
Cellular Responses to Widespread DNA Replication Stress

 ,
,  , , and
, , and 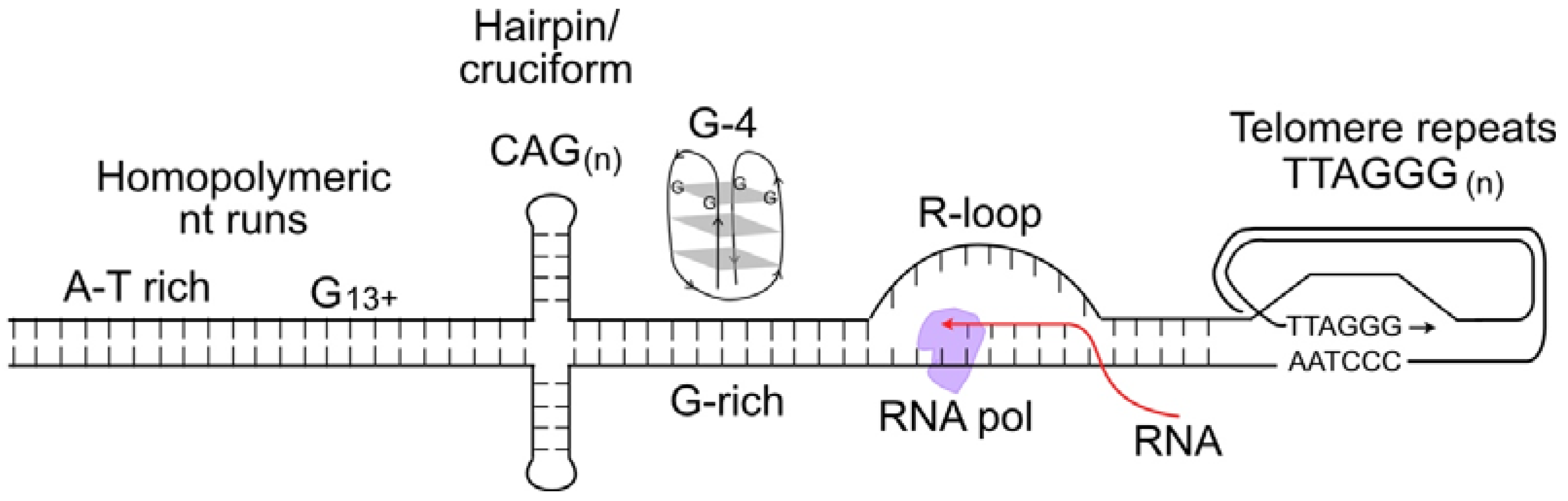{kind=link}
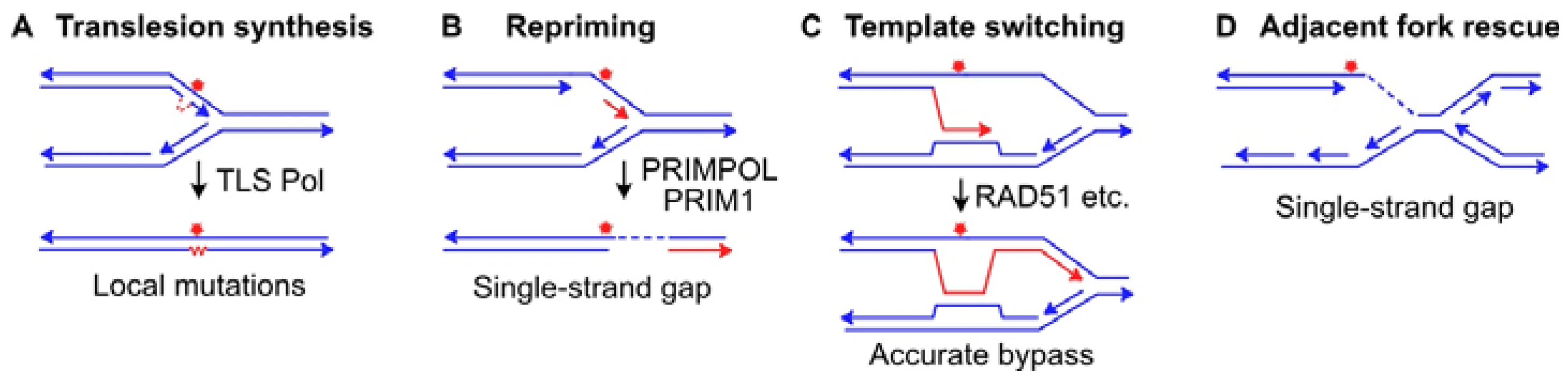{kind=link}
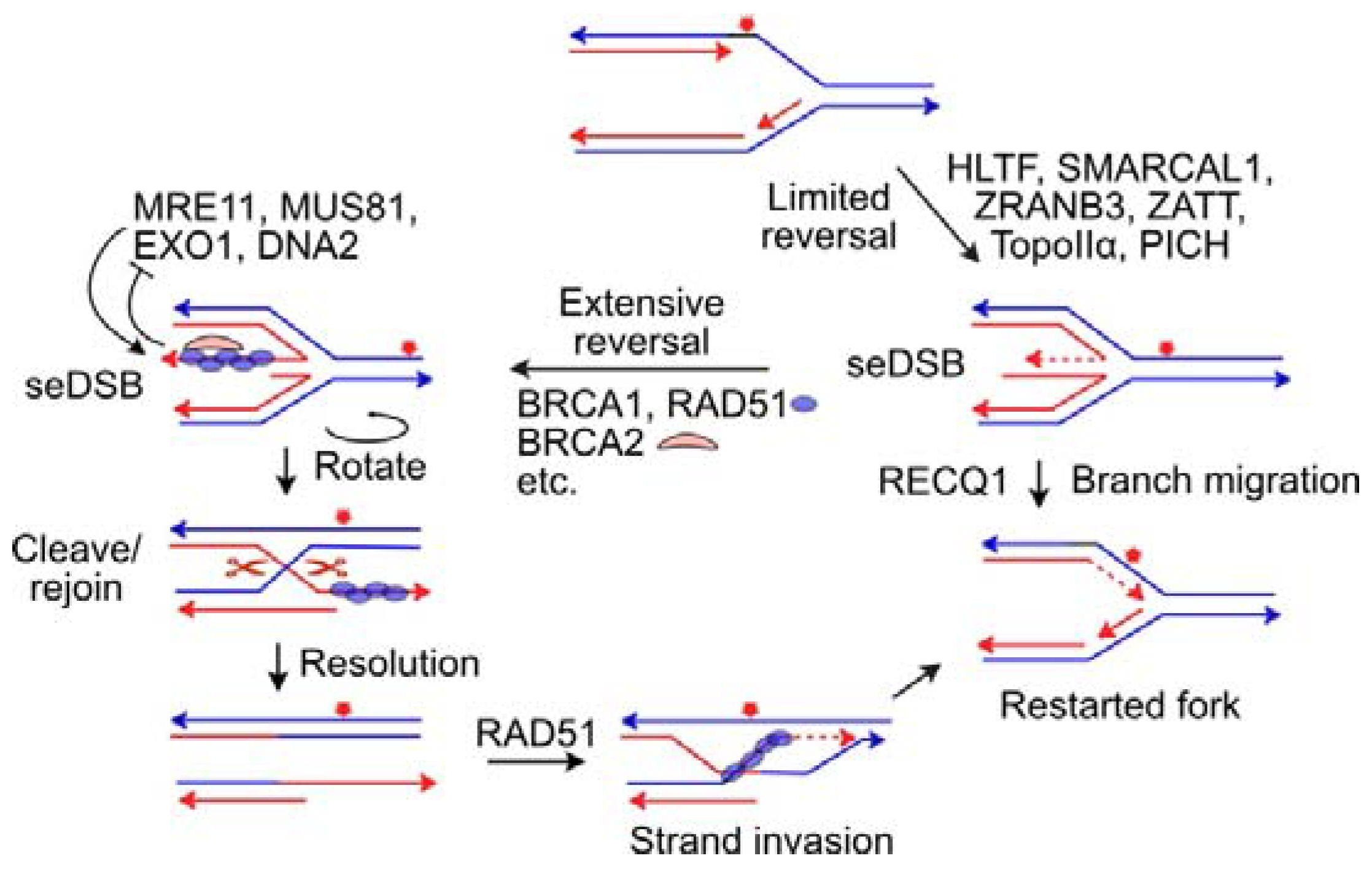{kind=link}
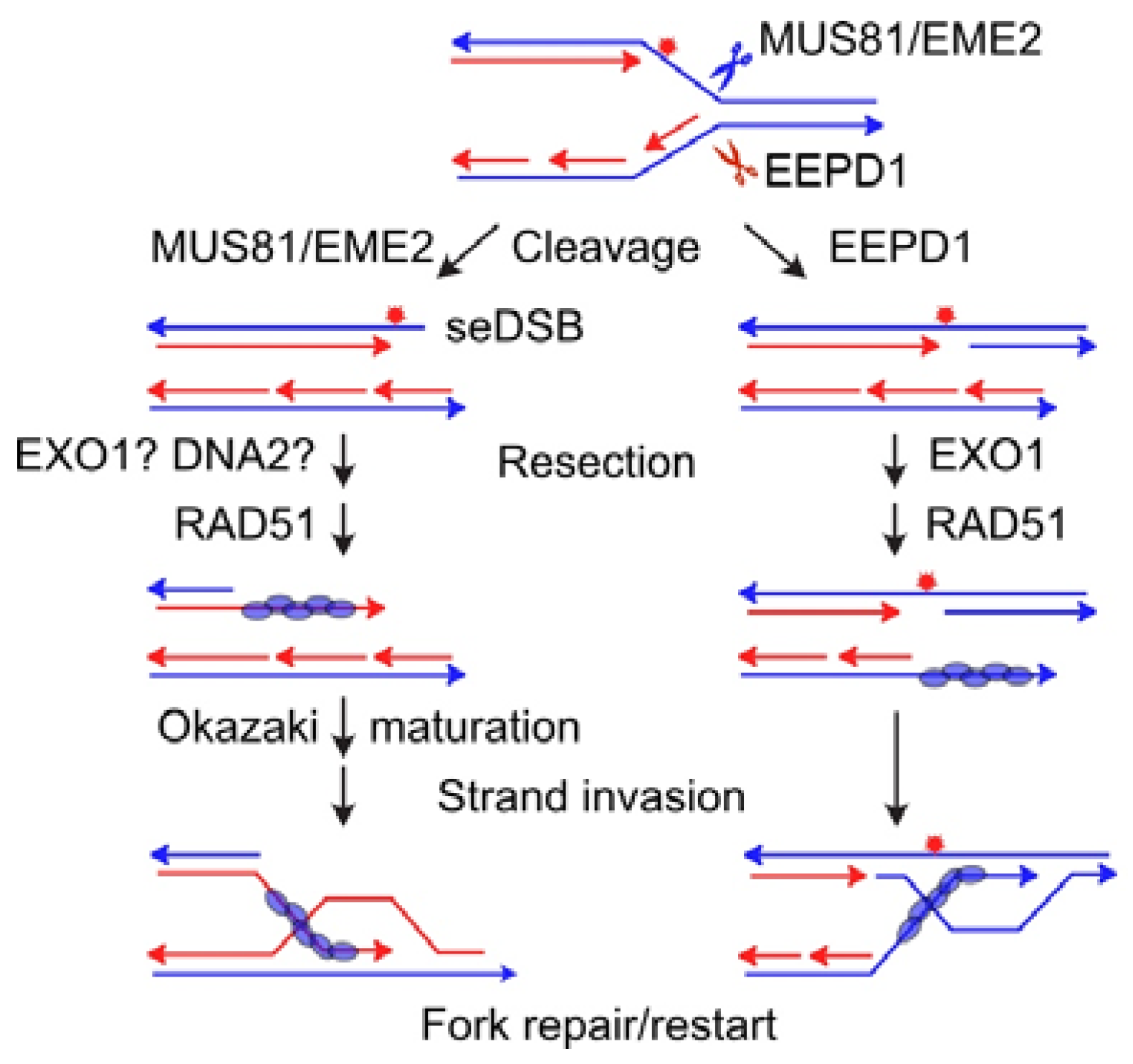{kind=link}
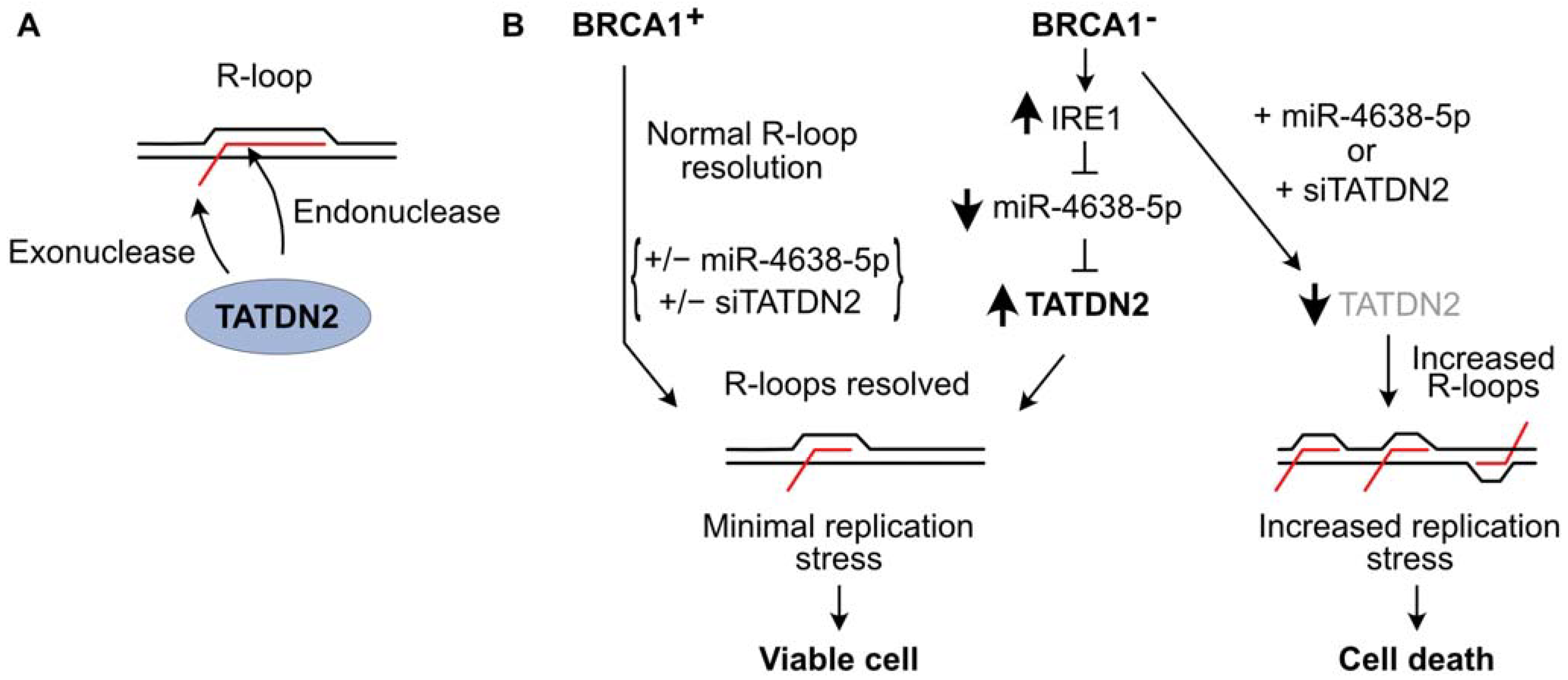{kind=link}
Abstract
1. Introduction
2. DNA Lesion Bypass and Stressed Fork Protection Mechanisms
3. Replication Fork Restart via Fork Cleavage by Structure-Specific Nucleases MUS81 and EEPD1
4. Other Nucleases Involved in the Replication Stress Response
5. TATDN2 Is a Structure-Specific RNA Nuclease That Degrades RNA in R-Loops
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Avalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. BioMed Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Manfredi, J.J. Another fork in the road-life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response--a double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, C.; Martoriati, A.; Cailliau, K. Proteins from the DNA damage response: Regulation, dysfunction, and anticancer strategies. Cancers 2021, 13, 3819. [Google Scholar] [CrossRef]
- Datta, A.; Brosh, R.M., Jr. New insights into DNA helicases as druggable targets for cancer therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Jo, U.; Kim, H. Exploiting the Fanconi anemia pathway for targeted anti-cancer therapy. Mol. Cells 2015, 38, 669–676. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA repair pathways in cancer therapy and resistance. Front. Pharmacol. 2020, 11, 629266. [Google Scholar] [CrossRef]
- Jurkovicova, D.; Neophytou, C.M.; Gasparovic, A.C.; Goncalves, A.C. DNA damage response in cancer therapy and resistance: Challenges and opportunities. Int. J. Mol. Sci. 2022, 23, 14672. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Herlihy, A.E.; Pennycook, B.R.; Kriston-Vizi, J.; de Bruin, R.A.M. Sustained E2F-dependent transcription Is a key mechanism to prevent replication-stress-induced DNA damage. Cell Rep. 2016, 15, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Belan, O.; Sebald, M.; Adamowicz, M.; Anand, R.; Vancevska, A.; Neves, J.; Grinkevich, V.; Hewitt, G.; Segura-Bayona, S.; Bellelli, R.; et al. POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Mol. Cell 2022, 82, 4664–4680.e4669. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef]
- Wang, Z.; Jia, R.; Wang, L.; Yang, Q.; Hu, X.; Fu, Q.; Zhang, X.; Li, W.; Ren, Y. The emerging roles of Rad51 in cancer and its potential as a therapeutic target. Front. Oncol. 2022, 12, 935593. [Google Scholar] [CrossRef]
- Beskow, C.; Skikuniene, J.; Holgersson, A.; Nilsson, B.; Lewensohn, R.; Kanter, L.; Viktorsson, K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br. J. Cancer 2009, 101, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Azzoli, C.G.; Santivasi, W.L.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.S. Mechanism of drug tolerant persister cancer cells: The landscape and clinical implication for therapy. J. Thorac. Oncol. 2021, 16, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-deficient cancers provide new opportunities for precision oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef] [PubMed]
- Hromas, R.; Williamson, E.; Lee, S.H.; Nickoloff, J. Preventing the chromosomal translocations that cause cancer. Trans. Am. Clin. Climatol. Assoc. 2016, 127, 176–195. [Google Scholar]
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the cancers addicted to DNA repair. J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef]
