
Identifying Explainable Machine Learning Models and a Novel SFRP2+ Fibroblast Signature as Predictors for Precision Medicine in Ovarian Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
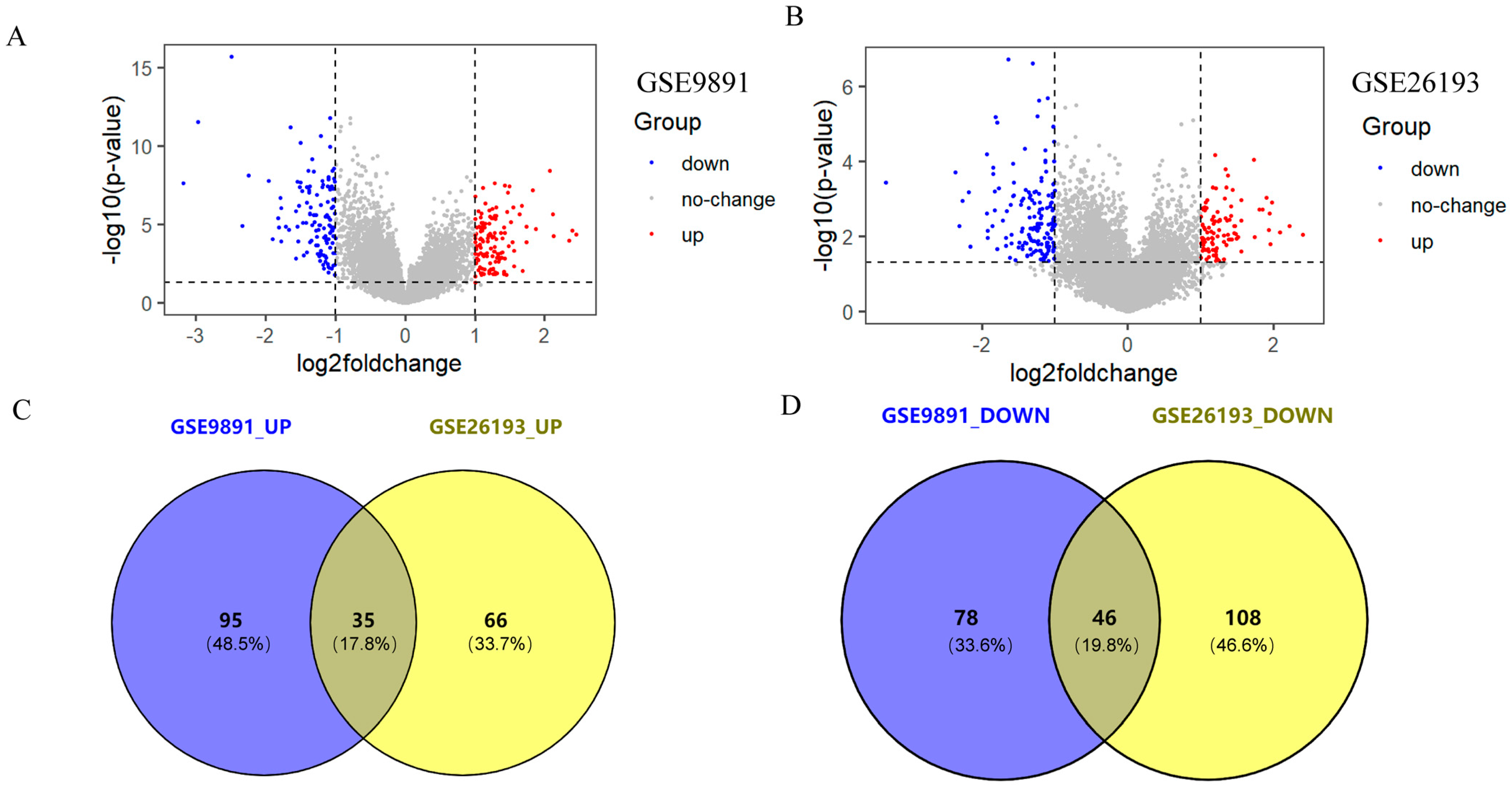
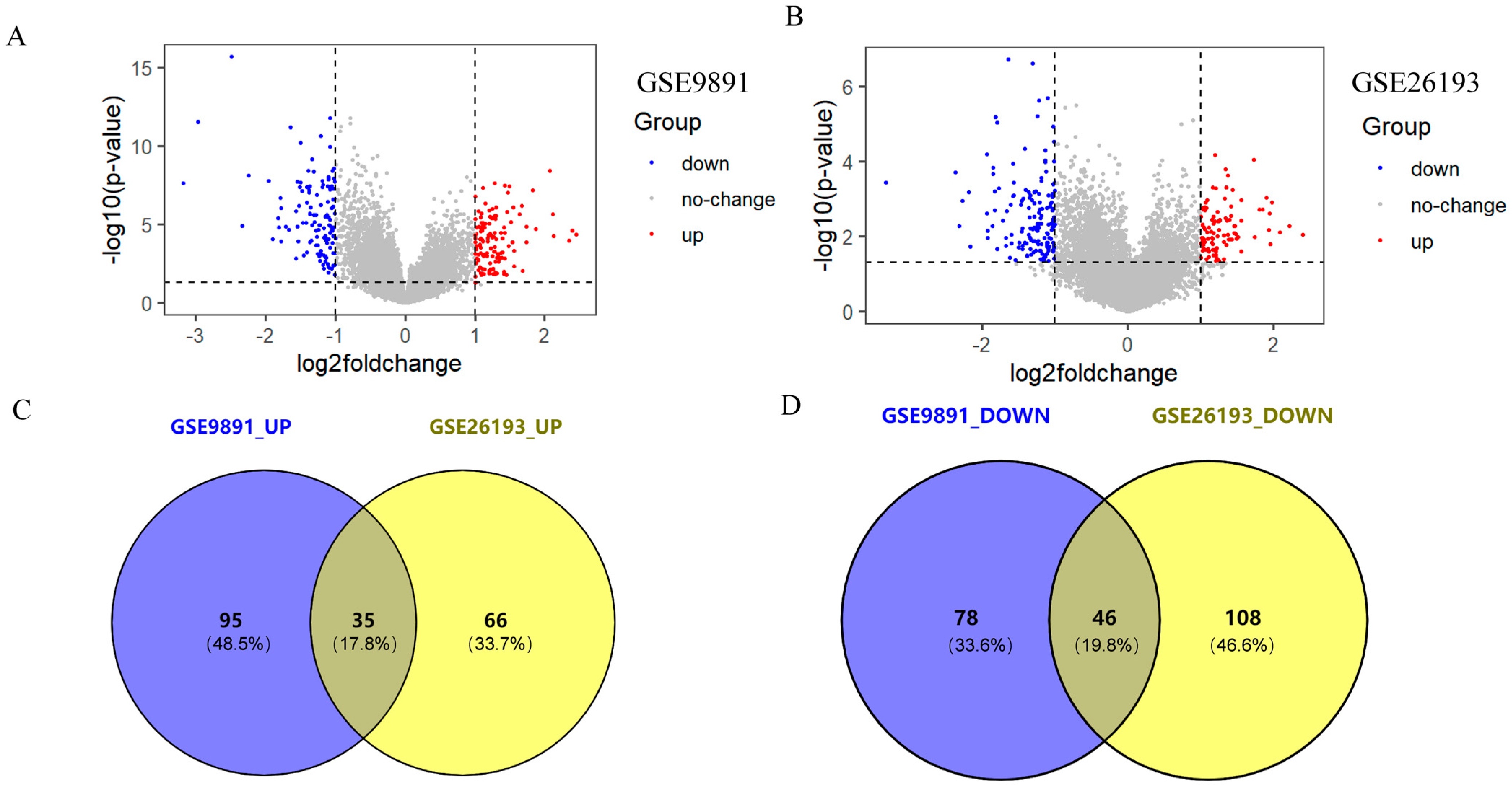
2.1. Acquisition of Differentially Expressed Genes (DEGs)
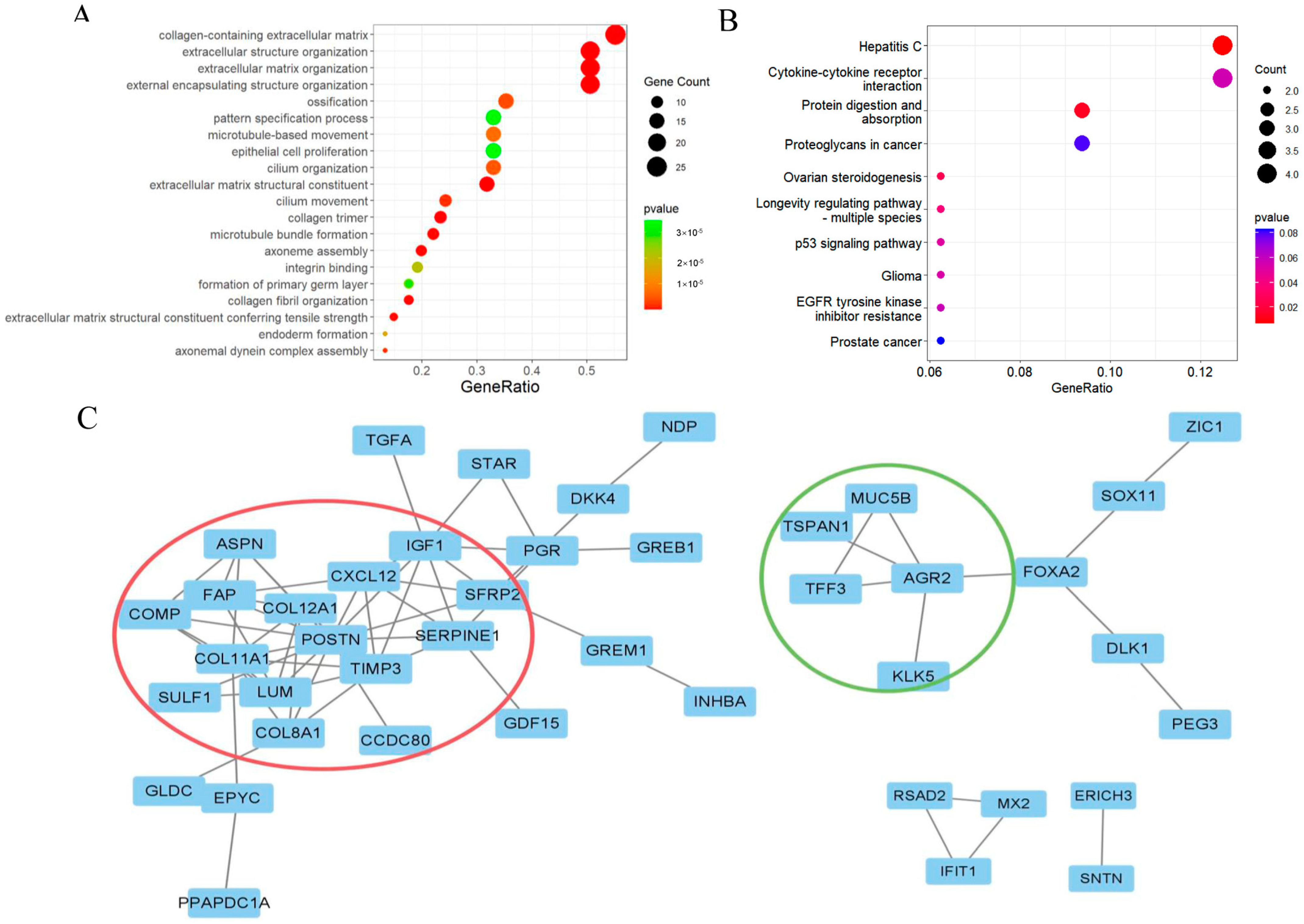
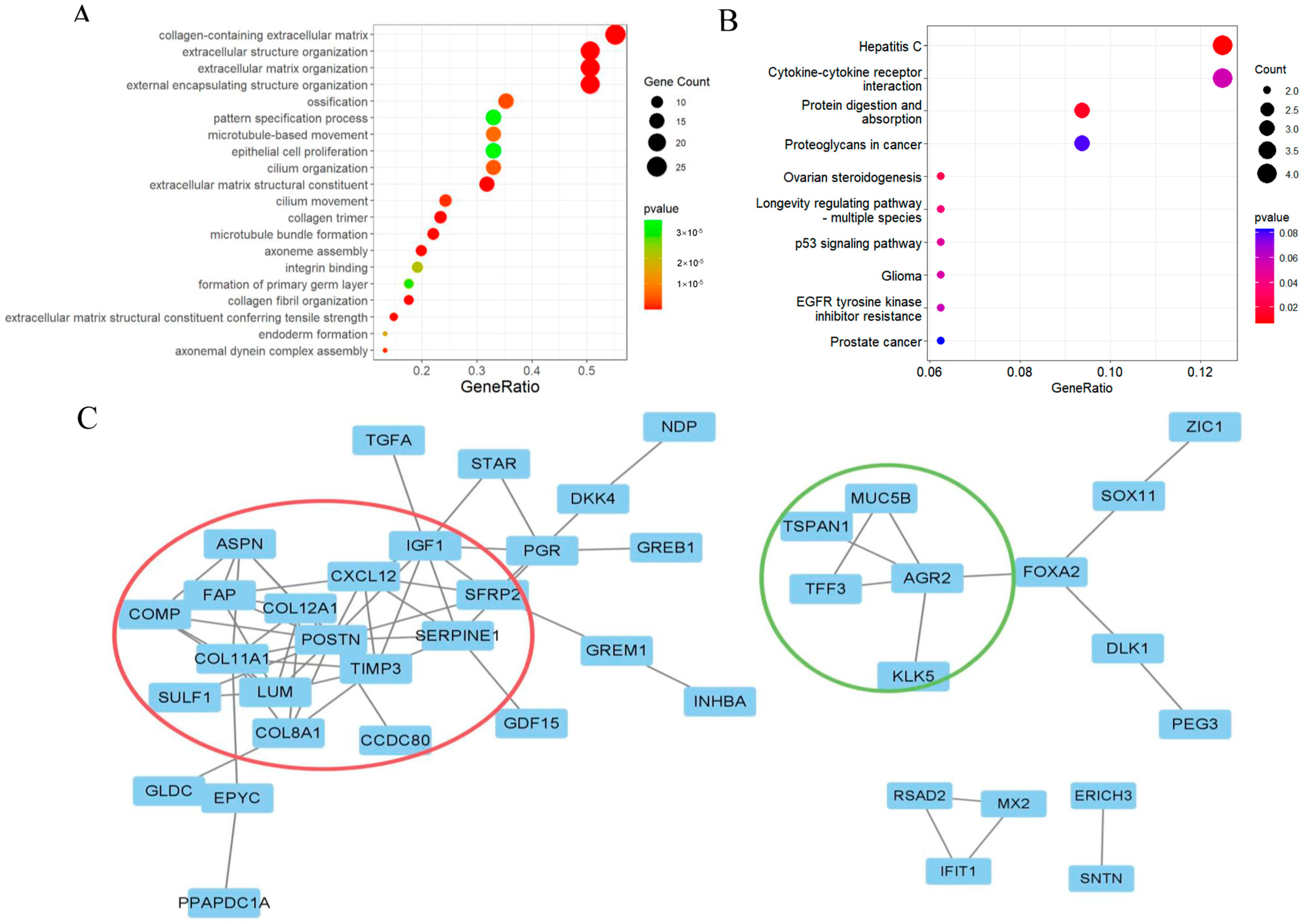
2.2. Functional Enrichment Analysis
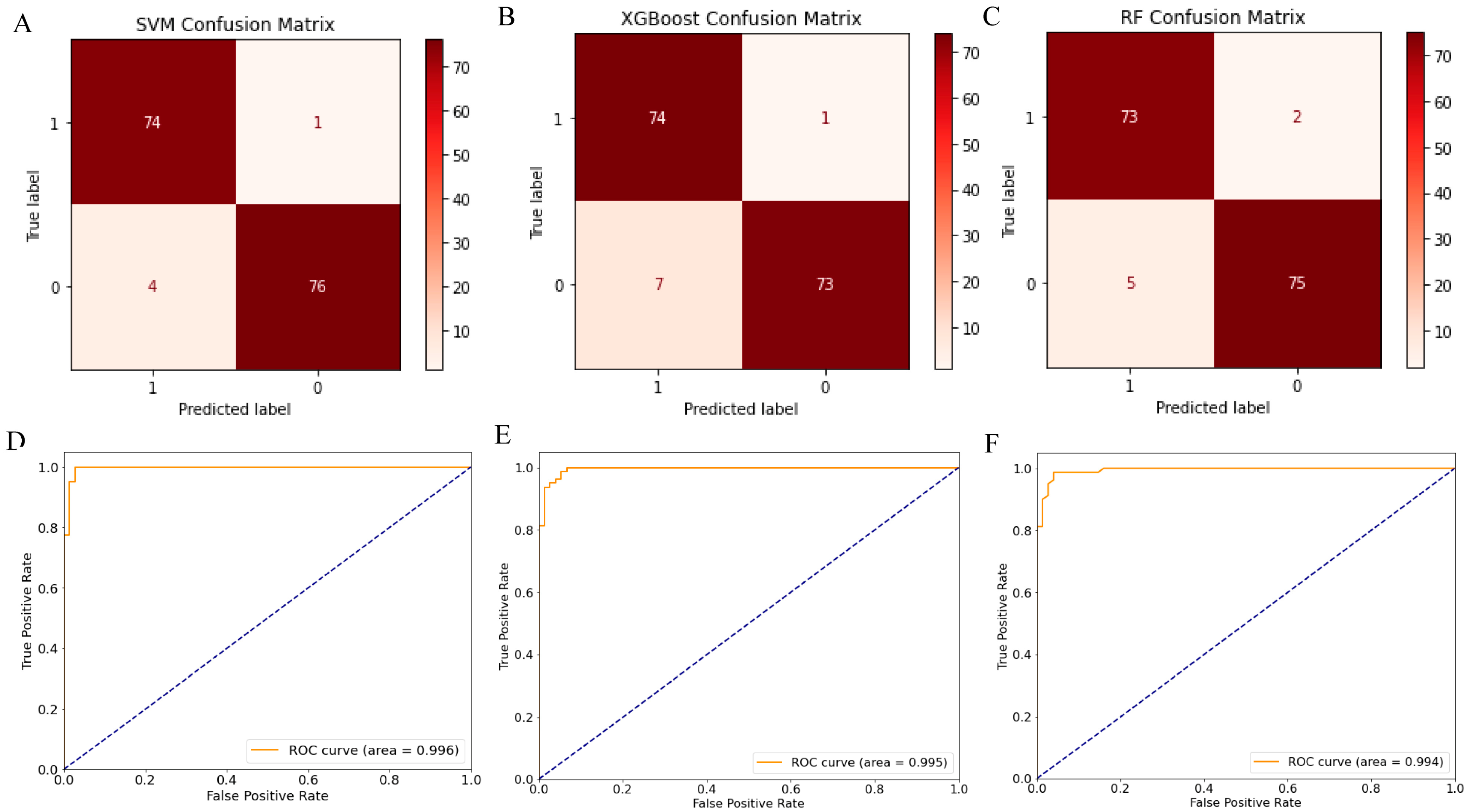
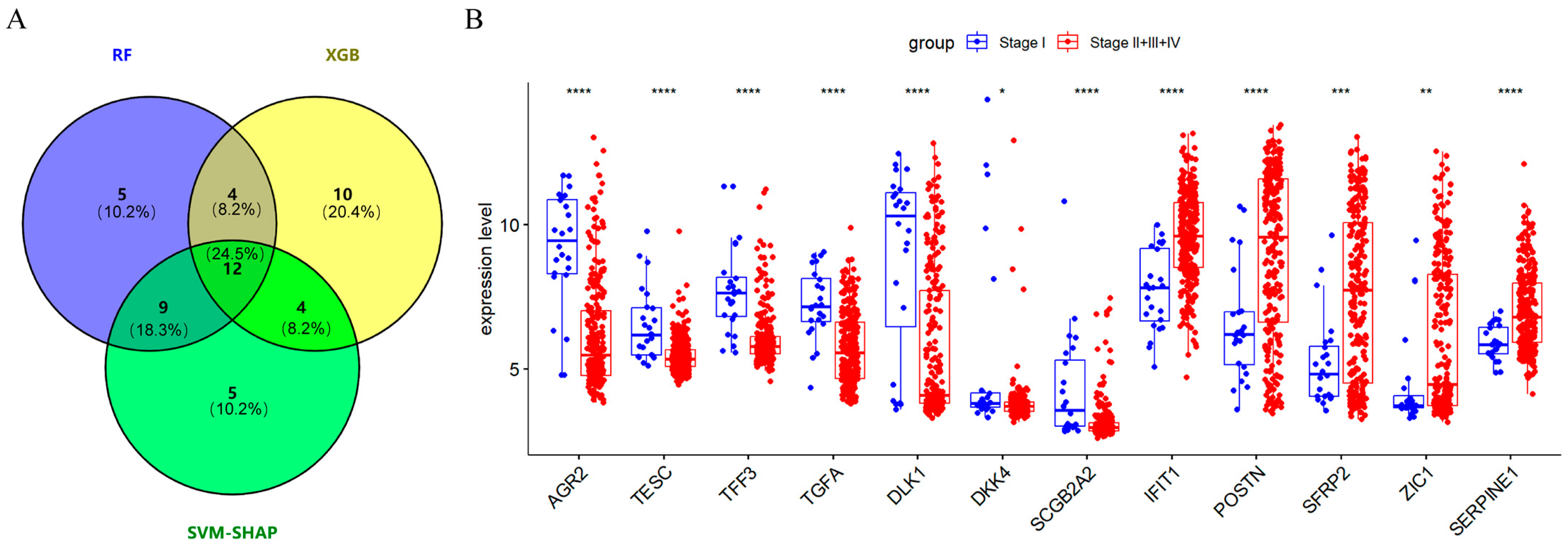
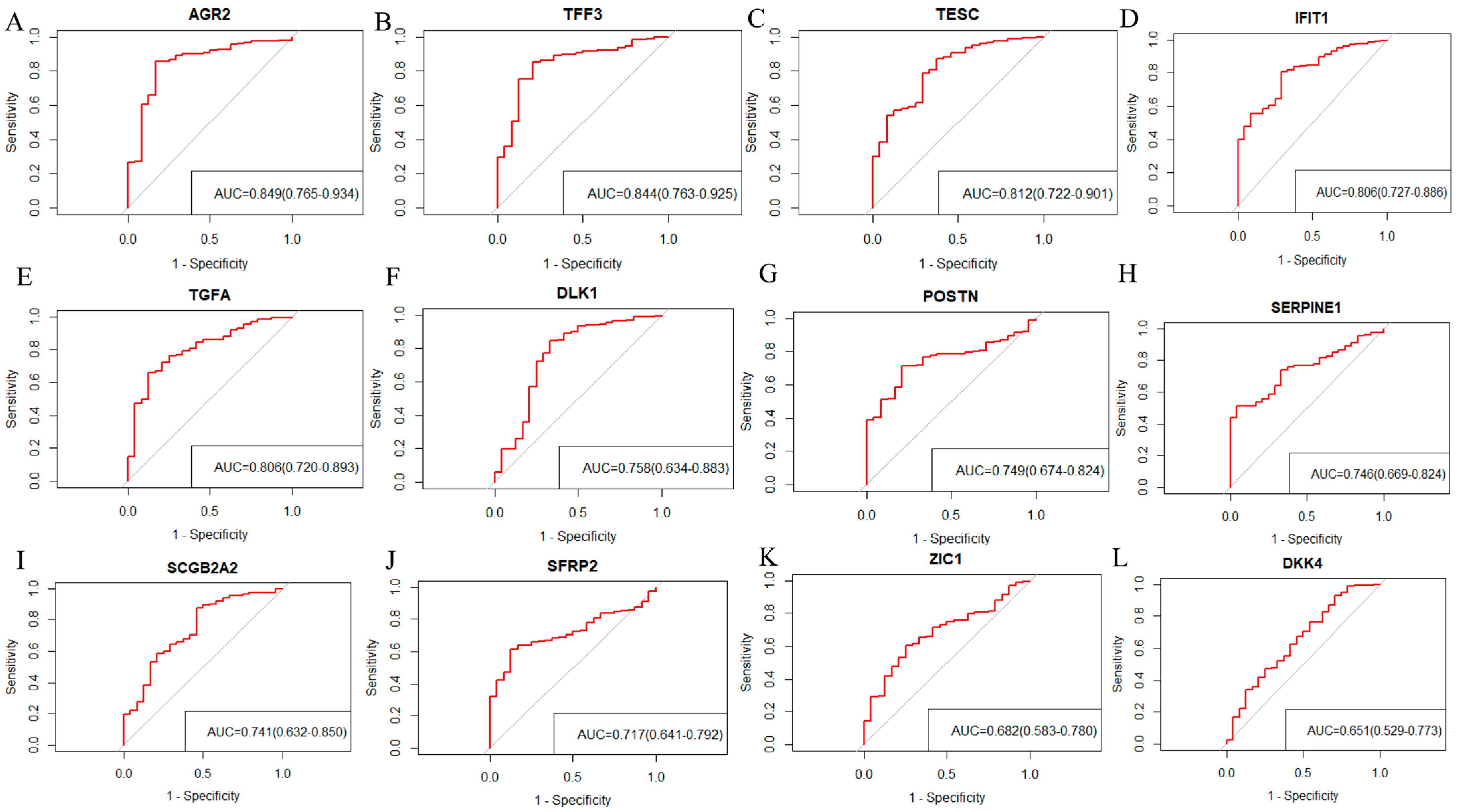
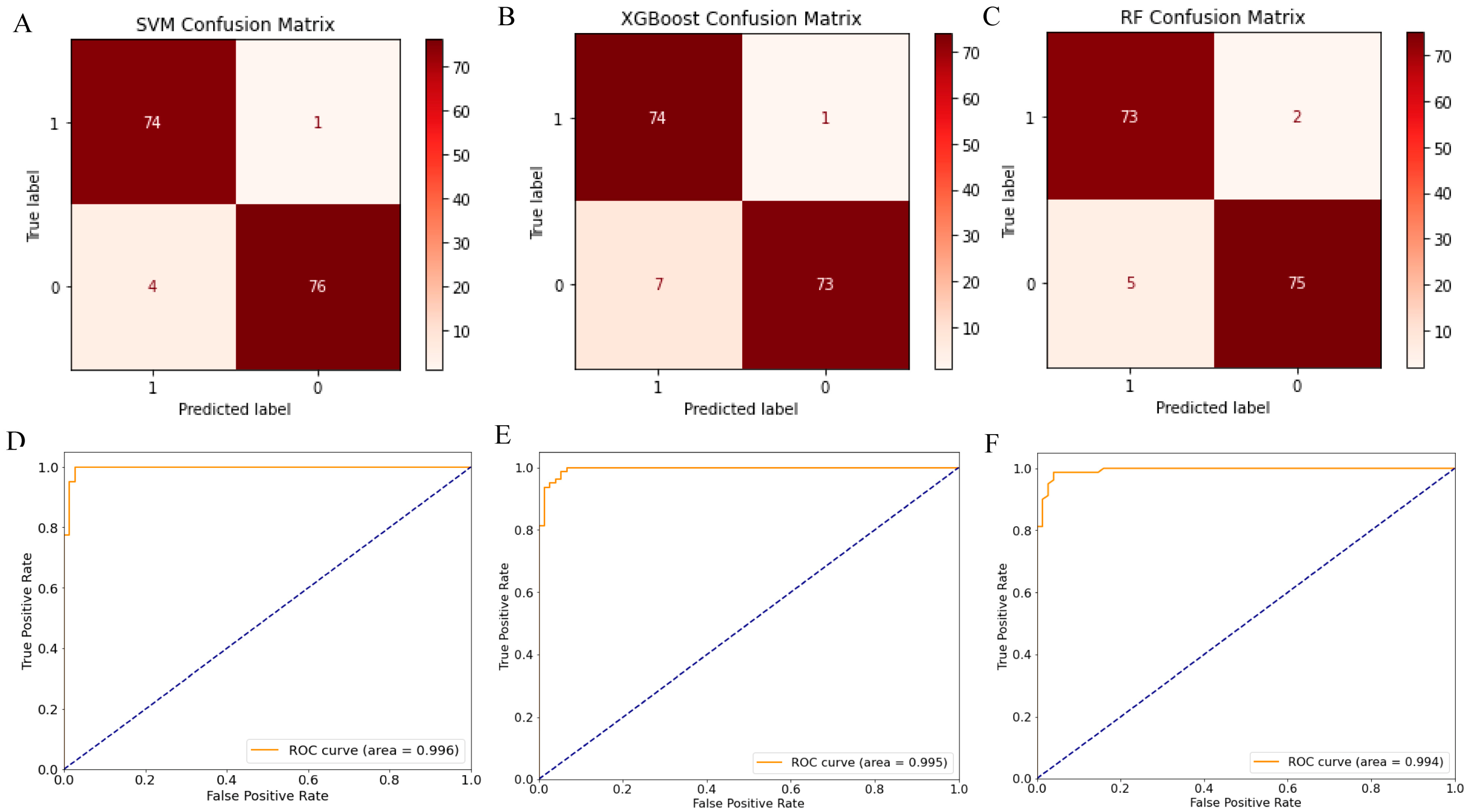
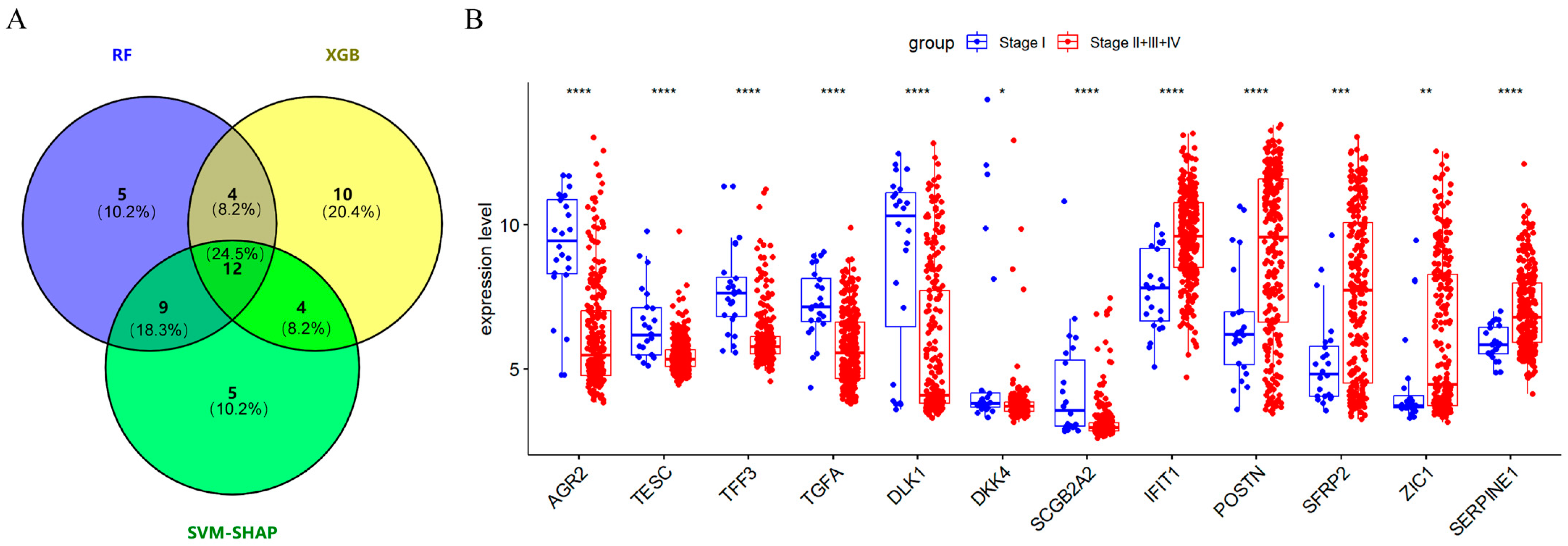
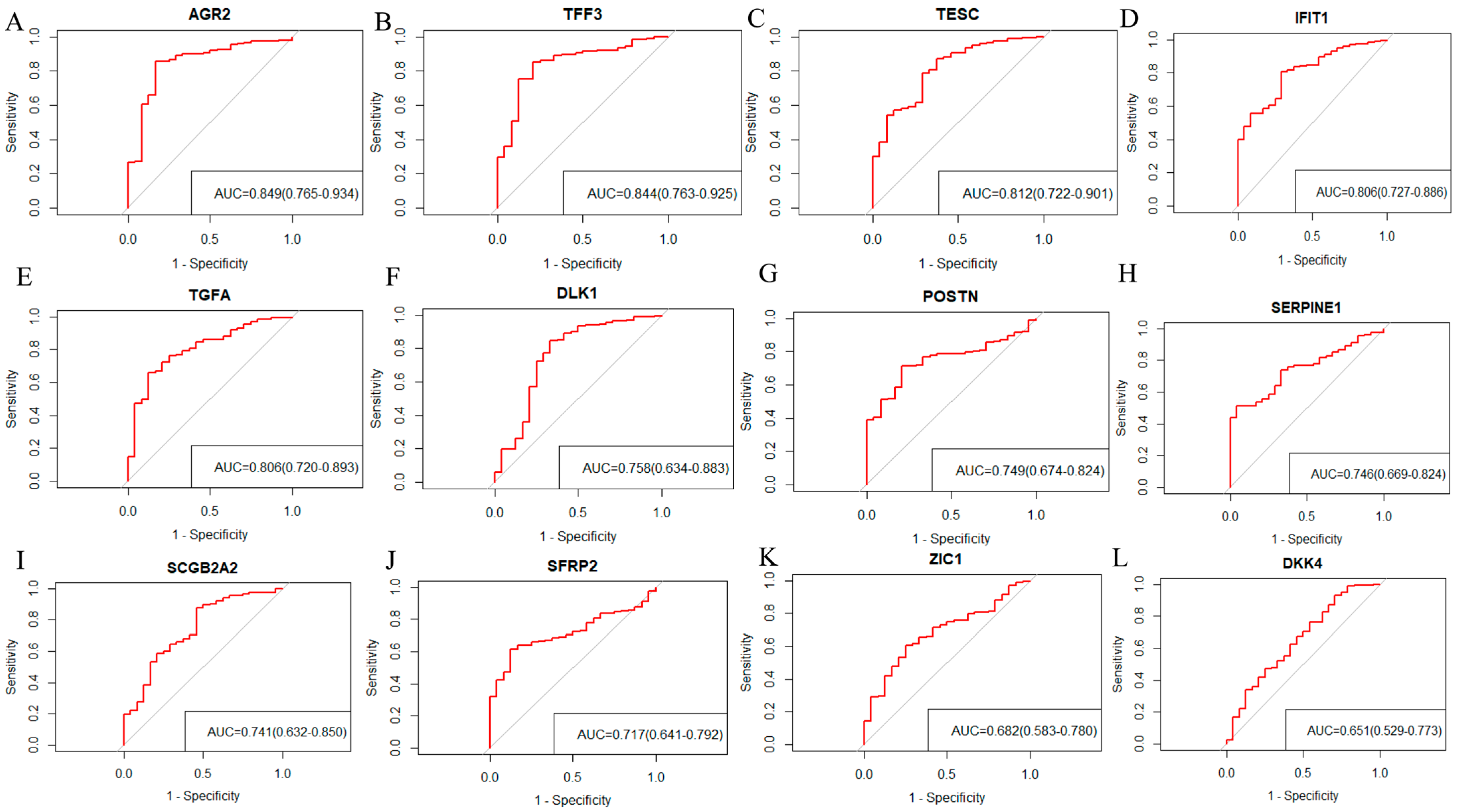
2.3. Screening 12 DEGs as Diagnostic Markers for Detecting Early-Stage OC Patients
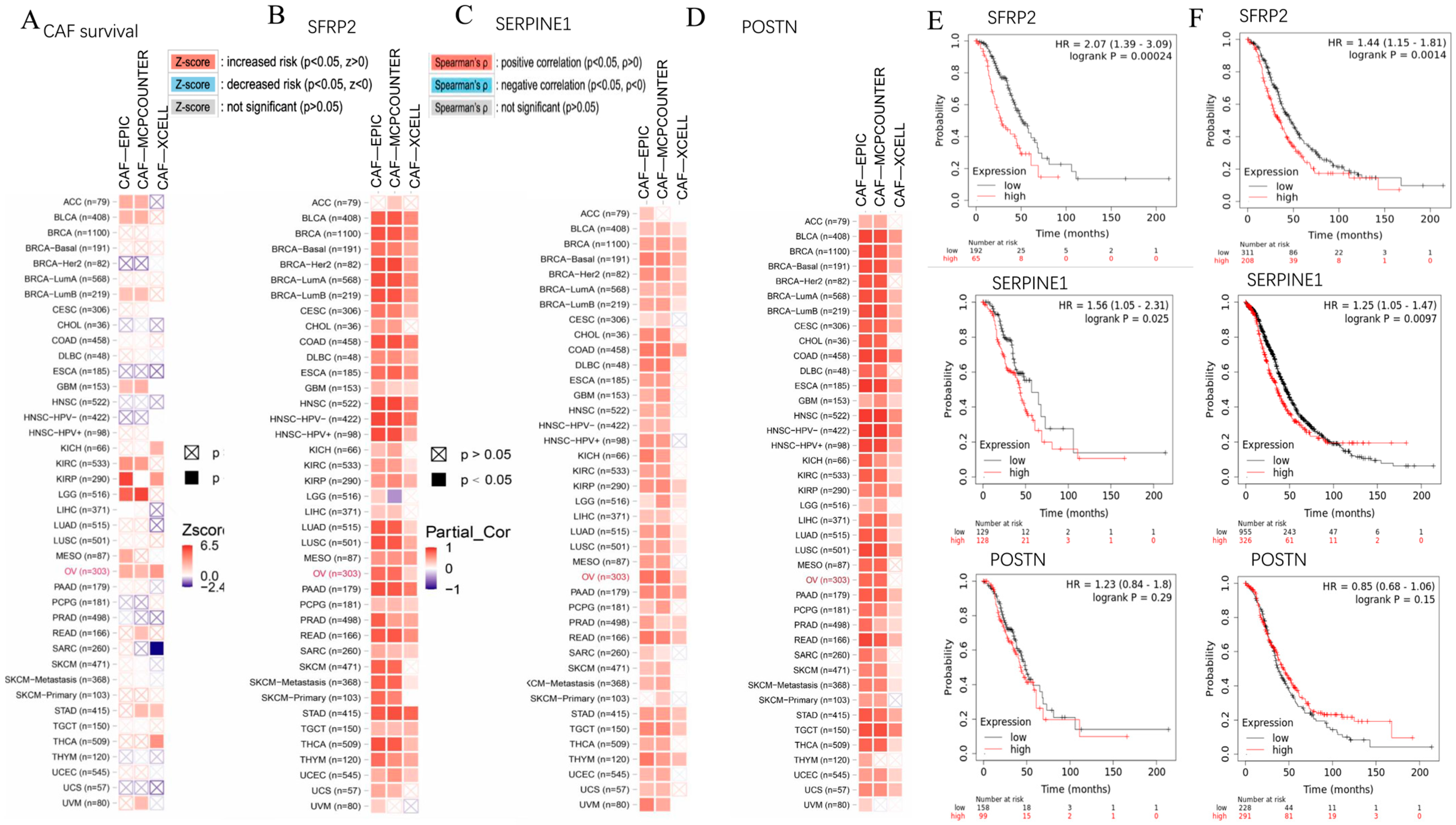
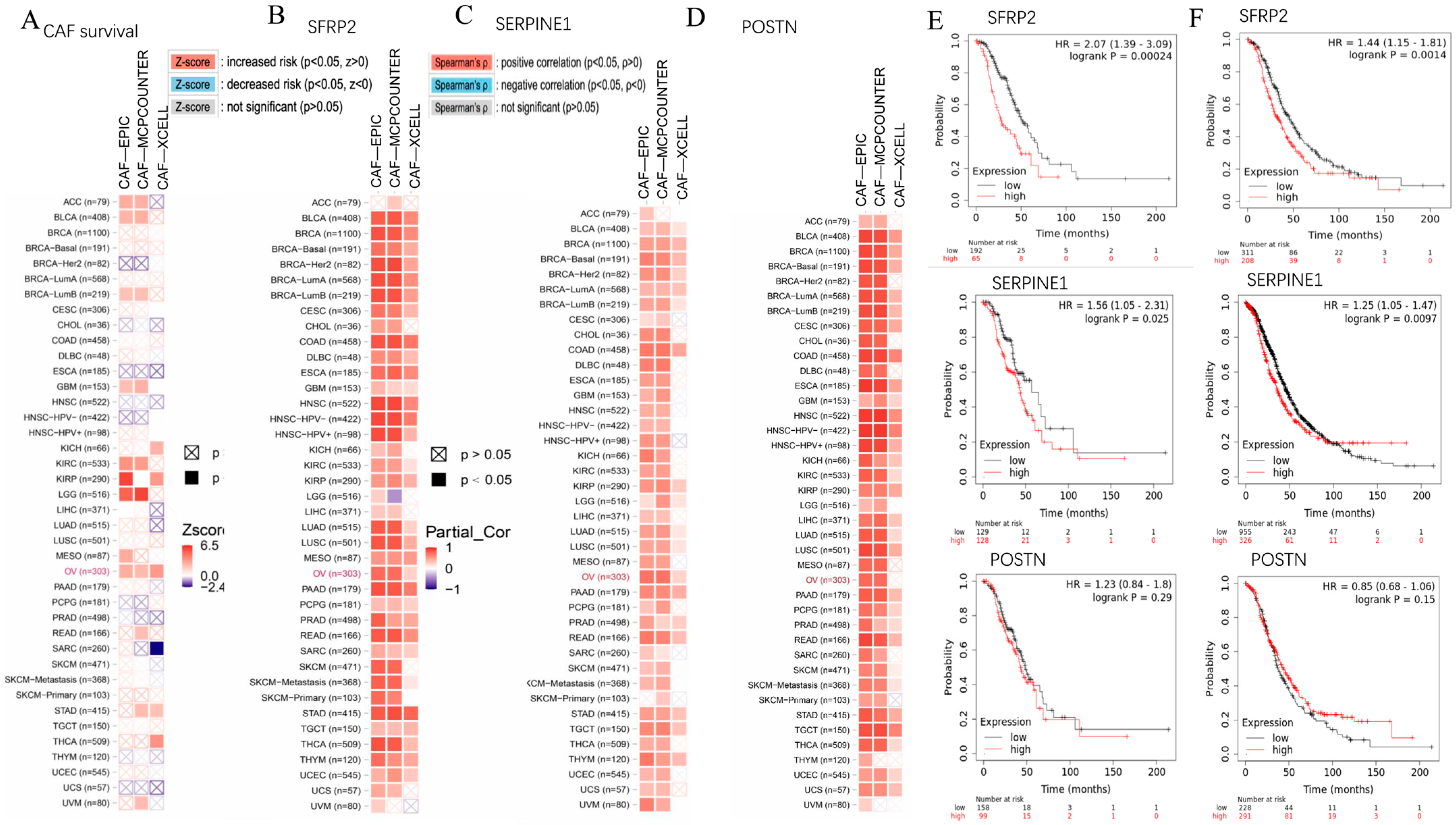
2.4. SFRP2 and SERPINE1 Were Intricately Linked to Cancer-Associated Fibroblasts and Associated with the Overall Survival of Patients in Moderate to Advanced-Stage OC
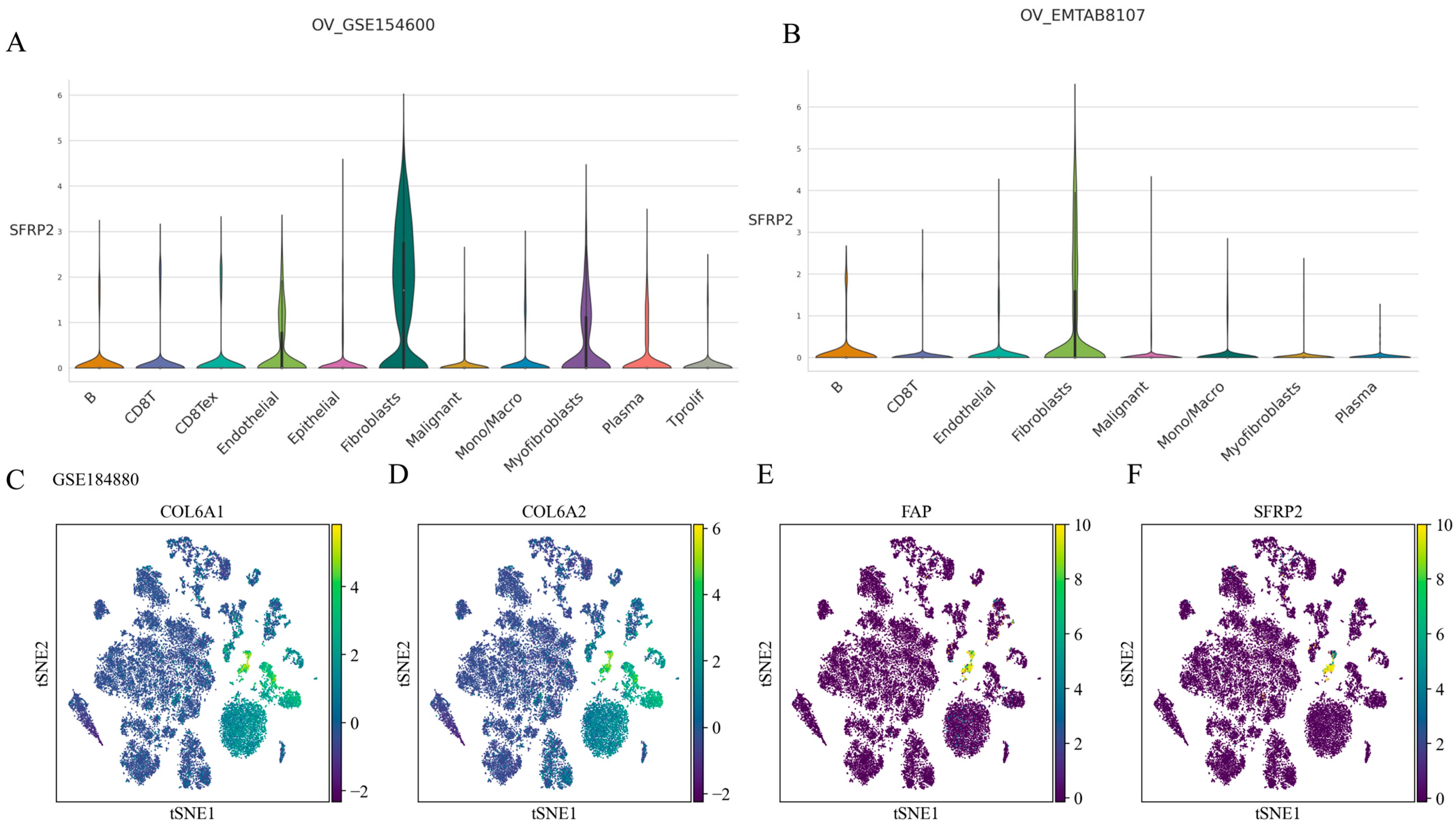
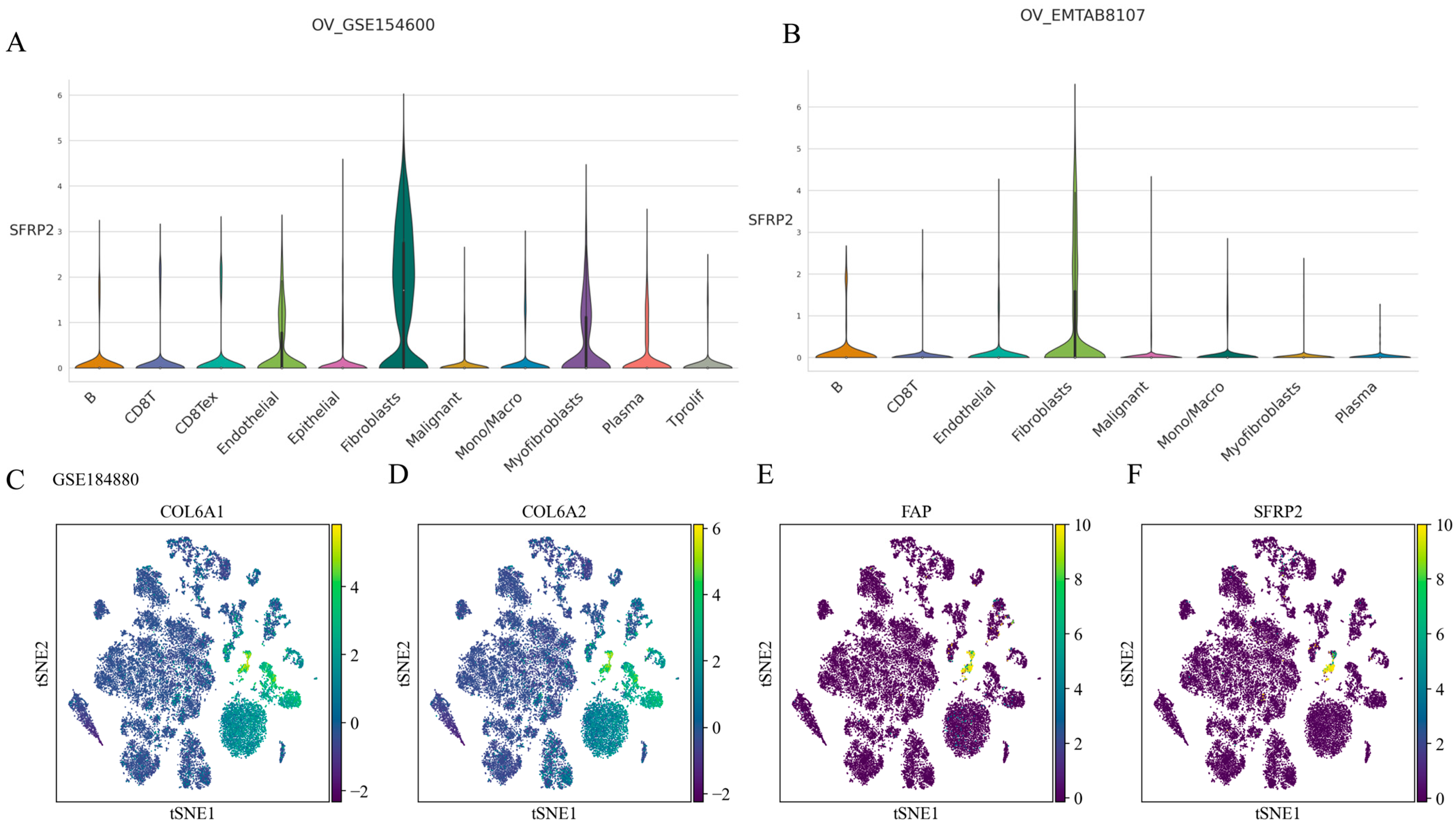
2.5. SFRP2 Might Define a Distinct CAF Subpopulation
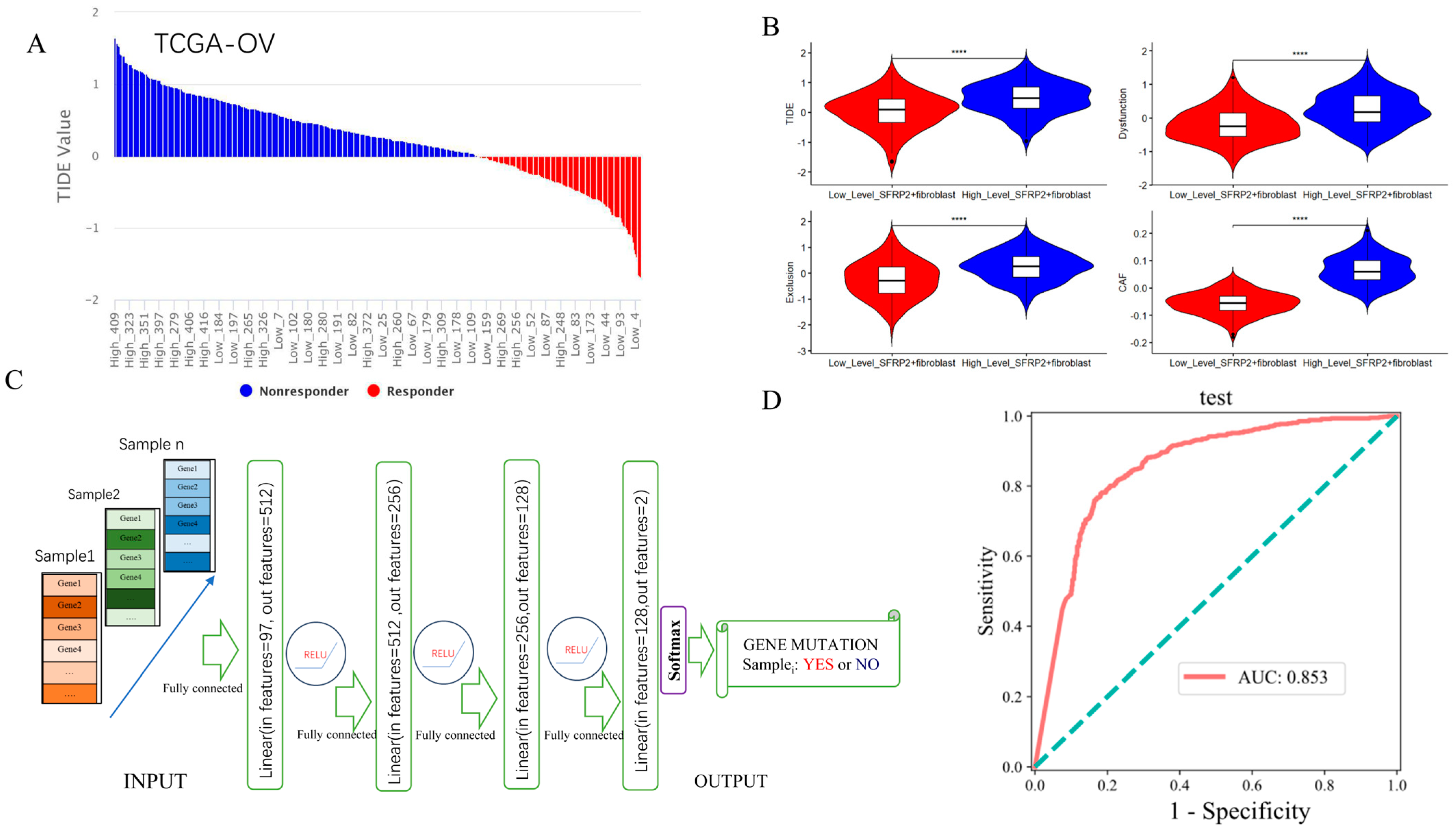
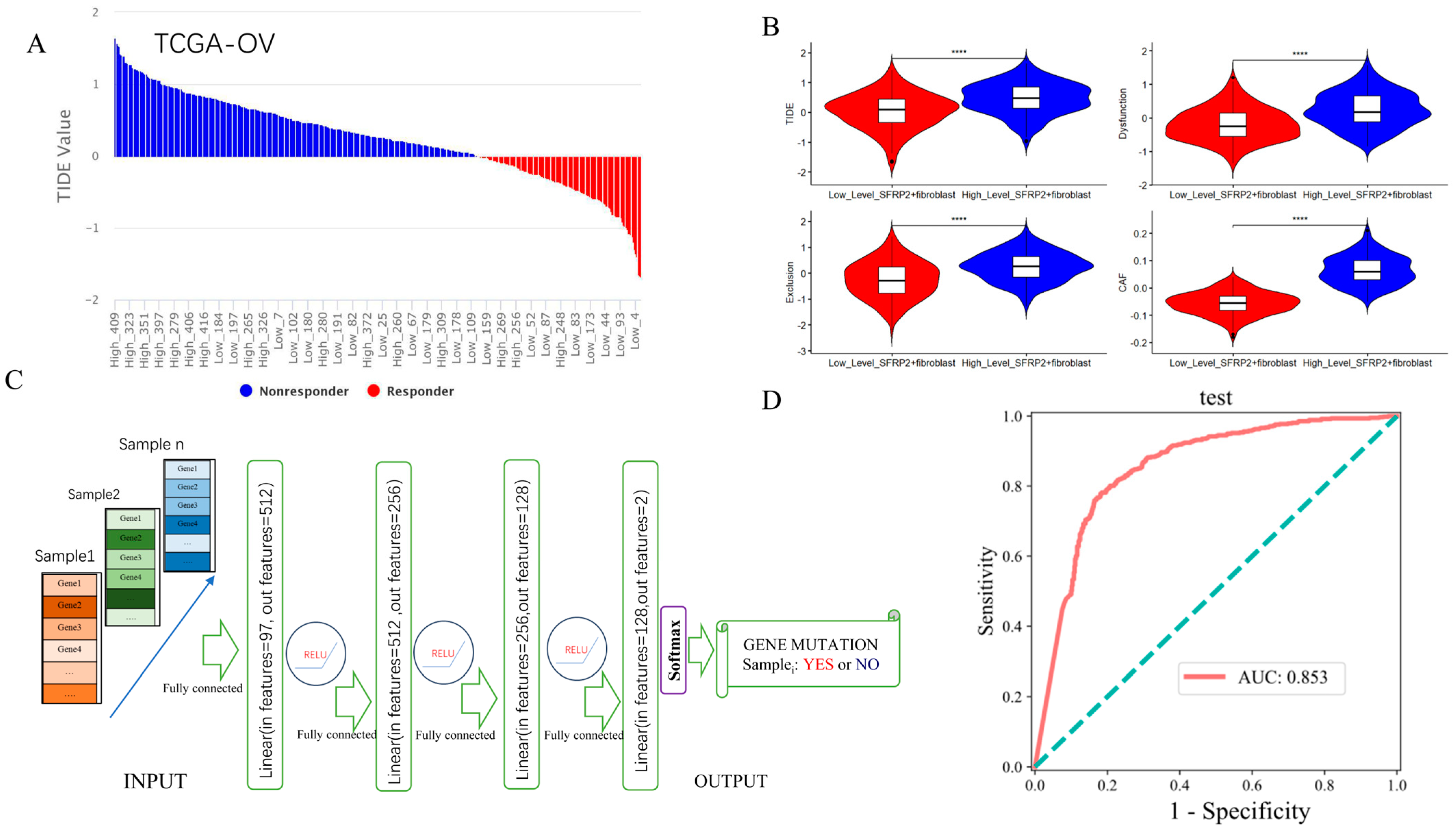
2.6. Contribution of SFRP2+ Fibroblast Signature in Predicting ICI Response and Detecting Pan-Cancer TP53 Mutation
3. Discussion
4. Materials and Methods
4.1. Data Collection and Processing
4.2. Screening and Analysis of Overlapping DEGs
4.3. Functional Analysis of Overlapping DEGs
4.4. Explainable Machine Learning
4.5. Pan-Cancer Bioinformatics Analysis Using the TIMER2 Website
4.6. Survival Analysis
4.7. Single-Cell RNA Sequencing Analysis
4.8. Single-Sample Gene Set Enrichment Analysis (ssGSEA)
4.9. Tumor Immune Dysfunction and Exclusion (TIDE) Analysis
4.10. Identification of Pan-Cancer TP53 Mutation by Artificial Neural Network
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fu, H.; Fu, Z.; Mao, M.; Si, L.; Bai, J.; Wang, Q.; Guo, R. Prevalence and prognostic role of PD-L1 in patients with gynecological cancers: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2023, 189, 104084. [Google Scholar] [CrossRef] [PubMed]
