancer Res. 2011, 17, 3551–3557. [Google Scholar] [CrossRef]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lønning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.A.; Kalev, D.; Egle, D.; et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.-K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef]
- Chin, Y.M.; Takahashi, Y.; Chan, H.T.; Otaki, M.; Fujishima, M.; Shibayama, T.; Miki, Y.; Ueno, T.; Nakamura, Y.; Low, S.K. Ultradeep targeted sequencing of circulating tumor DNA in plasma of early and advanced breast cancer. Cancer Sci. 2021, 112, 454–464. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Wu, A.; Attard, G. Plasma DNA analysis in prostate cancer: Opportunities for improving clinical management. Clin. Chem. 2019, 65, 100–107. [Google Scholar] [CrossRef]
- Mansukhani, S.; Barber, L.J.; Kleftogiannis, D.; Moorcraft, S.Y.; Davidson, M.; Woolston, A.; Proszek, P.Z.; Griffiths, B.; Fenwick, K.; Herman, B.; et al. Ultra-sensitive mutation detection and genome-wide DNA copy number reconstruction by error-corrected circulating tumor DNA sequencing. Clin. Chem. 2018, 64, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Noguchi, T.; Sakai, K.; Iwahashi, N.; Matsuda, K.; Matsukawa, H.; Yahata, T.; Toujima, S.; Nishio, K.; Ino, K. Changes in the gene mutation profiles of circulating tumor DNA detected using CAPPSeq in neoadjuvant chemotherapytreated advanced ovarian cancer. Oncol. Lett. 2020, 19, 2713–2720. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Hayashi, H.; Sakai, K.; Suzuki, S.; Haratani, K.; Takahama, T.; Tanizaki, J.; Nonagase, Y.; Tanaka, K.; Yoshida, T.; et al. CAPP-seq analysis of circulating tumor DNA from patients with EGFR T790M-positive lung cancer after osimertinib. Int. J. Clin. Oncol. 2021, 26, 1628–1639. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Cohen, J.; Hong, W.; Christie, M.; Wong, H.L.; Kosmider, S.; Wong, R.; Thomson, B. Circulating tumor DNA dynamics and recurrence risk in patients undergoing curative intent resection of colorectal cancer liver metastases: A prospective cohort study. PLoS Med. 2021, 18, e1003620. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.R.; Contente-Cuomo, T.; Sammut, S.-J.; Odenheimer-Bergman, A.; Ernst, B.; Perdigones, N.; Chin, S.-F.; Farooq, M.; Mejia, R.; Cronin, P.A.; et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 2019, 11, eaax7392. [Google Scholar] [CrossRef]
- Yi, Z.; Ma, F.; Rong, G.; Guan, Y.; Li, C.; Xu, B. Clinical spectrum and prognostic value of TP53 mutations in circulating tumor DNA from breast cancer patients in China. Cancer Commun. 2020, 40, 260–269. [Google Scholar] [CrossRef]
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Narayan, A.; Carriero, N.J.; Gettinger, S.N.; Kluytenaar, J.; Kozak, K.R.; Yock, T.I.; Muscato, N.E.; Ugarelli, P.; Decker, R.H.; Patel, A.A. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012, 72, 3492–3498. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Gale, D.; Lawson, A.R.J.; Howarth, K.; Madi, M.; Durham, B.; Smalley, S.; Calaway, J.; Blais, S.; Jones, G.; Clark, J.; et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS ONE 2018, 13, e0194630. [Google Scholar] [CrossRef]
- Balakrishnan, S.G.; Ahmad, M.R.; Koloor, S.S.R.; Petrů, M. Separation of ctDNA by superparamagnetic bead particles in microfluidic platform for early cancer detection. J. Adv. Res. 2021, 33, 109–116. [Google Scholar] [CrossRef]
- Rahman, M.; Niu, J.; Cui, X.; Zhou, C.; Tang, N.; Jin, H.; Cui, D. Electrochemical Biosensor Based on l-Arginine and rGO-AuNSs Deposited on the Electrode Combined with DNA Probes for Ultrasensitive Detection of the Gastric Cancer-Related PIK3CA Gene of ctDNA. ACS Appl. Bio. Mater. 2022, 5, 5094–5103. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Y.W.; Alemu, B.A.; Bekele, S.A.; Gizaw, S.T.; Zerihun, M.F.; Wabalo, E.K.; Teklemariam, M.D.; Mihrete, T.K.; Hanurry, E.Y.; Amogne, T.G.; et al. Epigenetic tumor heterogeneity in the era of single-cell profiling with nanopore sequencing. Clin. Epigenetics 2022, 14, 107. [Google Scholar] [CrossRef]
- Zhou, Q.; Gampenrieder, S.P.; Frantal, S.; Rinnerthaler, G.; Singer, C.F.; Egle, D.; Pfeiler, G.; Bartsch, R.; Wette, V.; Pichler, A.; et al. Persistence of ctDNA in Patients with Breast Cancer During Neoadjuvant Treatment Is a Significant Predictor of Poor Tumor Response. Clin. Cancer Res. 2022, 28, 697–707. [Google Scholar] [CrossRef]
- Cheng, L.; Gao, G.; Zhao, C.; Wang, H.; Yao, C.; Yu, H.; Yao, J.; Li, F.; Guo, L.; Jian, Q.; et al. Personalized circulating tumor DNA detection to monitor immunotherapy efficacy and predict outcome in locally advanced or metastatic non-small cell lung cancer. Cancer Med. 2023, 12, 14317–14326. [Google Scholar] [CrossRef]
- Stasik, S.; Mende, M.; Schuster, C.; Mahler, S.; Aust, D.; Tannapfel, A.; Reinacher-Schick, A.; Baretton, G.; Krippendorf, C.; Bornhäuser, M.; et al. Sensitive Quantification of Cell-Free Tumor DNA for Early Detection of Recurrence in Colorectal Cancer. Front. Genet. 2022, 12, 811291. [Google Scholar] [CrossRef]
- Fei, X.; Du, X.; Gong, Y.; Liu, J.; Fan, L.; Wang, J.; Wang, Y.; Zhu, Y.; Pan, J.; Dong, B.; et al. Early Plasma Circulating Tumor DNA as a Potential Biomarker of Disease Recurrence in Non-metastatic Prostate Cancer. Cancer Res. Treat. 2023, 55, 969–977. [Google Scholar] [CrossRef]
- Yang, J.; Gong, Y.; Lam, V.K.; Shi, Y.; Guan, Y.; Zhang, Y.; Ji, L.; Chen, Y.; Zhao, Y.; Qian, F.; et al. Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer. Cell Death Dis. 2020, 11, 346. [Google Scholar] [CrossRef]
- Hou, J.Y.; Chapman, J.S.; Kalashnikova, E.; Pierson, W.; Smith-McCune, K.; Pineda, G.; Vattakalam, R.M.; Ross, A.; Mills, M.; Suarez, C.J.; et al. Circulating tumor DNA monitoring for early recurrence detection in epithelial ovarian cancer. Gynecol. Oncol. 2022, 167, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yang, Z.; Hu, Z.; Zang, Z.; Pan, Y.; Chen, J.; Wang, J.; Hu, D.; Zhou, Z.; Xu, L.; et al. Preoperative serum ctDNA predicts early hepatocellular carcinoma recurrence and response to systemic therapies. Hepatol. Int. 2022, 16, 868–878. [Google Scholar] [CrossRef]
- Le Guin, C.H.D.; Bornfeld, N.; Bechrakis, N.E.; Jabbarli, L.; Richly, H.; Lohmann, D.R.; Zeschnigk, M. Early detection of metastatic uveal melanoma by the analysis of tumor-specific mutations in cell-free plasma DNA. Cancer Med. 2021, 10, 5974–5982. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.L.; Gorsia, D.N.; Collins, A.; Sandhu, P.; Foreman, N.; Gore, A.; Wood, J.; Kent, C.; Silcock, L.; Guttery, D.S. Utility of Circulating Tumor DNA for Detection and Monitoring of Endometrial Cancer Recurrence and Progression. Cancers 2020, 12, 2231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Bui, N.; Rathore, R.; Sudhaman, S.; George, G.V.; Malashevich, A.K.; Malhotra, M.; Liu, M.C.; Aleshin, A.; Ganjoo, K.N. Feasibility of Longitudinal ctDNA Assessment in Patients with Uterine and Extra-Uterine Leiomyosarcoma. Cancers 2022, 15, 157. [Google Scholar] [CrossRef]
- Wang, X.; Yu, N.; Cheng, G.; Zhang, T.; Wang, J.; Deng, L.; Li, J.; Zhao, X.; Xu, Y.; Yang, P.; et al. Prognostic value of circulating tumour DNA during post-radiotherapy surveillance in locally advanced esophageal squamous cell carcinoma. Clin. Transl. Med. 2022, 12, e1116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xie, K.; Zhu, G.; Ma, C.; Cheng, C.; Li, Y.; Xiao, X.; Li, C.; Tang, J.; Wang, H.; et al. Early detection and stratification of lung cancer aided by a cost-effective assay targeting circulating tumor DNA (ctDNA) methylation. Respir Res. 2023, 24, 163. [Google Scholar] [CrossRef]