- Nickoloff, J.A. Toward greater precision in cancer radiotherapy. Cancer Res. 2021, 81, 3156–3157. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Taylor, L.; Sharma, N.; Kato, T.A. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resist. 2021, 4, 244–263. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V. PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: An update of clinical trials. Curr. Drug Targets 2018, 19, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-ribose)polymerase (PARP) inhibitors and radiation therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Keyomarsi, K. PARP inhibitors as single agents and in combination therapy: The most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin. Investig. Drugs 2022, 31, 607–631. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.G.; Yeeles, J.T.P. The initial response of a eukaryotic replisome to DNA damage. Mol. Cell 2018, 70, 1067–1080.e1012. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Elledge, S.J.; Lehmann, A.R.; Lindahl, T.; Muzi-Falconi, M. (Eds.) DNA Repair, Mutagenesis, and Other Responses to DNA Damage, 1st ed.; Cold Spring Harbor Laboratory Press: Cold Sprng Harbor, NY, USA, 2014; p. 446. [Google Scholar]
- Morgan, M.A.; Lawrence, T.S. Molecular pathways: Overcoming radiation resistance by targeting DNA damage response pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [PubMed]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of response and resistance to DNA repair targeted therapies. Clin. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef]
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef]
- Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in resistance mechanism pathways for combination therapy. Cells 2019, 8, 1013. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespee, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Mognato, M.; Burdak-Rothkamm, S.; Rothkamm, K. Interplay between DNA replication stress, chromatin dynamics and DNA-damage response for the maintenance of genome stability. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108346. [Google Scholar] [CrossRef] [PubMed]
- Liptay, M.; Barbosa, J.S.; Rottenberg, S. Replication fork remodeling and therapy escape in DNA damage response-deficient cancers. Front. Oncol. 2020, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Mazouzi, A.; Stukalov, A.; Muller, A.C.; Chen, D.; Wiedner, M.; Prochazkova, J.; Chiang, S.C.; Schuster, M.; Breitwieser, F.P.; Pichlmair, A.; et al. A comprehensive analysis of the dynamic response to aphidicolin-mediated replication stress uncovers targets for ATM and ATMIN. Cell Rep. 2016, 15, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Xu, Y.J. The cell killing mechanisms of hydroxyurea. Genes 2016, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2019, 43, e20190138. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef]
- Stirling, P.C.; Chan, Y.A.; Minaker, S.W.; Aristizabal, M.J.; Barrett, I.; Sipahimalani, P.; Kobor, M.S.; Hieter, P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012, 26, 163–175. [Google Scholar] [CrossRef]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replicating large genomes: Divide and conquer. Mol. Cell 2016, 62, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Zorbas, H.; Price, G.B.; Zannis-Hadjopoulos, M. Inverted repeats, stem-loops, and cruciforms: Significance for initiation of DNA replication. J. Cell. Biochem. 1996, 63, 1–22. [Google Scholar] [CrossRef]
- Jain, A.; Wang, G.; Vasquez, K.M. DNA triple helices: Biological consequences and therapeutic potential. Biochimie 2008, 90, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Miglietta, G.; Russo, M.; Capranico, G. G-quadruplex-R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 2020, 48, 11942–11957. [Google Scholar] [CrossRef] [PubMed]
- Aksenova, A.Y.; Mirkin, S.M. At the beginning of the end and in the middle of the beginning: Structure and maintenance of telomeric DNA repeats and interstitial telomeric sequences. Genes 2019, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Rogacheva, M.V.; Nishant, K.T.; Zanders, S.; Bustamante, C.D.; Alani, E. Mutation hot spots in yeast caused by long-range clustering of homopolymeric sequences. Cell Rep. 2012, 1, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Qiu, L.; Mrasek, K.; Zhang, J.; Liehr, T.; Quintana, L.G.; Li, Z. Common fragile sites: Genomic hotspots of DNA damage and carcinogenesis. Int. J. Mol. Sci. 2012, 13, 11974–11999. [Google Scholar] [CrossRef]