- Esfandyari, S.; Elkafas, H.; Chugh, R.M.; Park, H.S.; Navarro, A.; Al-Hendy, A. Exosomes as Biomarkers for Female Reproductive Diseases Diagnosis and Therapy. Int. J. Mol. Sci. 2021, 22, 2165. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, X.Y.; Shan, W.L.; Chen, Y.; Zhu, Q.; Xia, B.R. Targeted therapy and immunotherapy: Diamonds in the rough in the treatment of epithelial ovarian cancer. Front. Pharmacol. 2023, 14, 1131342. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Kwolek, D.G.; Gerstberger, S.; Tait, S.; Qiu, J.M. Ovarian, Uterine, and Vulvovaginal Cancers: Screening, Treatment Overview, and Prognosis. Med. Clin. N. Am. 2023, 107, 329–355. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Bast, R.C., Jr. Minireview: Human ovarian cancer: Biology, current management, and paths to personalizing therapy. Endocrinology 2012, 153, 1593–1602. [Google Scholar] [CrossRef]
- Prat, J.; Oncology, F.C.O.G. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef]
- O’Shea, A.S. Clinical Staging of Ovarian Cancer. Methods Mol. Biol. 2022, 2424, 3–10. [Google Scholar] [CrossRef]
- Baldwin, L.A.; Huang, B.; Miller, R.W.; Tucker, T.; Goodrich, S.T.; Podzielinski, I.; DeSimone, C.P.; Ueland, F.R.; van Nagell, J.R.; Seamon, L.G. Ten-year relative survival for epithelial ovarian cancer. Obstet. Gynecol. 2012, 120, 612–618. [Google Scholar] [CrossRef]
- Arora, T.; Mullangi, S.; Lekkala, M.R. Ovarian Cancer. In StatPearls; StatPearls Publishing Copyright© 2023; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Akter, S.; Rahman, M.A.; Hasan, M.N.; Akhter, H.; Noor, P.; Islam, R.; Shin, Y.; Rahman, M.D.H.; Gazi, M.S.; Huda, M.N.; et al. Recent Advances in Ovarian Cancer: Therapeutic Strategies, Potential Biomarkers, and Technological Improvements. Cells 2022, 11, 650. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, L.; Guntupalli, S.R. Treatment of epithelial ovarian cancer. BMJ 2020, 371, m3773. [Google Scholar] [CrossRef]
- Bristow, R.E.; Tomacruz, R.S.; Armstrong, D.K.; Trimble, E.L.; Montz, F.J. Survival Effect of Maximal Cytoreductive Surgery for Advanced Ovarian Carcinoma During the Platinum Era: A Meta-Analysis. J. Clin. Oncol. 2002, 20, 1248–1259, reprinted in J. Clin. Oncol. 2023, 41, 4065–4076. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; Liu, P.Y.; Wilczynski, S.; Monk, B.; Copeland, L.J.; Alvarez, R.D.; Jiang, C.; Alberts, D. Phase III randomized trial of 12 versus 3 months of maintenance paclitaxel in patients with advanced ovarian cancer after complete response to platinum and paclitaxel-based chemotherapy: A Southwest Oncology Group and Gynecologic Oncology Group trial. J. Clin. Oncol. 2003, 21, 2460–2465. [Google Scholar] [CrossRef]