- Zhao, Y.; O’Keefe, C.M.; Hsieh, K.; Cope, L.; Joyce, S.C.; Pisanic, T.R.; Herman, J.G.; Wang, T.H. Multiplex Digital Methylation-Specific PCR for Noninvasive Screening of Lung Cancer. Adv. Sci. 2023, 10, e2206518. [Google Scholar] [CrossRef]
- Jiang, N.; Zhou, J.; Zhang, W.; Li, P.; Liu, Y.; Shi, H.; Zhang, C.; Wang, Y.; Zhou, C.; Peng, C.; et al. RNF213 gene mutation in circulating tumor DNA detected by targeted next-generation sequencing in the assisted discrimination of early-stage lung cancer from pulmonary nodules. Thorac. Cancer 2021, 12, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Dai, W.; Wang, H.; Lan, X.; Ma, C.; Su, Z.; Xiang, W.; Han, L.; Luo, W.; Zhang, L.; et al. Early detection and prognosis prediction for colorectal cancer by circulating tumour DNA methylation haplotypes: A multicentre cohort study. EClinicalMedicine 2022, 55, 101717. [Google Scholar] [CrossRef]
- Brenne, S.S.; Madsen, P.H.; Pedersen, I.S.; Hveem, K.; Skorpen, F.; Bygum Krarup, H.; Giskeødegård, G.F.; Laugsand, E.A. Colorectal cancer detected by liquid biopsy 2 years prior to clinical diagnosis in the HUNT study. Br. J. Cancer 2023, 129, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhu, D.; Shao, F.; Chen, S.; Guo, Y.; Li, K.; Wang, Y.; Ding, R.; Gao, L.; Ma, W.; et al. Efficient detection and post-surgical monitoring of colon cancer with a multi-marker DNA methylation liquid biopsy. Proc. Natl. Acad. Sci. USA 2021, 118, e2017421118. [Google Scholar] [CrossRef] [PubMed]
- Kodada, D.; Hyblova, M.; Krumpolec, P.; Janostiakova, N.; Barath, P.; Grendar, M.; Blandova, G.; Petrovic, O.; Janega, P.; Repiska, V.; et al. The Potential of Liquid Biopsy in Detection of Endometrial Cancer Biomarkers: A Pilot Study. Int. J. Mol. Sci. 2023, 24, 7811. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jeon, H.; Jeon, S.; Lee, S.H.; Kim, C.; Ahn, J.H.; Um, H.; Woo, Y.J.; Jeong, S.H.; Kim, Y.; et al. A method for early diagnosis of lung cancer from tumor originated DNA fragments using plasma cfDNA methylome and fragmentome profiles. Mol. Cell Probes 2022, 66, 101873. [Google Scholar] [CrossRef]
- Wei, Q.; Jin, C.; Wang, Y.; Guo, S.; Guo, X.; Liu, X.; An, J.; Xing, J.; Li, B. A computational framework to unify orthogonal information in DNA methylation and copy number aberrations in cell-free DNA for early cancer detection. Brief. Bioinform. 2022, 23, bbac200. [Google Scholar] [CrossRef]
- Nie, W.; Wang, Z.J.; Zhang, K.; Li, B.; Cai, Y.R.; Wen, F.C.; Zhang, D.; Bai, Y.Z.; Zhang, X.Y.; Wang, S.Y.; et al. ctDNA-adjusted bTMB as a predictive biomarker for patients with NSCLC treated with PD-(L)1 inhibitors. BMC Med. 2022, 20, 170. [Google Scholar] [CrossRef]
- Provencio, M.; Serna-Blasco, R.; Nadal, E.; Insa, A.; García-Campelo, M.R.; Casal Rubio, J.; Dómine, M.; Majem, M.; Rodríguez-Abreu, D.; Martínez-Martí, A.; et al. Overall Survival and Biomarker Analysis of Neoadjuvant Nivolumab Plus Chemotherapy in Operable Stage IIIA Non-Small-Cell Lung Cancer (NADIM phase II trial). J. Clin. Oncol. 2022, 40, 2924–2933. [Google Scholar] [CrossRef]
- Qiu, B.; Guo, W.; Zhang, F.; Lv, F.; Ji, Y.; Peng, Y.; Chen, X.; Bao, H.; Xu, Y.; Shao, Y.; et al. Dynamic recurrence risk and adjuvant chemotherapy benefit prediction by ctDNA in resected NSCLC. Nat. Commun. 2021, 12, 6770. [Google Scholar] [CrossRef]
- Gray, J.E.; Ahn, M.J.; Oxnard, G.R.; Shepherd, F.A.; Imamura, F.; Cheng, Y.; Okamoto, I.; Cho, B.C.; Lin, M.C.; Wu, Y.L.; et al. Early Clearance of Plasma Epidermal Growth Factor Receptor Mutations as a Predictor of Outcome on Osimertinib in Advanced Non-Small Cell Lung Cancer; Exploratory Analysis from AURA3 and FLAURA. Clin. Cancer Res. 2023, 29, 3340–3351. [Google Scholar] [CrossRef]
- Benhaim, L.; Bouché, O.; Normand, C.; Didelot, A.; Mulot, C.; Le Corre, D.; Garrigou, S.; Djadi-Prat, J.; Wang-Renault, S.F.; Perez-Toralla, K.; et al. Circulating tumor DNA is a prognostic marker of tumor recurrence in stage II and III colorectal cancer: Multicentric, prospective cohort study (ALGECOLS). Eur. J. Cancer 2021, 159, 24–33. [Google Scholar] [CrossRef]
- Musher, B.L.; Melson, J.E.; Amato, G.; Chan, D.; Hill, M.; Khan, I.; Kochuparambil, S.T.; Lyons, S.E.; Orsini, J., Jr.; Pedersen, S.K.; et al. Evaluation of Circulating Tumor DNA for Methylated BCAT1 and IKZF1 to Detect Recurrence of Stage II/Stage III Colorectal Cancer (CRC). Cancer Epidemiol. Biomark. Prev. 2020, 29, 2702–2709. [Google Scholar] [CrossRef]
- Londra, D.; Mastoraki, S.; Bournakis, E.; Zavridou, M.; Thanos, A.; Rampias, T.; Lianidou, E. USP44 Promoter Methylation in Plasma Cell-Free DNA in Prostate Cancer. Cancers 2021, 13, 4607. [Google Scholar] [CrossRef]
- Nitschke, C.; Markmann, B.; Walter, P.; Badbaran, A.; Tölle, M.; Kropidlowski, J.; Belloum, Y.; Goetz, M.R.; Bardenhagen, J.; Stern, L.; et al. Peripheral and Portal Venous KRAS ctDNA Detection as Independent Prognostic Markers of Early Tumor Recurrence in Pancreatic Ductal Adenocarcinoma. Clin. Chem. 2023, 69, 295–307. [Google Scholar] [CrossRef]
- Marsavela, G.; Johansson, P.A.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Khattak, M.A.; Meniawy, T.M.; Amanuel, B.; et al. The Prognostic Impact of Circulating Tumour DNA in Melanoma Patients Treated with Systemic Therapies-Beyond BRAF Mutant Detection. Cancers 2020, 12, 3793. [Google Scholar] [CrossRef] [PubMed]
- Bellone, S.; McNamara, B.; Mutlu, L.; Demirkiran, C.; Hartwich, T.M.P.; Harold, J.; Yang-Hartwich, Y.; Siegel, E.R.; Santin, A.D. Monitoring Treatment Response, Early Recurrence, and Survival in Uterine Serous Carcinoma and Carcinosarcoma Patients Using Personalized Circulating Tumor DNA Biomarkers. Int. J. Mol. Sci. 2023, 24, 8873. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.J.; Hossain, N.M.; Bukhari, A.; Dean, E.; Spiegel, J.Y.; Claire, G.K.; Kirsch, I.; Jacob, A.P.; Mullins, C.D.; Lee, L.W.; et al. Monitoring of Circulating Tumor DNA Improves Early Relapse Detection After Axicabtagene Ciloleucel Infusion in Large B-Cell Lymphoma: Results of a Prospective Multi-Institutional Trial. J. Clin. Oncol. 2021, 39, 3034–3043. [Google Scholar] [CrossRef] [PubMed]
- Azzi, G.; Tavallai, M.; Aushev, V.N.; Koyen Malashevich, A.; Botta, G.P.; Tejani, M.A.; Hanna, D.; Krinshpun, S.; Malhotra, M.; Jurdi, A.; et al. Using Tumor-Informed Circulating Tumor DNA (ctDNA)-Based Testing for Patients with Anal Squamous Cell Carcinoma. Oncologist 2023, 28, 220–229. [Google Scholar] [CrossRef]
- Jung, H.A.; Ku, B.M.; Kim, Y.J.; Park, S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Cho, J.H.; Kim, H.K.; Choi, Y.S.; et al. Longitudinal Monitoring of Circulating Tumor DNA From Plasma in Patients With Curative Resected Stage I-IIIA EGFR Mutant-Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2023, 33, S974. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Mei, J.; Kang, R.; Deng, S.; Chen, Y.; Yang, Y.; Feng, G.; Deng, Y.; Gan, F.; Lin, Y.; et al. Perioperative ctDNA-Based Molecular Residual Disease Detection for Non-Small Cell Lung Cancer: A Prospective Multicenter Cohort Study (LUNGCA-1). Clin. Cancer Res. 2022, 28, 3308–3317. [Google Scholar] [CrossRef]