- Lemmens, B.; van Schendel, R.; Tijsterman, M. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat. Commun. 2015, 6, 8909. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Lu, R.; Pickett, H.A. Telomeric replication stress: The beginning and the end for alternative lengthening of telomeres cancers. Open Biol. 2022, 12, 220011. [Google Scholar] [CrossRef]
- McDonald, M.J.; Yu, Y.H.; Guo, J.F.; Chong, S.Y.; Kao, C.F.; Leu, J.Y. Mutation at a distance caused by homopolymeric guanine repeats in Saccharomyces cerevisiae. Sci. Adv. 2016, 2, e1501033. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.T.C.; Yin, Y.; Morten, M.J.; Tonzi, P.; Gwo, P.P.; Odermatt, D.C.; Modesti, M.; Cantor, S.B.; Gari, K.; Huang, T.T.; et al. Single-molecule imaging reveals replication fork coupled formation of G-quadruplex structures hinders local replication stress signaling. Nat. Commun. 2021, 12, 2525. [Google Scholar] [CrossRef]
- Saxena, S.; Zou, L. Hallmarks of DNA replication stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef]
- Wu, Y.; Lee, S.H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. EEPD1 rescues stressed replication forks and maintains genome stability by promoting end resection and homologous recombination repair. PLoS Genet. 2015, 11, e1005675. [Google Scholar] [CrossRef]
- Jaiswal, A.S.; Dutta, A.; Srinivasan, G.; Yuan, Y.; Zhou, D.; Shaheen, M.; Sadideen, D.T.; Kirby, A.; Williamson, E.A.; Gupta, Y.K.; et al. TATDN2 resolution of R-loops is required for survival of BRCA1-mutant cancer cells. Nucleic Acids Res. 2023, gkad952. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Proteomic analyses of the eukaryotic replication machinery. Methods Enzymol. 2017, 591, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA breaks and end resection measured genome-wide by end sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef]
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callen, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual roles of poly(dA:dT) tracts in replicationo initiation and fork collapse. Cell 2018, 174, 1127–1142.e119. [Google Scholar] [CrossRef]
- Shimura, T.; Torres, M.J.; Martin, M.M.; Rao, V.A.; Pommier, Y.; Katsura, M.; Miyagawa, K.; Aladjem, M.I. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J. Mol. Biol. 2008, 375, 1152–1164. [Google Scholar] [CrossRef]
- De Haro, L.P.; Wray, J.; Williamson, E.A.; Durant, S.T.; Corwin, L.; Gentry, A.C.; Osheroff, N.; Lee, S.H.; Hromas, R.; Nickoloff, J.A. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010, 38, 5681–5691. [Google Scholar] [CrossRef]
- Jaiswal, A.S.; Kim, H.S.; Scharer, O.D.; Sharma, N.; Williamson, E.A.; Srinivasan, G.; Phillips, L.; Kong, K.; Arya, S.; Misra, A.; et al. EEPD1 promotes repair of oxidatively-stressed replication forks. NAR Cancer 2023, 5, zcac044. [Google Scholar] [CrossRef]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef]
- Agudelo Garcia, P.A.; Gardner, M.; Freitas, M.A.; Parthun, M.R. Isolation of proteins on nascent chromatin and characterization by quantitative mass spectrometry. Methods Mol. Biol. 2019, 1983, 17–27. [Google Scholar] [CrossRef]
- Quinet, A.; Carvajal-Maldonado, D.; Lemacon, D.; Vindigni, A. DNA fiber analysis: Mind the gap! Methods Enzymol. 2017, 591, 55–82. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Gohil, D.; Sarker, A.H.; Roy, R. Base excision repair: Mechanisms and impact in biology, disease, and medicine. Int. J. Mol. Sci. 2023, 24, 14186. [Google Scholar] [CrossRef] [PubMed]
- Compe, E.; Egly, J.M. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu. Rev. Biochem. 2016, 85, 265–290. [Google Scholar] [CrossRef]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA replication through natural impediments in the eukaryotic genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef]