- Ngoi, N.Y.; Syn, N.L.; Goh, R.M.; Goh, B.C.; Huang, R.Y.; Soon, Y.Y.; James, E.; Cook, A.; Clamp, A.; Tan, D.S. Weekly versus tri-weekly paclitaxel with carboplatin for first-line treatment in women with epithelial ovarian cancer. Cochrane Database Syst. Rev. 2022, 2, CD012007. [Google Scholar] [CrossRef] [PubMed]
- Luvero, D.; Milani, A.; Ledermann, J.A. Treatment options in recurrent ovarian cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2014, 6, 229–239. [Google Scholar] [CrossRef]
- Liu, J.F. Management of Advanced Ovarian, Fallopian Tube, and Primary Peritoneal Cancers. J. Natl. Compr. Cancer Netw. 2023, 21, 1–4. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Harter, P.; Leary, A.; Lorusso, D.; Miller, R.E.; Pothuri, B.; Ray-Coquard, I.; Tan, D.S.P.; Bellet, E.; Oaknin, A.; et al. Newly diagnosed and relapsed epithelial ovarian cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 833–848. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Li, N.; Zhang, Y.; Wang, J.; Zhu, J.; Wang, L.; Wu, X.; Yao, D.; Wu, Q.; Liu, J.; Tang, J.; et al. Fuzuloparib Maintenance Therapy in Patients With Platinum-Sensitive, Recurrent Ovarian Carcinoma (FZOCUS-2): A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase III Trial. J. Clin. Oncol. 2022, 40, 2436–2446. [Google Scholar] [CrossRef]
- Li, N.; Zhu, J.; Yin, R.; Wang, J.; Pan, L.; Kong, B.; Zheng, H.; Liu, J.; Wu, X.; Wang, L.; et al. Treatment With Niraparib Maintenance Therapy in Patients With Newly Diagnosed Advanced Ovarian Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2023, 9, 1230–1237. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Griffiths, R.W.; Zee, Y.K.; Evans, S.; Mitchell, C.L.; Kumaran, G.C.; Welch, R.S.; Jayson, G.C.; Clamp, A.R.; Hasan, J. Outcomes after multiple lines of chemotherapy for platinum-resistant epithelial cancers of the ovary, peritoneum, and fallopian tube. Int. J. Gynecol. Cancer 2011, 21, 58–65. [Google Scholar] [CrossRef]
- Takeiwa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Mechanisms of Apoptosis-Related Long Non-coding RNAs in Ovarian Cancer. Front. Cell Dev. Biol. 2021, 9, 641963. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Ogundipe, O.D.; Olajubutu, O.; Adesina, S.K. Targeted drug conjugate systems for ovarian cancer chemotherapy. Biomed. Pharmacother. 2023, 165, 115151. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Matulonis, U.A. Clinical and translational advances in ovarian cancer therapy. Nat. Cancer 2023, 4, 1239–1257. [Google Scholar] [CrossRef]