- Frank, M.S.; Andersen, C.S.A.; Ahlborn, L.B.; Pallisgaard, N.; Bodtger, U.; Gehl, J. Circulating Tumor DNA Monitoring Reveals Molecular Progression before Radiologic Progression in a Real-life Cohort of Patients with Advanced Non-small Cell Lung Cancer. Cancer Res. Commun. 2022, 2, 1174–1187. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Sharma, S.; Derouazi, M.; Murgioni, S.; Biason, P.; Rizzato, M.D.; Rasola, C.; Renner, D.; Shchegrova, S.; Koyen Malashevich, A.; et al. Detection of Molecular Residual Disease Using Personalized Circulating Tumor DNA Assay in Patients With Colorectal Cancer Undergoing Resection of Metastases. JCO Precis Oncol. 2021, 5, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Oikkonen, J.; Zhang, K.; Salminen, L.; Schulman, I.; Lavikka, K.; Andersson, N.; Ojanperä1, E.; Hietanen, S.; Grénman, S.; Lehtonen, R.; et al. Prospective Longitudinal ctDNA Workflow Reveals Clinically Actionable Alterations in Ovarian Cancer. JCO Precis Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Eisenhardt, A.E.; Brugger, Z.; Lausch, U.; Kiefer, J.; Zeller, J.; Runkel, A.; Schmid, A.; Bronsert, P.; Wehrle, J.; Leithner, A.; et al. Genotyping of Circulating Free DNA Enables Monitoring of Tumor Dynamics in Synovial Sarcomas. Cancers 2022, 14, 2078. [Google Scholar] [CrossRef]
- Cui, Y.; Kim, H.S.; Cho, E.S.; Han, D.; Park, J.A.; Park, J.Y.; Nam, W.; Kim, H.J.; Cha, I.H.; Cha, Y.H. Longitudinal detection of somatic mutations in saliva and plasma for the surveillance of oral squamous cell carcinomas. PLoS ONE 2021, 16, e0256979. [Google Scholar] [CrossRef] [PubMed]
- Vu, P.; Khagi, Y.; Riviere, P.; Goodman, A.; Kurzrock, R. Total Number of Alterations in Liquid Biopsies Is an Independent Predictor of Survival in Patients With Advanced Cancers. JCO Precis Oncol. 2020, 4, 192–201. [Google Scholar] [CrossRef]
- García-Saenz, J.A.; Ayllón, P.; Laig, M.; Acosta-Eyzaguirre, D.; García-Esquinas, M.; Montes, M.; Sanz, J.; Barquín, M.; Moreno, F.; Garcia-Barberan, V.; et al. Tumor Burden Moni-toring Using Cell-Free Tumor DNA Could be Limited by Tumor Heterogeneity in Advanced Breast Cancer and Should be Evaluated Together With Radiographic Imaging. BMC Cancer 2017, 17, 210. [Google Scholar] [CrossRef]
- Kojic, M.; Maybury, M.K.; Waddell, N.; Koufariotis, L.T.; Addala, V.; Millar, A.; Wood, S.; Pearson, J.V.; Hansford, J.R.; Hassall, T.; et al. Efficient detection and monitoring of pediatric brain malignancies with liquid biopsy based on patient-specific somatic mutation screening. Neuro Oncol. 2023, 25, 1507–1517. [Google Scholar] [CrossRef]
- Smith, C.G.; Moser, T.; Mouliere, F.; Field-Rayner, J.; Eldridge, M.; Riediger, A.L.; Chandrananda, D.; Heider, K.; Wan, J.C.M.; Warren, A.Y.; et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 2020, 12, 23. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Rodríguez, B.; Alba-Bernal, A.; López-López, E.; Quirós-Ortega, M.E.; Carbajosa, G.; Garrido-Aranda, A.; Álvarez, M.; Godoy-Ortiz, A.; Queipo-Ortuño, M.I.; Vicioso, L.; et al. Development of a Novel NGS Methodology for Ultrasensitive Circulating Tumor DNA Detection as a Tool for Early-Stage Breast Cancer Diagnosis. Int. J. Mol. Sci. 2022, 24, 146. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.M.Q.; Phan, T.H.; Jasmine, T.X.; Tran, T.T.T.; Huynh, L.A.K.; Vo, T.L.; Nai, T.H.T.; Tran, T.T.; Truong, M.H.; Tran, N.C.; et al. Multimodal analysis of genome-wide methylation, copy number aberrations, and end motif signatures enhances detection of early-stage breast cancer. Front. Oncol. 2023, 13, 1127086. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, B.J.; Córdoba, G.D.; Aranda, A.G.; lvarez, M.; Vicioso, L.; Pérez, C.L.; Hernando, C.; Bermejo, B.; Parreño, A.J.; Lluch, A.; et al. Detection of TP53 and PIK3CA Mutations in Circulating Tumor DNA Using Next-Generation Sequencing in the Screening Process for Early Breast Cancer Diagnosis. J. Clin. Med. 2019, 8, 1183. [Google Scholar] [CrossRef]
- Li, Z.; Guo, X.; Tang, L.; Peng, L.; Chen, M.; Luo, X.; Wang, S.; Xiao, Z.; Deng, Z.; Dai, L.; et al. Methylation Analysis of Plasma Cell-Free DNA for Breast Cancer Early Detection Using Bisulfite next-Generation Sequencing. Tumour Biol. 2016, 37, 13111–13119. [Google Scholar] [CrossRef] [PubMed]
- Fackler, M.J.; Lopez Bujanda, Z.; Umbricht, C.; Teo, W.W.; Cho, S.; Zhang, Z.; Visvanathan, K.; Jeter, S.; Argani, P.; Wang, C.; et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res. 2014, 74, 2160–2170. [Google Scholar] [CrossRef]
- Visvanathan, K.; Fackler, M.S.; Zhang, Z.; Lopez-Bujanda, Z.A.; Jeter, S.C.; Sokoll, L.J.; Garrett-Mayer, E.; Cope, L.M.; Umbricht, C.B.; Euhus, D.M.; et al. Monitoring of Serum DNA Methylation as an Early Independent Marker of Response and Survival in Metastatic Breast Cancer: TBCRC 005 Prospective Biomarker Study. J. Clin. Oncol. 2017, 35, 751–758. [Google Scholar] [CrossRef]
- Fackler, M.J.; Tulac, S.; Venkatesan, N.; Aslam, A.J.; Cope, L.M.; Lehman, J.; Denbow, R.; Reynolds, J.; Buckley, M.; Downs, B.M.; et al. Abstract PS4-03: An automated DNA methylation assay for monitoring treatment response in patients with metastatic breast cancer. Cancer Res. 2021, 81 (Suppl. S4), PS4-03. [Google Scholar] [CrossRef]
- Hai, L.; Li, L.; Liu., Z.; Tong, Z.; Sun, Y. Whole-genome circulating tumor DNA methylation landscape reveals sensitive biomarkers of breast cancer. Med. Comm. 2022, 3, e134. [Google Scholar] [CrossRef]
- Gerratana, L.; Basile, D.; Franzoni, A.; Allegri, L.; Viotto, D.; Corvaja, C.; Bortot, L.; Bertoli, E.; Buriolla, S.; Targato, G.; et al. Plasma-Based Longitudinal Evaluation of ESR1 Epigenetic Status in Hormone Receptor-Positive HER2-Negative Metastatic Breast Cancer. Front. Oncol. 2020, 10, 550185. [Google Scholar] [CrossRef]
- Turner, N.C.; Swift, C.; Jenkins, B.; Kilburn, L.; Coakley, M.; Beaney, M.; Fox, L.; Goddard, K.; Garcia-Murillas, I.; Proszek, P.; et al. Results of the c-TRAK TN trial: A clinical trial utilising ctDNA mutation tracking to detect molecular residual disease and trigger intervention in patients with moderate- and high-risk early-stage triple-negative breast cancer. Ann. Oncol. 2023, 34, 200–211. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Hancock, B.A.; Solzak, J.P.; Brinza, D.; Scafe, C.; Miller, K.D.; Radovich, M. Next-generation sequencing of circulating tumor DNA to predict recurrence in triple-negative breast cancer patients with residual disease after neoadjuvant chemotherapy. NPJ Breast Cancer 2017, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kagara, N.; Tanei, T.; Naoi, Y.; Shimoda, M.; Shimomura, A.; Shimazu, K.; Kim, S.J.; Noguchi, S. Correlation of Methylated Circulating Tumor DNA With Response to Neoadjuvant Chemotherapy in Breast Cancer Patients. Clin. Breast Cancer. 2017, 17, 61–69.e3. [Google Scholar] [CrossRef]
- Connolly, R.M.; Fackler, M.J.; Zhang, Z.; Zhou, X.C.; Goetz, M.P.; Boughey, J.C.; Walsh, B.; Carpenter, J.T.; Storniolo, A.M.; Watkins, S.P.; et al. Tumor and serum DNA methylation in women receiving preoperative chemotherapy with or without vorinostat in TBCRC008. Breast Cancer Res. Treat. 2018, 167, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.H.; Xu, C.S.; Han, H.; Wang, C.; Lin, S.G. Value of the level of methylation of RASSF1A and WIF-1 in tissue and serum in neoadjuvant chemotherapeutic assessment for advanced breast cancer. Oncol. Lett. 2017, 14, 4499–4504. [Google Scholar] [CrossRef]
- Jank, P.; Gehlhaar, C.; Bianca, L.; Caterina, F.; Andreas, S.; Karn, T.; Marmé, F.; Sinn, H.P.; van Mackelenbergh, M.; Sinn, B.; et al. MGMT promoter methylation in triple negative breast cancer of the GeparSixto trial. PLoS ONE 2020, 15, e0238021. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-H.; Wang, M.-Y.; Lo, C.; Tsai, L.W.; Yen, T.C.; Huang, T.Y.; Huang, W.C.; Yang, K.; Chen, C.K.; Fan, S.C.; et al. Circulating Tumor DNA as a Predictive Marker of Recurrence for Patients With Stage II-III Breast Cancer Treated With Neoadjuvant Therapy. Front. Oncol. 2021, 11, 736769. [Google Scholar] [CrossRef]