- Allen, C.; Ashley, A.K.; Hromas, R.; Nickoloff, J.A. More forks on the road to replication stress recovery. J. Mol. Cell Biol. 2011, 3, 4–12. [Google Scholar] [CrossRef]
- Budzowska, M.; Kanaar, R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem. Biophys. 2009, 53, 17–31. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Mullins, E.A.; Rodriguez, A.A.; Bradley, N.P.; Eichman, B.F. Emerging roles of DNA glycosylases and the base excision repair pathway. Trends Biochem. Sci. 2019, 44, 765–781. [Google Scholar] [CrossRef]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef]
- Povirk, L.F. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: Bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 1996, 355, 71–89. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA double-strand breaks: Biological effects and relevance to cancer radiotherapy. Genes 2020, 11, 99–116. [Google Scholar] [CrossRef]
- Parker, M.W.; Botchan, M.R.; Berger, J.M. Mechanisms and regulation of DNA replication initiation in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 107–144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Pozo, P.N.; Oh, S.; Stone, H.M.; Cook, J.G. Distinct and sequential re-replication barriers ensure precise genome duplication. PLoS Genet. 2020, 16, e1008988. [Google Scholar] [CrossRef]
- Blow, J.J.; Dutta, A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 2005, 6, 476–486. [Google Scholar] [CrossRef]
- Thakur, B.L.; Ray, A.; Redon, C.E.; Aladjem, M.I. Preventing excess replication origin activation to ensure genome stability. Trends Genet. 2022, 38, 169–181. [Google Scholar] [CrossRef]
- Ler, A.A.L.; Carty, M.P. DNA damage tolerance pathways in human cells: A potential therapeutic target. Front. Oncol. 2021, 11, 822500. [Google Scholar] [CrossRef]
- Yang, W. An overview of Y-family DNA polymerases and a case study of human DNA polymerase η. Biochemistry 2014, 53, 2793–2803. [Google Scholar] [CrossRef]
- Goodman, M.F.; Woodgate, R. Translesion DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef]
- Ma, X.; Tang, T.S.; Guo, C. Regulation of translesion DNA synthesis in mammalian cells. Environ. Mol. Mutagen. 2020, 61, 680–692. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Boehm, E.M.; Spies, M.; Washington, M.T. PCNA tool belts and polymerase bridges form during translesion synthesis. Nucleic Acids Res. 2016, 44, 8250–8260. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.D.; Gakhar, L.; Ramaswamy, S.; Washington, M.T. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nat. Struct. Mol. Biol. 2010, 17, 479–484. [Google Scholar] [CrossRef]
- Gonzalez-Magana, A.; Blanco, F.J. Human PCNA structure, function and interactions. Biomolecules 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Tirman, S.; Cybulla, E.; Quinet, A.; Meroni, A.; Vindigni, A. PRIMPOL ready, set, reprime! Crit. Rev. Biochem. Mol. Biol. 2021, 56, 17–30. [Google Scholar] [CrossRef]
- Kang, Z.; Fu, P.; Alcivar, A.L.; Fu, H.; Redon, C.; Foo, T.K.; Zuo, Y.; Ye, C.; Baxley, R.; Madireddy, A.; et al. BRCA2 associates with MCM10 to suppress PRIMPOL-mediated repriming and single-stranded gap formation after DNA damage. Nat. Commun. 2021, 12, 5966. [Google Scholar] [CrossRef]
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF promotes fork reversal, limiting replication stress resistance and preventing multiple mechanisms of unrestrained DNA synthesis. Mol. Cell 2020, 78, 1237–1251.e1237. [Google Scholar] [CrossRef]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To skip or not to skip: Choosing repriming to tolerate DNA damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef]
- Diamant, N.; Hendel, A.; Vered, I.; Carell, T.; Reissner, T.; de Wind, N.; Geacinov, N.; Livneh, Z. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 2012, 40, 170–180. [Google Scholar] [CrossRef]