- Zhou, J.; Cao, W.; Wang, L.; Pan, Z.; Fu, Y. Application of artificial intelligence in the diagnosis and prognostic prediction of ovarian cancer. Comput. Biol. Med. 2022, 146, 105608. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, J.; Rutledge, H.; Acharya, U.R.; Loh, H.W.; Gururajan, R.; Tao, X.; Zhou, X.; Li, Y.; Gurney, T.; Kondalsamy-Chennakesavan, S. Gynecological cancer prognosis using machine learning techniques: A systematic review of the last three decades (1990–2022). Artif. Intell. Med. 2023, 139, 102536. [Google Scholar] [CrossRef] [PubMed]
- Breen, J.; Allen, K.; Zucker, K.; Adusumilli, P.; Scarsbrook, A.; Hall, G.; Orsi, N.M.; Ravikumar, N. Artificial intelligence in ovarian cancer histopathology: A systematic review. NPJ Precis. Oncol. 2023, 7, 83. [Google Scholar] [CrossRef]
- Corti, C.; Cobanaj, M.; Dee, E.C.; Criscitiello, C.; Tolaney, S.M.; Celi, L.A.; Curigliano, G. Artificial intelligence in cancer research and precision medicine: Applications, limitations and priorities to drive transformation in the delivery of equitable and unbiased care. Cancer Treat Rev. 2023, 112, 102498. [Google Scholar] [CrossRef]
- Wang, R.; Pan, W.; Jin, L.; Li, Y.; Geng, Y.; Gao, C.; Chen, G.; Wang, H.; Ma, D.; Liao, S. Artificial intelligence in reproductive medicine. Reproduction 2019, 158, R139–R154. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, W.; Tu, H. Big data and artificial intelligence in cancer research. Trends Cancer 2023. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Serum proteomic pattern analysis for early cancer detection. Technol. Cancer Res. Treat. 2006, 5, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.; Kanter, F.; Kulbe, H.; Jank, P.; Denkert, C.; Nebrich, G.; Schmitt, W.D.; Wu, Z.; Kunze, C.A.; Sehouli, J.; et al. MALDI-Imaging for Classification of Epithelial Ovarian Cancer Histotypes from a Tissue Microarray Using Machine Learning Methods. Proteom. Clin. Appl. 2019, 13, e1700181. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; De Smet, F.; Van Gorp, T.; Pochet, N.; Engelen, K.; Amant, F.; De Moor, B.; Timmerman, D.; Vergote, I. Expression profiling to predict the clinical behaviour of ovarian cancer fails independent evaluation. BMC Cancer 2008, 8, 18. [Google Scholar] [CrossRef]