- Liu, B.; Yi, Z.; Guan, Y.; Ouyang, Q.; Li, C.; Guan, X.; Lv, D.; Li, L.; Zhai, J.; Qian, H.; et al. Molecular landscape of TP53 mutations in breast cancer and their utility for predicting the response to HER-targeted therapy in HER2 amplification-positive and HER2 mutation-positive amplification-negative patients. Cancer Med. 2022, 11, 2767–2778. [Google Scholar] [CrossRef]
- Guan, X.; Liu, B.; Niu, Y.; Dong, X.; Dong, X.; Zhu, X.; Li, C.; Li, L.; Yi, Z.; Sun, X.; et al. Longitudinal HER2 amplification tracked in circulating tumor DNA for therapeutic effect monitoring and prognostic evaluation in patients with breast cancer. Breast 2020, 49, 261–266. [Google Scholar] [CrossRef]
- Rothé, F.; Silva, M.J.; Venet, D.; Campbell, C.; Bradburry, I.; Rouas, G.; de Azambuja, E.; Maetens, M.; Fumagalli, D.; Rodrik-Outmezguine, V.; et al. Circulating Tumor DNA in HER2-Amplified Breast Cancer: A Translational Research Substudy of the NeoALTTO Phase III Trial. Clin. Cancer Res. 2019, 25, 3581–3588. [Google Scholar] [CrossRef]
- Li, X.; Lu, J.; Zhang, L.; Luo, Y.; Zhao, Z.; Li, M. Clinical Implications of Monitoring ESR1 Mutations by Circulating Tumor DNA in Estrogen Receptor Positive Metastatic Breast Cancer: A Pilot Study. Transl. Oncol. 2020, 13, 321–328. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Rugo, H.S.; Im, S.A.; Slamon, D.J.; Harbeck, N.; Bondarenko, I.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Women with HR+/HER2- ABC: Updated Exploratory Analyses of PALOMA-3, a Double-blind, Phase III Randomized Study. Clin. Cancer Res. 2022, 28, 3433–3442. [Google Scholar] [CrossRef]
- Berger, F.; Marce, M.; Delaloge, S.; Hardy-Bessard, A.C.; Bachelot, T.; Bièche, I.; Pradines, A.; De La Motte Rouge, T.; Canon, J.L.; André, F.; et al. Randomised, open-label, multicentric phase III trial to evaluate the safety and efficacy of palbociclib in combination with endocrine therapy, guided by ESR1 mutation monitoring in oestrogen receptor-positive, HER2-negative metastatic breast cancer patients: Study design of PADA-1. BMJ Open 2022, 12, e055821. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Lyu, D.; Liu, B.; Lan, B.; Sun, X.; Li, L.; Zhai, J.; Qian, H.; Ma, F. Clinical value of next-generation sequencing in guiding decisions regarding endocrine therapy for advanced HR-positive/HER-2-negative breast cancer. Chin. J. Cancer Res. 2022, 34, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Mastoraki, S.; Strati, A.; Tzanikou, E.; Chimonidou, M.; Politaki, E.; Voutsina, A.; Psyrri, A.; Georgoulias, V.; Lianidou, E. ESR1 Methylation: A Liquid Biopsy-Based Epigenetic Assay for the Follow-up of Patients with Metastatic Breast Cancer Receiving Endocrine Treatment. Clin. Cancer Res. 2018, 24, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Chimonidou, M.; Strati, A.; Malamos, N.; Kouneli, S.; Georgoulias, V.; Lianidou, E. Direct comparison study of DNA methylation markers in EpCAM-positive circulating tumour cells, corresponding circulating tumour DNA, and paired primary tumours in breast cancer. Oncotarget 2017, 8, 72054–72068. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E.; et al. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Coombes, R.C.; Page, K.; Salari, R.; Hastings, R.K.; Armstrong, A.; Ahmed, S.; Ali, S.; Cleator, S.; Kenny, L.; Stebbing, J.; et al. Personalized Detection of Circulating Tumor DNA Antedates Breast Cancer Metastatic Recurrence. Clin. Cancer Res. 2019, 25, 4255–4263. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Chopra, N.; Comino-Mendez, I.; Beaney, M.; Tovey, H.; Cutts, R.J.; Swift, C.; Kriplani, D.; Afentakis, M.; Hrebien, S.; et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol. 2019, 5, 1473–1478. [Google Scholar] [CrossRef]
- Radovich, M.; Jiang, G.; Hancock, B.A.; Chitambar, C.; Nanda, R.; Falkson, C.; Lynce, F.C.; Gallagher, C.; Isaacs, C.; Blaya, M.; et al. Association of Circulating Tumor DNA and Circulating Tumor Cells After Neoadjuvant Chemotherapy with Disease Recurrence in Patients With Triple-Negative Breast Cancer: Preplanned Secondary Analysis of the BRE12-158 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1410–1415. [Google Scholar] [CrossRef]
- Yi, Z.; Ma, F.; Rong, G.; Liu, B.; Guan, Y.; Li, J.; Sun, X.; Wang, W.; Guan, X.; Mo, H. The molecular tumor burden index as a response evaluation criterion in breast cancer. Signal Transduct. Target. Ther. 2021, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Jelovac, D.; Balukrishna, S.; Cochran, R.L.; Croessmann, S.; Zabransky, D.J.; Wong, H.Y.; Toro, P.V.; Cidado, J.; Blair, B.G.; et al. Detection of Cancer DNA in Plasma of Patients with Early-Stage Breast Cancer. Clin. Cancer Res. 2014, 20, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.H.; Dahlgren, M.; Schulz, R.; Grabau, D.; van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Shaw, J.A.; Page, K.; Blighe, K.; Hava, N.; Guttery, D.; Ward, B.; Brown, J.; Ruangpratheep, C.; Stebbing, J.; Payne, R.; et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 2012, 22, 220–231. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures 2019; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Gradishar, W.J.; Anderson, B.O.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D.; et al. Breast cancer, version 3.2020, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Can. Netw. 2020, 18, 452–478. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Magnanini, S.; Rinaldini, M.; Berardi, F.; Di Biagio, G.; Testare, F.; Tavoni, N.; Schittulli, F.; D’Amico, C.; Pedicini, T.; et al. Impact of follow-up testing on survival and health-related quality of life in breast cancer patients. A multicenter randomized controlled trial. JAMA 1994, 271, 1587–1592. [Google Scholar] [CrossRef]
- Lu, W.L.; Jansen, L.; Post, W.J.; Bonnema, J.; Van de Velde, J.C.; De Bock, G.H. Impact on survival of early detection of isolated breast recurrences after the primary treatment for breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2009, 114, 403–412. [Google Scholar] [CrossRef]
- Wang, M.; Hou, L.; Chen, M.; Zhou, Y.; Liang, Y.; Wang, S.; Jiang, J.; Zhang, Y. Neoadjuvant Chemotherapy Creates Surgery Opportunities For Inoperable Locally Advanced Breast Cancer. Sci. Rep. 2017, 7, 44673. [Google Scholar] [CrossRef]
- Derks, M.G.M.; van de Velde, C.J.H. Neoadjuvant chemotherapy in breast cancer: More than just downsizing. Lancet Oncol. 2018, 19, 2–3. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Long-term outcomes for neoadjuvant versus adjuvant chemotherapy in early breast cancer: Meta-analysis of individual patient data from ten randomised trials. Lancet Oncol. 2018, 19, 27–39. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Untch, M.; Blohmer, J.U.; Costa, S.D.; Eidtmann, H.; Fasching, P.A.; Gerber, B.; Eiermann, W.; Hilfrich, J.; Huober, J.; et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 2012, 30, 1796–1804. [Google Scholar] [CrossRef]
- Symmans, W.F.; Wei, C.; Gould, R.; Yu, X.; Zhang, Y.; Liu, M.; Walls, A.; Bousamra, A.; Ramineni, M.; Sinn, B.; et al. Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J. Clin. Oncol. 2017, 35, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Symmans, W.F.; Peintinger, F.; Hatzis, C.; Rajan, R.; Kuerer, H.; Valero, V.; Assad, L.; Poniecka, A.; Hennessy, B.; Green, M.; et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J. Clin. Oncol. 2007, 25, 4414–4422. [Google Scholar] [CrossRef]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Consortium, I.S.T.; Yee, D.; DeMichele, A.M.; Yau, C.; Isaacs, C.; Symmans, W.F.; Albain, K.S.; Chen, Y.Y.; Krings, G.; Wei, S.; et al. Association of Event-Free and Distant Recurrence-Free Survival With Individual-Level Pathologic Complete Response in Neoadjuvant Treatment of Stages 2 and 3 Breast Cancer: Three-Year Follow-up Analysis for the I-SPY2 Adaptively Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1355–1362. [Google Scholar] [CrossRef]