- Jansen, J.G.; Tsaalbi-Shtylik, A.; Hendriks, G.; Gali, H.; Hendel, A.; Johansson, F.; Erixon, K.; Livneh, Z.; Mullenders, L.H.; Haracska, L.; et al. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol. Cell. Biol. 2009, 29, 3113–3123. [Google Scholar] [CrossRef]
- Lehmann, C.P.; Jimenez-Martin, A.; Branzei, D.; Tercero, J.A. Prevention of unwanted recombination at damaged replication forks. Curr. Genet. 2020, 66, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Ripley, B.M.; Gildenberg, M.S.; Washington, M.T. Control of DNA damage bypass by ubiquitylation of PCNA. Genes 2020, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. The safe path at the fork: Ensuring replication-associated DNA double-strand breaks are repaired by homologous recombination. Front. Genet. 2021, 12, 748033. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.A.; Smogorzewska, A. Mechanisms of direct replication restart at stressed replisomes. DNA Repair 2020, 95, 102947. [Google Scholar] [CrossRef] [PubMed]
- Courtot, L.; Hoffmann, J.S.; Bergoglio, V. The protective role of dormant origins in response to replicative stress. Int. J. Mol. Sci. 2018, 19, 3569. [Google Scholar] [CrossRef]
- Brambati, A.; Zardoni, L.; Achar, Y.J.; Piccini, D.; Galanti, L.; Colosio, A.; Foiani, M.; Liberi, G. Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res. 2018, 46, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Jossen, R.; Bermejo, R. The DNA damage checkpoint response to replication stress: A Game of Forks. Front. Genet. 2013, 4, 26. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Zou, L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef]
- Qiu, S.; Jiang, G.; Cao, L.; Huang, J. Replication fork reversal and protection. Front. Cell Dev. Biol. 2021, 9, 670392. [Google Scholar] [CrossRef]
- Cejka, P.; Symington, L.S. DNA end resection: Mechanism and control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Krishnamoorthy, A.; Zhao, R.; Cortez, D. Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Sci. Adv. 2020, 6, eabc3598. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 2017, 68, 414–430.e418. [Google Scholar] [CrossRef] [PubMed]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Techer, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell 2017, 67, 867–881.e867. [Google Scholar] [CrossRef] [PubMed]
- Lemacon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, M.E.; Sharma, N.; Maxwell, P.; Selemenakis, P.; Wiese, C. RAD54L regulates replication fork progression and nascent strand degradation in BRCA1/2-deficient cells. bioRxiv 2023, bioRxiv:07.26.550704. [Google Scholar] [CrossRef]
- Fugger, K.; Mistrik, M.; Neelsen, K.J.; Yao, Q.; Zellweger, R.; Kousholt, A.N.; Haahr, P.; Chu, W.K.; Bartek, J.; Lopes, M.; et al. FBH1 catalyzes regression of stalled replication forks. Cell Rep. 2015, 10, 1749–1757. [Google Scholar] [CrossRef]
- Chu, W.K.; Payne, M.J.; Beli, P.; Hanada, K.; Choudhary, C.; Hickson, I.D. FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat. Commun. 2015, 6, 5931. [Google Scholar] [CrossRef]
- Simandlova, J.; Zagelbaum, J.; Payne, M.J.; Chu, W.K.; Shevelev, I.; Hanada, K.; Chatterjee, S.; Reid, D.A.; Liu, Y.; Janscak, P.; et al. FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J. Biol. Chem. 2013, 288, 34168–34180. [Google Scholar] [CrossRef]
- Fugger, K.; Mistrik, M.; Danielsen, J.R.; Dinant, C.; Falck, J.; Bartek, J.; Lukas, J.; Mailand, N. Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. J. Cell Biol. 2009, 186, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, A.; Osman, F.; Folkyte, V.; Sofueva, S.; Whitby, M.C. Fbh1 limits Rad51-dependent recombination at blocked replication forks. Mol. Cell. Biol. 2009, 29, 4742–4756. [Google Scholar] [CrossRef] [PubMed]