- Chaddad, A.; Niazi, T.; Probst, S.; Bladou, F.; Anidjar, M.; Bahoric, B. Predicting Gleason Score of Prostate Cancer Patients Using Radiomic Analysis. Front. Oncol. 2018, 8, 630. [Google Scholar] [CrossRef]
- Wei, W.; Li, Y.; Huang, T. Using Machine Learning Methods to Study Colorectal Cancer Tumor Micro-Environment and Its Biomarkers. Int. J. Mol. Sci. 2023, 24, 11133. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Salehi, A.; Wang, L.; Coates, P.J.; Norberg Spaak, L.; Gu, X.; Sgaramella, N.; Nylander, K. Reiterative modeling of combined transcriptomic and proteomic features refines and improves the prediction of early recurrence in squamous cell carcinoma of head and neck. Comput. Biol. Med. 2022, 149, 105991. [Google Scholar] [CrossRef]
- Li, W.; Yin, Y.; Quan, X.; Zhang, H. Gene Expression Value Prediction Based on XGBoost Algorithm. Front. Genet. 2019, 10, 1077. [Google Scholar] [CrossRef]
- Hathaway, Q.A.; Roth, S.M.; Pinti, M.V.; Sprando, D.C.; Kunovac, A.; Durr, A.J.; Cook, C.C.; Fink, G.K.; Cheuvront, T.B.; Grossman, J.H.; et al. Machine-learning to stratify diabetic patients using novel cardiac biomarkers and integrative genomics. Cardiovasc. Diabetol. 2019, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Sorayaie Azar, A.; Babaei Rikan, S.; Naemi, A.; Bagherzadeh Mohasefi, J.; Pirnejad, H.; Bagherzadeh Mohasefi, M.; Wiil, U.K. Application of machine learning techniques for predicting survival in ovarian cancer. BMC Med. Inform. Decis. Mak. 2022, 22, 345. [Google Scholar] [CrossRef] [PubMed]
- Yagin, F.H.; Cicek, I.B.; Alkhateeb, A.; Yagin, B.; Colak, C.; Azzeh, M.; Akbulut, S. Explainable artificial intelligence model for identifying COVID-19 gene biomarkers. Comput. Biol. Med. 2023, 154, 106619. [Google Scholar] [CrossRef]
- Lai, Y.; Lin, X.; Lin, C.; Lin, X.; Chen, Z.; Zhang, L. Identification of endoplasmic reticulum stress-associated genes and subtypes for prediction of Alzheimer’s disease based on interpretable machine learning. Front. Pharmacol. 2022, 13, 975774. [Google Scholar] [CrossRef] [PubMed]