- Ignatova, E.O.; Frolova, M.A.; Petrovsky, A.B.; Stenina, M.B.; Glazkova, E.V.; Krokhina, O.V.; Tjulandin, C.A. Evaluation of efficacy and toxicity of neoadjuvant chemotherapy with dose-dense doxorubicin, cisplatin, and paclitaxel in patients with early triple-negative breast cancer. Malig. Tumours 2016, 4, 49–57. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef]
- de Ruijter, T.C.; van der Heide, F.; Smits, K.M.; Aarts, M.J.; van Engeland, M.; Heijnen, V.C.G. Prognostic DNA methylation markers for hormone receptor breast cancer: A systematic review. Breast Cancer Res. 2020, 22, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, Y.; Gong, Y.; Zhang, Y.; Lu, Y.; Wang, C.; Yao, R.; Li, P.; Guan, Y.; Wang, J.; et al. Clinical factors associated with circulating tumor DNA (ctDNA ) in primary breast cancer. Mol. Oncol. 2019, 13, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A. AKT inhibition in solid tumors with AKT1 mutations. J. Clin. Oncol. 2017, 35, 2251–2259. [Google Scholar] [CrossRef]
- Tan, G.; Chu, C.; Gui, X.; Li, J.; Chen, Q. The prognostic value of circulating cell-free DNA in breast cancer: A meta-analysis. Medicine 2018, 97, e0197. [Google Scholar] [CrossRef]

{kind=link}
| Target | Method | Example Assay | Sensitivity (%) | Advantages | Limitations |
|---|---|---|---|---|---|
| Single locus | Digital PCR | ddPCR, BEAMing | 0.01 | High sensitivity | Detects only known mutations |
| Low DNA input | |||||
| Provides quantification and monitoring of recurrent mutations | |||||
| Gene panel | Targeted panel sequencing | TAM-Seq, Safe-SeqS | 0.01–1 | High sensitivity | Less comprehensive than other NGS methods |
| Fast | |||||
| Cost-effective compared with other NGS methods | |||||
| Targeted digital sequencing | TARDIS | 0.03–1 | High sensitivity | More complex workflow | |
| Hybrid capture sequencing | CAPP-Seq | 0.02 | Able to detect copy number variations and rearrangements | Requires high cfDNA input | |
| Less comprehensive | |||||
| More complex workflow | |||||
| Comprehensive | Whole-exome sequencing Whole-genome sequencing | 1–10 | Identifies novel mutations | Less sensitive | |
| Does not require prior information about the tumor mutation | Expensive | ||||
| Longer turnaround time |
| Authors | Date of Publication | Analysis | Reference |
|---|---|---|---|
| ctDNA Studies Using Point Mutation Analysis | |||
| Fribbens, C. et al. | 2016 | ESR1 mutations | [20] |
| Riva, F. et al. | 2017 | TP53 mutations | [54] |
| Tolaney, S. et al. | 2022 | PIK3CA and ESR1 mutations | [56] |
| Sabatier, R. et al. | 2022 | PI3KCA, AKT1, and TP53 mutations | [57] |
| Di Leo, A. et al. | 2018 | PIK3CA mutations | [59] |
| Chin, Y. et al. | 2021 | 57 SNV * the majority were in TP53 (37%), PIK3CA (21%), AKT1 (7%), EGFR (5%) and KRAS (5%) | [61] |
| Yi, Z. et al. | 2020 | TP53 mutations | [70] |
| Visvanathan, K. et al. | 2017 | Cumulative methylation index (CMI) of a 6-gene panel (AKR1B1, HOXB4, RASGRF2, RASSF1, HIST1H3C and TM6SF1) | [128] |
| Fackler, M. et al. | 2021 | 9-gene panel of breast-cancer-specific DNA methylation markers | [129] |
| Gerratana, L. et al. | 2020 | Methylation status of ESR1 main promoters (promA and promB) | [131] |
| Turner, N. et al. | 2023 | One or two mutations from panel of genes | [132] |
| Chen, Y. et al. | 2017 | TP53, PIK3CA, CDKN2A from panel of genes | [133] |
| Takahashi, H. et al. | 2017 | Methylation of the promoter region of RASSF1A | [134] |
| Connolly, R. et al. | 2018 | 10-gene panel; cumulative methylation index (CMI) | [135] |
| Han, Z. et al. | 2017 | Methylation of RASSF1A and WIF-1 | [136] |
| Jank, P. et al. | 2020 | Methylation of 5 CpG islands of MGMT promoter | [137] |
| Lin, P. et al. | 2021 | Target gene panel (14 genes) | [138] |
| Liu, B. et al. | 2022 | TP53 mutations | [139] |
| Guan, X. et al. | 2020 | HER2 amplification | [140] |
| Rothé, F. et al. | 2019 | PIK3CA and/or TP53 mutations | [141] |
| Li, X. et al. | 2020 | ESR1 mutations | [142] |
| Cristofanilli, M. et al. | 2022 | ESR1, PIK3CA, and TP53 mutations | [143] |
| Bidard, F. et al. Berger, F. et al. | 2022 2022 | ESR1 mutations | [55,144] |
| Turner, N. et al. | 2020 | PIK3CA, ESR1, HER2, PTEN, and AKT1 mutations | [145] |
| Lyu, D., et al. | 2022 | Mutations in PI3K/AKT/mTOR signaling pathway, ESR1, HER2 mutations | [146] |
| Mastoraki, S. et al. | 2018 | ESR1 methylation | [147] |
| Chimonidou, M. et al. | 2017 | Methylation of the promoter region of SOX17 | [148] |
| Panagopoulou, M. et al. | 2019 | Methylation status of a panel of cancer-related genes (KLK10, SOX17, WNT5A, MSH2, GATA3) | [149] |
| ctDNA studies using whole-genome sequencing | |||
| Parsons, H. et al. | 2020 | Exome sequencing to identify patient-specific SNVs, design custom minimal residual disease tests | [18] |
| Magbanua, M. et al. | 2021 | Whole-exome sequencing, design of individual mutation panels | [19] |
| Moss, J. et al. | 2020 | 3 target regions specifically hypermethylated or hypomethylated uniquely in breast cancer from whole-genome bioinformatic analysis | [43] |
| Widschwendter, M. et al. | 2017 | Representation bisulfite sequencing (RRBS), found specific region EFC#93 analysis via ultra-high coverage bisulfite sequencing | [48] |
| McDonald, B. et al. | 2019 | Exome sequencing of tumor biopsies and analysis of dozens to hundreds of mutations in serial plasma samples | [69] |
| Garcia-Murillas, I. et al. | 2015 | Targeted NGS of exons of 273 genes | [150] |
| Coombes, R. et al. | 2019 | Ultra-deep sequencing | [151] |
| Garcia-Murillas, I. et al. | 2019 | Identify somatic mutations by breast cancer driver gene panel, design of individual mutation panels | [152] |
| Radovich, M. et al. | 2020 | Big commercial platform covering multiple genes | [153] |
| Yi, Z. et al. | 2021 | Target-capture deep sequencing of 1021 genes, calculation of molecular tumor burden index | [154] |
| Author, Year [Reference] | Number of Patients | Characteristic of Patients, Trial | Studied Parameter | Method, Tumor/ctDNA | Main Results |
|---|---|---|---|---|---|
| Parsons, H., 2020 [18] | 158 | Metastatic breast cancer ER + HER2- (n = 16) and non-metastatic breast cancer 0-III stage (n = 142) | Exome sequencing to identify patient-specific SNVs 2, custom test | NGS | Whole-exome sequencing of tumors was performed and individualized MRD 3 tests were designed. MRD detection at 1 year was strongly associated with distant recurrence (HR = 20.8; 95% CI 7.3–58.9). Median lead time from first positive sample to recurrence was 18.9 months (range = 3.4–39.2 months) |
| Fackler, M., 2021 [42] | 72 | Metastatic breast cancer (n = 46), benign breast disease (n = 17), healthy normal controls (n = 9) | 9-gene panel of breast-cancer-specific DNA methylation markers | Methylation specific-PCR in automated Liquid Biopsy for Breast Cancer Methylation (LBx-BCM) prototype | 9-gene panel of methylated DNA markers that discriminates stage IV BC from benign breast disease and healthy normal subjects using ctDNA was identified. This assay has potential clinical utility in monitoring therapeutic response and predicting disease recurrence. |