- Rickman, K.A.; Noonan, R.J.; Lach, F.P.; Sridhar, S.; Wang, A.T.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX modulates RAD51 activity to control replication fork protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; de Boer, H.R.; Demmers, J.; van Vugt, M.; Ray Chaudhuri, A. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Bennett, L.G.; Wilkie, A.M.; Antonopoulou, E.; Ceppi, I.; Sanchez, A.; Vernon, E.G.; Gamble, A.; Myers, K.N.; Collis, S.J.; Cejka, P.; et al. MRNIP is a replication fork protection factor. Sci. Adv. 2020, 6, eaba5974. [Google Scholar] [CrossRef]
- Lim, K.S.; Li, H.; Roberts, E.A.; Gaudiano, E.F.; Clairmont, C.; Sambel, L.A.; Ponnienselvan, K.; Liu, J.C.; Yang, C.; Kozono, D.; et al. USP1 Is required for replication fork protection in BRCA1-deficient tumors. Mol. Cell 2018, 72, 925–941.e924. [Google Scholar] [CrossRef]
- Berti, M.; Teloni, F.; Mijic, S.; Ursich, S.; Fuchs, J.; Palumbieri, M.D.; Krietsch, J.; Schmid, J.A.; Garcin, E.B.; Gon, S.; et al. Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nat. Commun. 2020, 11, 3531. [Google Scholar] [CrossRef]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef]
- Tye, S.; Ronson, G.E.; Morris, J.R. A fork in the road: Where homologous recombination and stalled replication fork protection part ways. Semin. Cell Dev. Biol. 2021, 113, 14–26. [Google Scholar] [CrossRef]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef]
- Porebski, B.; Wild, S.; Kummer, S.; Scaglione, S.; Gaillard, P.L.; Gari, K. WRNIP1 protects reversed DNA replication forks from SLX4-dependent nucleolytic cleavage. iScience 2019, 21, 31–41. [Google Scholar] [CrossRef]
- Weston, R.; Peeters, H.; Ahel, D. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 2012, 26, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Bu, M.; Chen, X.; Ding, L.; Yang, Y.; Han, J.; Feng, X.H.; Xu, P.; Liu, T.; Ying, S.; et al. The ZATT-TOP2A-PICH axis drives extensive replication fork reversal to promote genome stability. Mol. Cell 2021, 81, 198–211.e196. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef]
- Interthal, H.; Heyer, W.D. MUS81 encodes a novel helix-hairpin-helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet. 2000, 263, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Boddy, M.N.; Gaillard, P.H.L.; McDonald, W.H.; Shanahan, P.; Yates, J.R., 3rd; Russell, P. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 2001, 107, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Melchionna, R.; Denis, C.M.; Gaillard, P.H.; Blasina, A.; Van de Weyer, I.; Boddy, M.N.; Russell, P.; Vialard, J.; McGowan, C.H. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell 2001, 8, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Dehe, P.M.; Coulon, S.; Scaglione, S.; Shanahan, P.; Takedachi, A.; Wohlschlegel, J.A.; Yates, J.R., 3rd; Llorente, B.; Russell, P.; Gaillard, P.H. Regulation of Mus81-Eme1 Holliday junction resolvase in response to DNA damage. Nat. Struct. Mol. Biol. 2013, 20, 598–603. [Google Scholar] [CrossRef]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Wyatt, H.D.; Sarbajna, S.; Matos, J.; West, S.C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 2013, 52, 234–247. [Google Scholar] [CrossRef]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef]
- Amangyeld, T.; Shin, Y.K.; Lee, M.; Kwon, B.; Seo, Y.S. Human MUS81-EME2 can cleave a variety of DNA structures including intact Holliday junction and nicked duplex. Nucleic Acids Res. 2014, 42, 5846–5862. [Google Scholar] [CrossRef]
- Pepe, A.; West, S.C. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef]
- Pepe, A.; West, S.C. Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 2014, 42, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chen, X.B.; McGowan, C.H. Mus81 endonuclease localizes to nucleoli and to regions of DNA damage in human S-phase cells. Mol. Biol. Cell 2003, 14, 4826–4834. [Google Scholar] [CrossRef] [PubMed]