- Ahamad, M.M.; Aktar, S.; Uddin, M.J.; Rahman, T.; Alyami, S.A.; Al-Ashhab, S.; Akhdar, H.F.; Azad, A.; Moni, M.A. Early-Stage Detection of Ovarian Cancer Based on Clinical Data Using Machine Learning Approaches. J. Pers. Med. 2022, 12, 1211. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhao, Y.; Dong, X.; Jin, Y.; Cheng, S.; Zhang, N.; Xu, S.; Gu, S.; Wu, Y.; Yang, J.; et al. Artificial intelligence-based preoperative prediction system for diagnosis and prognosis in epithelial ovarian cancer: A multicenter study. Front. Oncol. 2022, 12, 975703. [Google Scholar] [CrossRef]
- Liu, J.; Liu, L.; Antwi, P.A.; Luo, Y.; Liang, F. Identification and Validation of the Diagnostic Characteristic Genes of Ovarian Cancer by Bioinformatics and Machine Learning. Front. Genet. 2022, 13, 858466. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Lee, S.W.; Kwak, H.S.; Kang, M.H.; Park, Y.Y.; Jeong, G.S. Fibroblast-associated tumour microenvironment induces vascular structure-networked tumouroid. Sci. Rep. 2018, 8, 2365. [Google Scholar] [CrossRef]
- Cai, J.; Tang, H.; Xu, L.; Wang, X.; Yang, C.; Ruan, S.; Guo, J.; Hu, S.; Wang, Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 2012, 33, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Shani, O.; Vorobyov, T.; Monteran, L.; Lavie, D.; Cohen, N.; Raz, Y.; Tsarfaty, G.; Avivi, C.; Barshack, I.; Erez, N. Fibroblast-Derived IL33 Facilitates Breast Cancer Metastasis by Modifying the Immune Microenvironment and Driving Type 2 Immunity. Cancer Res. 2020, 80, 5317–5329. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, Z.; Wang, Y.; Zhao, H.; Du, Y. The Role of Cancer-Associated Fibroblasts in Ovarian Cancer. Cancers 2022, 14, 2637. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yu, Q.; Pang, J.; Chen, Y.; Sheng, M.; Tang, W. The Value of the Stemness Index in Ovarian Cancer Prognosis. Genes 2022, 13, 993. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.S.W. Chimeric oncolytic Ad5/3 virus replicates and lyses ovarian cancer cells through desmoglein-2 cell entry receptor. J. Med. Virol. 2020, 92, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Moufarrij, S.; Dandapani, M.; Arthofer, E.; Gomez, S.; Srivastava, A.; Lopez-Acevedo, M.; Villagra, A.; Chiappinelli, K.B. Epigenetic therapy for ovarian cancer: Promise and progress. Clin. Epigenetics 2019, 11, 7. [Google Scholar] [CrossRef]
- Wang, L.; Sun, T.; Li, S.; Zhang, Z.; Jia, J.; Shan, B. Protein anabolism is key to long-term survival in high-grade serous ovarian cancer. Transl. Oncol. 2021, 14, 100885. [Google Scholar] [CrossRef]