| Widschwendter, M., 2017 [48] | 419 | Breast cancer, SUCCESS trial | Representation bisulfite sequencing (RRBS) in tissue, ultra-high coverage bisulfite sequencing in serum; specific region EFC#93 (a pattern of five, linked CpGs methylated in BC) analysis | NGS | EFC#93 was an independent poor prognostic marker in pre-chemotherapy samples (HR for death = 7.689) and superior to circulating tumor cells (CTCs) (HR for death = 5.681). More than 70% of patients with both CTCs and EFC#93 serum DNAme positivity in their pre-chemotherapy samples relapsed within five years. EFC#93 positivity after chemotherapy is significantly (p = 0.014) less frequently observed in early stage (T1) compared to late stage (T2–4) cancers. EFC#93 serum positivity before chemotherapy was a very strong marker of poor prognosis, for both DFS and OS 4. |
| Riva, F., 2017, [54] | 46 | Nonmetastatic triple-negative breast cancer | TP53 | NGS/ddPCR | Correlation with mitotic index (p = 0.003), tumor grade (p = 0.003), and stage (p = 0.03) |
| Chin, Y., 2021 [61] | 109 | I–IV stages; 83%—luminal HER2 negative, 6%—HER2 positive, 11%—triple-negative | Oncomine Pan-Cancer Cell-Free Assay panel (52 genes) TP53, PIK3CA, AKT1, EGFR, KRAS | NGS | Correlation of the frequency of detection of ctDNA with the prevalence of the disease: stage (p = 0.00026), involvement of lymph nodes (p = 0.00649) and presence of distant metastases (p = 0.0005) |
| Gerratana, L., 2020 [131] | 49 | Metastatic breast cancer ER + HER2- | ESR1 epigenetic status was defined by assessing the methylation of its main promoters (promA and promB) in ct DNA | Methylation- specific digital droplet PCR (MS-ddPCR) | No significant impact on PFS was observed for main promoters of ESR1: promA (p = 0.3777) and promB (p = 0.7455) dichotomized at the median while a ≥2-fold increase in promB or in either promA or promB after 3 months hormonotherapy resulted in a significantly worse prognosis (p = 0.0189, p = 0.0294, respectively). A significant increase after 3 months hormonotherapy was observed for promB among patients with PIK3CA mutation (p = 0.0173). Significantly lower promA levels at baseline were observed in patients with liver metastases (p = 0.0212). |
| Turner, N., 2023 [132] | 208 | Triple-negative breast cancer after primary treatment, c-TRAK TN trial | RMH200 gene panel (200 cancer genes), or the ABC-BIO panel (41 gene) for tumor, using one or two mutations in cfDNA | NGS/dPCR | 71.9% patients (23/32, 95% CI 53.3–86.3%) had metastatic disease on staging at the time of ctDNA detection. Median lead time between ctDNA detection and disease recurrence in the intervention group was 1.6 months (95% CI 1.2–4.9 months) |
| Chen, Y., 2017 [133] | 38 | Triple-negative breast cancer after primary treatment | Oncomine Research Panel consisting of 134 cancer genes (TP53, PIK3CA, CDKN2A) | NGS | ctDNA mutations in the plasma were detected of four patients (three TP53 mutations, one AKT1 mutation, one CDKN2A mutation). All four patients had recurrence of their disease (100% specificity), but sensitivity was limited to detecting only 4 of 13 patients who clinically relapsed (31% sensitivity). The analysis did not identify any de novo mutations exclusively in the plasma, suggesting that only mutations first identified in the primary tumor were detectable in the plasma Patients with detectable circulating tumor DNA had an inferior DFS (p < 0.0001; median DFS: 4.6 mos. vs. not reached; HR = 12.6, 95% CI: 3.06–52.2) |
| Garcia-Murillas, I., 2015 [150] | 55 | Early breast cancer | Panel targeting 14 breast cancer driver genes for tumor, mutation-specific dPCR assays for cfDNA | NGS/ddPCR | Detection of ctDNA in patients who received therapy before surgery was predictive of early relapse (HR = 12.0): the median DFS 1 was 13.6 months (ctDNA detected) versus median not reached (ctDNA not detected). In total, 50% of the patients who relapsed in the study had ctDNA detected in a single postsurgical sample and 80% had ctDNA detected in serial samples. Of the patients who did not relapse, 96% did not have ctDNA detected in either a single postsurgical sample (p = 0.0038) or serial samples (p < 0.0001) |
| Coombes, R., 2019 [151] | 49 | Breast cancer after primary treatment | Whole genome sequencing of tumor, ultra-deep sequencing of 16 individual somatic variants in cfDNA | NGS | Plasma ctDNA was detected ahead of clinical or radiologic relapse in 16 of the 18 relapsed patients (sensitivity of 89%); metastatic relapse was predicted with a lead time of up to 2 years (median 8.9 months; range 0.5–24.0 months). None of the 31 non-relapsing patients were ctDNA-positive at any time point across 156 plasma samples (specificity of 100%) |
| Author, Year [Reference] | Number of Patients | Characteristics of Patients, Trial | Studied Parameter | Method, Tumor/ctDNA | Main Results |
|---|---|---|---|---|---|
| Magbanua, M., 2021 [19] | 84 | Early breast cancer | Whole-exome sequencing, design patient-specific custom test | NGS | Patients who remained ctDNA positive at T1 (3 week after initiation of paclitaxel) were significantly more likely to have residual disease after NAC (83% non-pCR) compared with those who cleared ctDNA (52% non-pCR; OR = 4.33, p = 0.012). After NAC, all patients who achieved pCR were ctDNA negative (n = 17, 100%). Patients who did not achieve pCR but were ctDNA negative (86%) had excellent outcomes, similar to those who achieved pCR (HR = 1.4; 95% CI 0.15–13.5). |
| Moss, J., 2020 [43] | 29 | stage IIA–IIIC | 3 target regions specifically hypermethylated or hypomethylated uniquely in breast cancer received via bioinformatic analysis | NGS | Levels of methylation of ctDNA during the last month of NAC could predict the presence of residual disease (p = 0.006) and were significantly lower than at the start of treatment for patients with a pCR but not for patients with residual disease (p = 0.008 and p = 0.58, respectively). The association between methylation of ctDNA and residual disease is strong even when taking into account other factors such as age, receptor status, and stage. |
| Riva, F., 2017 [54] | 46 | Nonmetastatic triple-negative breast cancer | TP53 | NGS | ctDNA positivity after 1 cycle of NAC 1 was correlated with shorter DFS 2 (p < 0.001) and overall (p = 0.006) survival. |
| McDonald, B., 2019 [69] | 33 | Nonmetastatic breast cancer | Exome sequencing of tumor biopsies and analysis of dozens to hundreds of mutations in serial plasma samples | Targeted digital sequencing (TARDIS) | TARDIS results were informative in 100% of the samples. Patients with pCR 4 showed a large decrease in ctDNA concentration during therapy. |
| Turner, N., 2023 [132] | 208 | Triple-negative breast cancer after primary treatment, c-TRAK TN trial | RMH200 gene panel (200 cancer genes), or the ABC-BIO panel (41 gene) for tumor, using one or two mutations in cfDNA | NGS/dPCR | 71.9% patients (23/32, 95% CI 53.3–86.3%) had metastatic disease on staging at the time of ctDNA detection. Median lead time between ctDNA detection and disease recurrence in the intervention group was 1.6 months (95% CI 1.2–4.9 months). The rapid relapsing nature of high-risk triple-negative BC challenged implementation of MRD 3 detection. |
| Takahashi, H., 2017 [134] | 87 | Breast cancer II–III stage | Methylated ctDNA (met-ctDNA) for the promoter region of RASSF1A | One-step methylation-specific PCR (OS-MSP) | In the patients with positive met-ctDNA before NAC, met-ctDNA significantly decreased after NAC in those with disease that responded to therapy (p = 0.006), but not in patients whose disease did not respond to therapy. Met-ctDNA after NAC was found to be significantly (p = 0.008) correlated to the extent of residual tumor burden. |