- Kramara, J.; Osia, B.; Malkova, A. Break-induced replication: The where, the why, and the how. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Moriel-Carretero, M.; Vicat, T.; Aguilera, A.; Pasero, P. Homologous recombination and Mus81 promote replication completion in response to replication fork blockage. EMBO Rep. 2020, 21, e49367. [Google Scholar] [CrossRef] [PubMed]
- Dendouga, N.; Gao, H.; Moechars, D.; Janicot, M.; Vialard, J.; McGowan, C.H. Disruption of murine Mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol. Cell. Biol. 2005, 25, 7569–7579. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zheng, H.; Wen, X.; Sun, J.; Wang, Y.; Gao, X.; Guo, L.; Lu, R. MUS81 is associated with cell proliferation and cisplatin sensitivity in serous ovarian cancer. Biochem. Biophys. Res. Commun. 2016, 476, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Zhong, A.; Zhang, H.; Xie, S.; Deng, M.; Zheng, H.; Wang, Y.; Chen, M.; Lu, R.; Guo, L. Inhibition of MUS81 improves the chemical sensitivity of olaparib by regulating MCM2 in epithelial ovarian cancer. Oncol. Rep. 2018, 39, 1747–1756. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef]
- Bixel, K.; Hays, J.L. Olaparib in the management of ovarian cancer. Pharmgenomics Pers. Med. 2015, 8, 127–135. [Google Scholar] [CrossRef]
- Cadoo, K.; Simpkins, F.; Mathews, C.; Liu, Y.L.; Provencher, D.; McCormick, C.; ElNaggar, A.C.; Altman, A.D.; Gilbert, L.; Black, D.; et al. Olaparib treatment for platinum-sensitive relapsed ovarian cancer by BRCA mutation and homologous recombination deficiency status: Phase II LIGHT study primary analysis. Gynecol. Oncol. 2022, 166, 425–431. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef] [PubMed]
- Van Wietmarschen, N.; Sridharan, S.; Nathan, W.J.; Tubbs, A.; Chan, E.M.; Callen, E.; Wu, W.; Belinky, F.; Tripathi, V.; Wong, N.; et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Speed, M.C.; Allen, C.P.; Maranon, D.G.; Williamson, E.; Singh, S.; Hromas, R.; Nickoloff, J.A. Distinct roles of structure-specific endonucleases EEPD1 and Metnase in replication stress responses. NAR Cancer 2020, 2, zcaa008. [Google Scholar] [CrossRef]
- Yamada, K.; Miyata, T.; Tsuchiya, D.; Oyama, T.; Fujiwara, Y.; Ohnishi, T.; Iwasaki, H.; Shinagawa, H.; Ariyoshi, M.; Mayanagi, K.; et al. Crystal structure of the RuvA-RuvB complex: A structural basis for the Holliday junction migrating motor machinery. Mol. Cell 2002, 10, 671–681. [Google Scholar] [CrossRef]
- Khan, S.R.; Kuzminov, A. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J. Biol. Chem. 2012, 287, 6250–6265. [Google Scholar] [CrossRef]
- Donaldson, J.R.; Courcelle, C.T.; Courcelle, J. RuvABC is required to resolve holliday junctions that accumulate following replication on damaged templates in Escherichia coli. J. Biol. Chem. 2006, 281, 28811–28821. [Google Scholar] [CrossRef]
- Ragu, S.; Droin, N.; Matos-Rodrigues, G.; Barascu, A.; Caillat, S.; Zarkovic, G.; Siberchicot, C.; Dardillac, E.; Gelot, C.; Guirouilh-Barbat, J.; et al. A noncanonical response to replication stress protects genome stability through ROS production, in an adaptive manner. Cell Death Differ. 2023, 30, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Nickoloff, J.A.; Wu, Y.; Williamson, E.A.; Sidhu, G.S.; Reinert, B.L.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Kong, K.; et al. Endonuclease EEPD1 is a gatekeeper for repair of stressed replication forks. J. Biol. Chem. 2017, 292, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Chun, C.; Wu, Y.; Lee, S.H.; Williamson, E.A.; Reinert, B.L.; Jaiswal, A.S.; Nickoloff, J.A.; Hromas, R.A. The homologous recombination component EEPD1 is required for genome stability in response to developmental stress of vertebrate embryogenesis. Cell Cycle 2016, 15, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Ensembl.org. EEPD1 Gene Tree. Available online: https://www.ensembl.org/Homo_sapiens/Gene/Compara_Tree?db=core;g=ENSG00000122547 (accessed on 15 October 2023).