- Wang, K.J.; Adrian, A.M.; Chen, K.H.; Wang, K.M. A hybrid classifier combining Borderline-SMOTE with AIRS algorithm for estimating brain metastasis from lung cancer: A case study in Taiwan. Comput. Methods Programs Biomed. 2015, 119, 63–76. [Google Scholar] [CrossRef]
- Lin, H.; Angeli, M.; Chung, K.J.; Ejimadu, C.; Rosa, A.R.; Lee, T. sFRP2 activates Wnt/β-catenin signaling in cardiac fibroblasts: Differential roles in cell growth, energy metabolism, and extracellular matrix remodeling. Am. J. Physiol. Cell Physiol. 2016, 311, C710–C719. [Google Scholar] [CrossRef]
- Duan, H.; Yan, Z.; Chen, W.; Wu, Y.; Han, J.; Guo, H.; Qiao, J. TET1 inhibits EMT of ovarian cancer cells through activating Wnt/β-catenin signaling inhibitors DKK1 and SFRP2. Gynecol. Oncol. 2017, 147, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ye, Z.; Wan, J.; Liang, J.; Liu, M.; Xu, X.; Li, L. Secreted Frizzled-Related Protein 2 Is Associated with Disease Progression and Poor Prognosis in Breast Cancer. Dis. Markers 2019, 2019, 6149381. [Google Scholar] [CrossRef]
- Lai, H.Y.; Chiu, C.C.; Kuo, Y.H.; Tsai, H.H.; Wu, L.C.; Tseng, W.H.; Liu, C.L.; Hsing, C.H.; Huang, S.K.; Li, C.F. High Stromal SFRP2 Expression in Urothelial Carcinoma Confers an Unfavorable Prognosis. Front. Oncol. 2022, 12, 834249. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Plazyo, O.; Billi, A.C.; Tsoi, L.C.; Xing, X.; Wasikowski, R.; Gharaee-Kermani, M.; Hile, G.; Jiang, Y.; Harms, P.W.; et al. Single cell and spatial sequencing define processes by which keratinocytes and fibroblasts amplify inflammatory responses in psoriasis. Nat. Commun. 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Liu, J.; Wang, D.R.; Yu, H.F.; Li, Y.K.; Zhang, J.Q. Diagnostic and prognostic value of the methylation status of secreted frizzled-related protein 2 in colorectal cancer. Clin. Investig. Med. 2011, 34, E88–E95. [Google Scholar] [CrossRef]
- Kasashima, H.; Duran, A.; Martinez-Ordonez, A.; Nakanishi, Y.; Kinoshita, H.; Linares, J.F.; Reina-Campos, M.; Kudo, Y.; L’Hermitte, A.; Yashiro, M.; et al. Stromal SOX2 Upregulation Promotes Tumorigenesis through the Generation of a SFRP1/2-Expressing Cancer-Associated Fibroblast Population. Dev. Cell 2021, 56, 95–110.e10. [Google Scholar] [CrossRef]
- Lu, X.; Ji, C.; Jiang, L.; Zhu, Y.; Zhou, Y.; Meng, J.; Gao, J.; Lu, T.; Ye, J.; Yan, F. Tumour microenvironment-based molecular profiling reveals ideal candidates for high-grade serous ovarian cancer immunotherapy. Cell Prolif. 2021, 54, e12979. [Google Scholar] [CrossRef]