| Connolly, R., 2018 [135] | 62 | Breast cancer | 10-gene panel; cumulative methylation index (CMI) | Methylation- specific PCR | High tissue CMI levels at 15th day of treatment may predict poor response. Increase in tissue CMI levels at 15th day of treatment was associated with 40% lower chance of obtaining pCR (OR = 0.60, 95% CI 0.37–0.97; p = 0.037). |
| Han, Z., 2017 [136] | 126 | Advanced breast cancer | Methylation of RASSF1A and WIF-1 | Methylation- specific PCR | Positive rates of RASSF1A methylation and WIF-1 in serum of the patients in the effective group were significantly lower than those in the ineffective group (p = 0.002 and p = 0.001, respectively), the mRNA of RASSF1A and WIF-mRNA was significantly higher than the ineffective group (p < 0.05). |
| Jank, P., 2020 [137] | 174 | Triple-negative breast cancer II-III stage, GeparSixto trial | Methylation of 5 CpG islands of MGMT promoter | Pyrosequencing | MGMT promoter methylation was not significantly associated with pCR rate, and was not related to different chemotherapy response rates in the triple-negative BC. |
| Lin, P., 2021 [138] | 60 | Breast cancer II–III stage | Deep sequencing of a target gene panel (14 genes) | NGS | The presence of ctDNA after NAC was a robust marker for predicting relapse in stage II-to-III BC patients (HR = 4.29, 95% CI 2.06–8.92, p < 0.0001) |
| Garcia-Murillas, I., 2015 [150] | 55 | Early breast cancer | Panel targeting 14 breast cancer driver genes for tumor, mutation-specific dPCR assays for cfDNA | NGS/ddPCR | Detection of ctDNA in patients who received NAC before surgery in serial samples was predictive of early relapse (HR = 12.0): the median disease-free survival was 13.6 months (ctDNA detected) versus median not reached (ctDNA not detected). In total, 50% of the patients who relapsed in the study had ctDNA detected in a single postsurgical sample and 80% had ctDNA detected in serial samples. Of the patients who did not relapse, 96% did not have ctDNA detected in either a single postsurgical sample (p = 0.0038) or serial samples (p < 0.0001). |
| Garcia-Murillas, I., 2019 [152] | 101 | Early breast cancer | Breast cancer driver gene panel, design of individual mutation panels | NGS | ctDNA detection during follow-up was associated with a high rate of relapse. |
| Radovich, M., 2020 [153] | 142 | Early triple-negative breast cancer, BRE12-158 | Commercial platform covering multiple genes (FoundationACT® or FoundationOneLiquid Assay®) | NGS | Detection of ctDNA and circulating tumor cells in triple-negative BC patients after NAC was associated with disease recurrence. |
| Author, Year [Reference] | Number of Patients | Characteristics of Patients, Trial | Studied Parameter | Method | Main Results |
|---|---|---|---|---|---|
| Fribbens, C., 2016 [20] | 161 + 360 | Metastatic ER+ breast cancer, SoFEA trial, PALOMA3 trial | ESR1 mutation | ddPCR | SoFEA: ESR1 mutations were detected in the ctDNA of 39.1% of the patients (63 of 161). ESR1 mutation in their ctDNA had worse PFS than those with the ESR1 wild type when treated with aromatase inhibitor exemestane (2.6 months versus 8.0 months, HR = 2.12; p = 0.01), but not fulvestrant. In ESR1 mutations, fulvestrant resulted in higher PFS compared to exemestane (HR 0.52; 95% CI 0.30–0.92; p = 0.02). PALOMA3: ESR1 mutations were detected in the ctDNA of 25.3% of the patients (91 of 360). Presence of ESR1 mutations in ctDNA of advanced BC patients showed worse PFS compared with those with the ESR1 wild type (HR = 1.46, p = 0.02). |
| Bidard, F., 2022 [55] | 1017 | Metastatic ER+ HER2- breast cancer, PADA-1 trial | ESR1 | ddPCR | Earlier detection of ESR1 mutation growth as a marker of progression and early (before the appearance of traditional clinical and radiological signs) change in therapy ensured a gain in PFS: 11.9 and 5.7 months. (HR = 0.61, 95% CI 0.43–0.86; p = 0.004). |
| Tolaney, S., 2022 [56] | 669 | Advanced ER+ HER2- breast cancer, MONARCH 2 trial | Mutations PIK3CA, ESR1 | ddPCR | Increase in median PFS with the addition of abemaciclib to fulvestrant (vs. placebo + fulvestrant) in both wild-type PIK3CA (median 16.9 vs. 12.3 months; HR = 0.51, 95% CI 0.33–0.78) and PIK3CA mutation (median 17.1 vs. 5.7 months, HR = 0.53; 95% CI 0.33–0.84); as well as with wild-type ESR1 (median 15.3 vs. 11.2 months, HR = 0.44, 95% CI 0.27–0.71) and with ESR1 mutation (median 20.7 vs. 13.1 months; HR 0.54; 95% CI 0.37–0.79). |
| Sabatier, R., 2022 [57] | HER2-negative advanced breast cancer, TAKTIC trial | Low-coverage whole-genome sequencing for all plasma samples; ddPCR for some patients with driver mutations of PI3KCA, AKT1, and TP53 in their tumors | NGS + ddPCR | The presence of ctDNA upon inclusion was correlated with PFS (6-month PFS was 92% for ctDNA-negative patients versus 68% for ctDNA-positive cases (HR = 3.45, p = 0.007)). Copy number alterations were associated with disease progression under paclitaxel-LY2780301. Therefore, ctDNA detection at baseline was associated with shorter PFS, while plasma-based copy number analysis helped to identify alterations involved in resistance to AKT/p70S6K inhibitor plus paclitaxel treatment. | |
| Di Leo, A., 2018 [59] | 348 | Locally advanced and metastatic ER+ HER2- breast cancer, BELLE-3 trial | PIK3CA mutation | Inostics BEAMing assay | Median PFS was significantly longer in the buparlisib versus placebo group (3.9 months vs. 1.8 month (HR = 0.67, 95% CI 0.53–0.84, p = 0.0003) in patients with PIK3CA mutations detected in tumor tissue or ctDNA isolated upon study entry. |
| Yi, Z., 2020 [70] | 804 | Metastatic breast cancer | TP53 | NGS | TP53 mutations were associated with a shorter DFS vs. wild-type TP53 (HR = 1.32, 95% CI = 1.09–1.61, p = 0.005); TP53 mutations in exons 5–8 were associated with worse outcome (HR = 1.50, 95% CI = 1.11–2.03, p = 0.009); TP53 mutation status was not significantly associated with PFS in HER2-positive patients who received first-line trastuzumab-based therapy (p = 0.966). In the taxane combination group, patients with TP53 mutations exhibited longer PFS than those without TP53 mutations (HR = 0.08, 95% CI = 0.02–0.30, p < 0.001). In the non-taxane combination group, patients with TP53 mutations displayed shorter PFS than those with wild-type TP53 (HR = 4.84, p = 0.005). |
| Visvanathan, K., 2017 [128] | 141 | Metastatic breast cancer | Cumulative methylation index (CMI) of a minimal 6-gene subset (AKR1B1, HOXB4, RASGRF2, RASSF1, HIST1H3C and TM6SF1) | Quantitative multiplex assay based on multiplex nested real-time PCR (cMethDNA) | Median PFS and OS were significantly shorter in women with a high CMI (PFS = 2.1 months; OS = 12.3 months) versus a low CMI (PFS = 5.8 months; OS = 21.7 months). In multivariable models, among women with metastatic BC, a high versus low CMI at week 4 was independently associated with worse PFS (HR = 1.79; 95% CI 1.23–2.60; p = 0.002) and OS (HR = 1.75; 95% CI 1.21–2.54; p = 0.003). |
| Chen, Y., 2017 [133] | 38 | Triple-negative breast cancer after primary treatment | Oncomine Research Panel consisting of 134 cancer genes for tumor; TP53, AKT, CDKN2A in cfDNA | NGS | Patients with detectable ctDNA had an inferior DFS (p < 0.0001; median DFS: 4.6 mos. vs. not reached; HR = 12.6, 95% CI: 3.06–52.2). |
| Liu, B., 2022 [139] | 1184 | HER2-positive breast cancer, Geneplus cohort | TP53 | NGS | TP53 mutations were associated with a shorter PFS 1 (p = 0.004) on anti-HER2 antibody therapy; the value of TP53 mutation in predicting HER2 tyrosine kinase inhibitor response was inconsistent. |
| Guan, X., 2020 [140] | 105 | HER2-positive breast cancer | HER2 copy numbers | NGS | Correlation of the number of copies of the HER2 gene before treatment with the frequency of objective effects (p = 0.010); consistently high copy number after 6 weeks was associated with a decrease in DFS 2 (p < 0.001). |
| Rothé, F., 2019 [141] | 69 | Early HER2+ breast cancer, NeoALTTO trial | PIK3CA and/or TP53 mutations | ddPCR | ctDNA detection before neoadjuvant anti-HER2 therapy was associated with low pCR 3 rates. Patients with HER2-enriched tumors and undetectable ctDNA at baseline had the highest pCR rates. |
| Li, X., 2020 [142] | 45 | Metastatic ER+ breast cancer | Targeted NGS panel of 425 genes; TP53 mutation; ESR1 mutation | NGS | Six genes: TP53 (64.4%), PIK3CA (46.7%), ESR1 (20%), ERBB2 (15.6%), ATM (15.6%), and BRCA1 (13.3%), were mutated in more than 13% of the patients. Patients with TP53 mutations (29 patients of 45) had significantly worse OS 4 than the carriers of wild-type alleles (p = 0.0094). ESR1 mutations were recurrently enriched in ER+ metastatic BC patients but were rarely present in primary tumor tissues. The median time from aromatase inhibitor endocrine therapy to the first detection of ESR1 mutations was 39 months (95% CI 21.3–57.6). The change in allele frequency of ESR1 mutations (observed in 9 of 45 patients) was an important biomarker, which could predict endocrine resistance of ER+ BC. Therapy with everolimus in four cases with acquired ESR1 mutations showed longer PFS. |
| Cristofanilli, M., 2022 [143] | 331 | Metastatic ER+ HER2- breast cancer, PALOMA3 trial | Panel of 17 driver and CDK4/6-related genes; analysis of ESR1, PIK3CA, TP53 mutations | NGS | Favorable OS in the palbociclib (+fulvestrant) vs. placebo (+fulvestrant) group was observed regardless of ESR1, PIK3CA, or TP53 mutation status; ESR1, PIK3CA and or TP53 mutations were prognostic for OS (HR = 1.58, 1.44 and 2.19, consequently). |
| Turner, N., 2020 [145] | 1034 | Advanced breast cancer, PlasmaMATCH trial | Mutations PIK3CA, ESR1, HER2, PTEN, and AKT1 | dPCR + NGS | Neratinib for HER2-mutant BC and capivasertib for AKT1-mutant BC identified by ctDNA testing had comparable activity to that observed when guided by tissue testing in previous study, respectively. ctDNA testing enables the selection of mutation-directed therapies for patients with BC. |
| Lyu, D., 2022 [146] | 113 | Metastatic ER+ breast cancer | Whole genome sequencing, PI3K/AKT/mTOR signaling pathway, ESR1, HER2 mutations | NGS | The risk of progression was lower in groups of patients who received treatment in accordance with the mutational status (HR = 0.55, p = 0.023) |
| Mastoraki, S., 2018 [147] | 58 | ER+ HER2- metastatic breast cancer | ESR1 methylation in CTCs and paired plasma ctDNA | Methylation- specific PCR | ESR1 methylation in CTCs and a high concordance with paired plasma ctDNA were reported. In serial peripheral blood samples of patients treated with everolimus/exemestane, ESR1 methylation was observed in 10/36 (27.8%) CTC-positive samples, and was associated with lack of response to treatment (p = 0.023). |
| Chimonidou, M., 2017 [148] | 153 | Breast cancer patients and healthy individuals | DNA methylation status of SOX17, CST6 and BRMS1 promoters in CTCs and ctDNA | Methylation- specific PCR | Association between the EpCAM (epithelial cell adhesion molecule)-positive CTC-fraction and ctDNA for SOX17 promoter methylation both for patients with early (p = 0.001) and metastatic BC (p = 0.046) was reported but not for CST6 and BRMS1. In early BC, SOX17 promoter methylation in the EpCAM-positive CTC-fraction was associated with CK-19 mRNA expression (p = 0.006) and worse OS (p = 0.044). In the metastatic setting, SOX17 promoter methylation in ctDNA was highly correlated with CK-19 (p = 0.04) and worse OS (p = 0.016). |
| Panagopoulou, M., 2019 [149] | 235 | 150 and 16 breast cancer patients under adjuvant and neoadjuvant therapy, respectively, 34 patients with metastatic disease and 35 healthy volunteers | Methylation status of a panel of cancer-related genes (KLK10, SOX17, WNT5A, MSH2, GATA3) | Methylation- specific PCR | Methylation of at least 3 or 4 genes was significantly correlated to shorter OS and no pharmacotherapy response, respectively. Classification analysis by a fully automated, machine learning software (JADBio software, Gnosis Data Analysis) produced a single-parametric linear model using cfDNA plasma concentration values, with great discriminating power to predict response to therapy (AUC 0.803, 95% CI 0.606–1.000) in the metastatic group. Two more multi-parametric signatures were produced for the metastatic group, predicting survival and disease outcome. A multiple logistic regression model was constructed, discriminating between patient groups and healthy individuals. cfDNA emerged as a highly potent predictive classifier in metastatic BC. |
| Yi, Z., 2021 [154] | 125 | Metastatic breast cancer, CAMELLIA trial | Target-capture deep sequencing of 1021 genes to detect somatic variants in ctDNA; determining the molecular tumor burden index (mTBI) | NGS | High-level pretreatment mTBI was correlated with shorter OS (p = 0.011). Patients with an mTBI decrease to less than 0.02% at the first tumor evaluation had longer PFS and OS (p < 0.001 and p = 0.007, respectively). The patients classified as molecular responders had longer PFS and OS than the nonmolecular responders (p < 0.001 and p = 0.036, respectively). |
| Murtaza, M., 2015 [172] | 1 | Metastatic ER+ HER2+ breast cancer | Genomic architecture and infer clonal evolution in eight tumor biopsies and nine plasma samples collected over 1193 days of clinical follow-up were characterized using exome and targeted amplicon sequencing | NGS | Ubiquitous stem mutations (common to all tumor biopsies) have the highest circulating levels in plasma followed by metastatic-clade and private mutations. In addition, serial changes during treatment in circulating levels of private somatic mutations correlate with disease progression in their respective tumor lesions on imaging. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavarykina, T.M.; Lomskova, P.K.; Pronina, I.V.; Khokhlova, S.V.; Stenina, M.B.; Sukhikh, G.T. Circulating Tumor DNA Is a Variant of Liquid Biopsy with Predictive and Prognostic Clinical Value in Breast Cancer Patients. Int. J. Mol. Sci. 2023, 24, 17073. https://doi.org/10.3390/ijms242317073
Zavarykina TM, Lomskova PK, Pronina IV, Khokhlova SV, Stenina MB, Sukhikh GT. Circulating Tumor DNA Is a Variant of Liquid Biopsy with Predictive and Prognostic Clinical Value in Breast Cancer Patients. International Journal of Molecular Sciences. 2023; 24(23):17073. https://doi.org/10.3390/ijms242317073
Chicago/Turabian StyleZavarykina, Tatiana M., Polina K. Lomskova, Irina V. Pronina, Svetlana V. Khokhlova, Marina B. Stenina, and Gennady T. Sukhikh. 2023. "Circulating Tumor DNA Is a Variant of Liquid Biopsy with Predictive and Prognostic Clinical Value in Breast Cancer Patients" International Journal of Molecular Sciences 24, no. 23: 17073. https://doi.org/10.3390/ijms242317073
APA StyleZavarykina, T. M., Lomskova, P. K., Pronina, I. V., Khokhlova, S. V., Stenina, M. B., & Sukhikh, G. T. (2023). Circulating Tumor DNA Is a Variant of Liquid Biopsy with Predictive and Prognostic Clinical Value in Breast Cancer Patients. International Journal of Molecular Sciences, 24(23), 17073. https://doi.org/10.3390/ijms242317073