- Geng, T.; Zheng, M.; Wang, Y.; Reseland, J.E.; Samara, A. An artificial intelligence prediction model based on extracellular matrix proteins for the prognostic prediction and immunotherapeutic evaluation of ovarian serous adenocarcinoma. Front. Mol. Biosci. 2023, 10, 1200354. [Google Scholar] [CrossRef]
- Ding, J.; Wang, C.; Sun, Y.; Guo, J.; Liu, S.; Cheng, Z. Identification of an Autophagy-Related Signature for Prognosis and Immunotherapy Response Prediction in Ovarian Cancer. Biomolecules 2023, 13, 339. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef]
- Way, G.P.; Sanchez-Vega, F.; La, K.; Armenia, J.; Chatila, W.K.; Luna, A.; Sander, C.; Cherniack, A.D.; Mina, M.; Ciriello, G.; et al. Machine Learning Detects Pan-cancer Ras Pathway Activation in The Cancer Genome Atlas. Cell Rep. 2018, 23, 172–180.e3. [Google Scholar] [CrossRef] [PubMed]
- Nulsen, J.; Misetic, H.; Yau, C.; Ciccarelli, F.D. Pan-cancer detection of driver genes at the single-patient resolution. Genome Med. 2021, 13, 12. [Google Scholar] [CrossRef]
- Bolboaca, S.D. Medical Diagnostic Tests: A Review of Test Anatomy, Phases, and Statistical Treatment of Data. Comput. Math. Methods Med. 2019, 2019, 1891569. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Zhou, D.; Huang, J. Identifying Explainable Machine Learning Models and a Novel SFRP2+ Fibroblast Signature as Predictors for Precision Medicine in Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 16942. https://doi.org/10.3390/ijms242316942
Yang Z, Zhou D, Huang J. Identifying Explainable Machine Learning Models and a Novel SFRP2+ Fibroblast Signature as Predictors for Precision Medicine in Ovarian Cancer. International Journal of Molecular Sciences. 2023; 24(23):16942. https://doi.org/10.3390/ijms242316942
Chicago/Turabian StyleYang, Ziyi, Dandan Zhou, and Jun Huang. 2023. "Identifying Explainable Machine Learning Models and a Novel SFRP2+ Fibroblast Signature as Predictors for Precision Medicine in Ovarian Cancer" International Journal of Molecular Sciences 24, no. 23: 16942. https://doi.org/10.3390/ijms242316942
APA StyleYang, Z., Zhou, D., & Huang, J. (2023). Identifying Explainable Machine Learning Models and a Novel SFRP2+ Fibroblast Signature as Predictors for Precision Medicine in Ovarian Cancer. International Journal of Molecular Sciences, 24(23), 16942. https://doi.org/10.3390/ijms242316942