- Panopoulou, G.; Hennig, S.; Groth, D.; Krause, A.; Poustka, A.J.; Herwig, R.; Vingron, M.; Lehrach, H. New evidence for genome-wide duplications at the origin of vertebrates using an amphioxus gene set and completed animal genomes. Genome Res. 2003, 13, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. Nucleases and co-factors in DNA replication stress responses. DNA 2022, 2, 68–85. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, S.K.; Hromas, R.; Lee, S.H. The SET domain Is essential for Metnase functions in replication restart and the 5’ end of SS-overhang cleavage. PLoS ONE 2015, 10, e0139418. [Google Scholar] [CrossRef]
- Kim, H.-S.; Chen, Q.; Kim, S.-K.; Nickoloff, J.A.; Hromas, R.; Georgiadis, M.M.; Lee, S.-K. The DDN catalytic motif is required for Metnase functions in NHEJ repair and replication restart. J. Biol. Chem. 2014, 289, 10930–10938. [Google Scholar] [CrossRef]
- Fabre, F.; Chan, A.; Heyer, W.D.; Gangloff, S. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. USA 2002, 99, 16887–16892. [Google Scholar] [CrossRef]
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Donigan, K.A.; Sun, K.W.; Nemec, A.A.; Murphy, D.L.; Cong, X.; Northrup, V.; Zelterman, D.; Sweasy, J.B. Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 2012, 287, 23830–23839. [Google Scholar] [CrossRef] [PubMed]
- Makridakis, N.M.; Reichardt, J.K. Translesion DNA polymerases and cancer. Front. Genet. 2012, 3, 174. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Yoon, B.H.; Kim, S.K.; Kim, S.Y. GENT2: An updated gene expression database for normal and tumor tissues. BMC Med. Genom. 2019, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef]
- Hromas, R.; Kim, H.S.; Sidhu, G.; Williamson, E.A.; Jaiswal, A.; Totterdale, T.A.; Nole, J.; Lee, S.-H.; Nickoloff, J.A.; Kong, K.Y. The endonuclease EEPD1 mediates synthetic lethality in RAD52-depleted BRCA1-mutant breast cancer cells. Breast Cancer Res. BCR 2017, 19, 122. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Przetocka, S.; Porro, A.; Bolck, H.A.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 2018, 72, 568–582.e566. [Google Scholar] [CrossRef]
- Kim, H.S.; Williamson, E.A.; Nickoloff, J.A.; Hromas, R.A.; Lee, S.H. Metnase mediates loading of Exonuclease 1 onto single-strand overhang DNA for end resection at stalled replication forks. J. Biol. Chem. 2016, 292, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Payliss, B.J.; Patel, A.; Sheppard, A.C.; Wyatt, H.D.M. Exploring the structures and functions of macromolecular SLX4-nuclease complexes in genome stability. Front. Genet. 2021, 12, 784167. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.H.; Gaillard, P.H. SLX4: Multitasking to maintain genome stability. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 475–514. [Google Scholar] [CrossRef] [PubMed]
- Young, S.J.; West, S.C. Coordinated roles of SLX4 and MutSβ in DNA repair and the maintenance of genome stability. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 157–177. [Google Scholar] [CrossRef]
- Betous, R.; Goullet de Rugy, T.; Pelegrini, A.L.; Queille, S.; de Villartay, J.P.; Hoffmann, J.S. DNA replication stress triggers rapid DNA replication fork breakage by Artemis and XPF. PLoS Genet. 2018, 14, e1007541. [Google Scholar] [CrossRef]
- Trego, K.S.; Groesser, T.; Davalos, A.R.; Parplys, A.C.; Zhao, W.; Nelson, M.R.; Hlaing, A.; Shih, B.; Rydberg, B.; Pluth, J.M.; et al. Non-catalytic roles for XPG with BRCA1 and BRCA2 in homologous recombination and genome stability. Mol. Cell 2016, 61, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, M.; Chai, Q.; Parrish, J.; Xue, D.; Patrick, S.M.; Turchi, J.J.; Yannone, S.M.; Chen, D.; Shen, B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005, 6, 83–89. [Google Scholar] [CrossRef]
- Zheng, L.; Jia, J.; Finger, L.D.; Guo, Z.; Zer, C.; Shen, B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011, 39, 781–794. [Google Scholar] [CrossRef]
- Jaiswal, A.S.; Williamson, E.A.; Jaiswal, A.S.; Kong, K.; Hromas, R.A. In vitro reconstitutive base excision repair (BER) assay. Methods Mol. Biol. 2023, 2701, 91–112. [Google Scholar] [CrossRef]
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef]
- Mackay, R.P.; Xu, Q.; Weinberger, P.M. R-loop physiology and pathology: A brief review. DNA Cell Biol. 2020, 39, 1914–1925. [Google Scholar] [CrossRef]
- Chen, P.B.; Chen, H.V.; Acharya, D.; Rando, O.J.; Fazzio, T.G. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015, 22, 999–1007. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Alt, F.W. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J. Biol. Chem. 2000, 275, 24163–24172. [Google Scholar] [CrossRef] [PubMed]
- So, C.C.; Martin, A. DSB structure impacts DNA recombination leading to class switching and chromosomal translocations in human B cells. PLoS Genet. 2019, 15, e1008101. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Yadav, T.; Duan, M.; Tan, J.; Xiang, Y.; Gao, B.; Xu, J.; Liang, Z.; Liu, Y.; Nakajima, S.; et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018, 9, 4115. [Google Scholar] [CrossRef]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell 2018, 175, 558–570.e511. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J. R-loops and mitochondrial DNA metabolism. Methods Mol. Biol. 2022, 2528, 173–202. [Google Scholar] [CrossRef] [PubMed]
- Posse, V.; Al-Behadili, A.; Uhler, J.P.; Clausen, A.R.; Reyes, A.; Zeviani, M.; Falkenberg, M.; Gustafsson, C.M. RNase H1 directs origin-specific initiation of DNA replication in human mitochondria. PLoS Genet. 2019, 15, e1007781. [Google Scholar] [CrossRef]
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-loop-associated DNA damage to safeguard genome stability. Front. Cell Dev. Biol. 2020, 8, 618157. [Google Scholar] [CrossRef]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Pozo, M.; Santos-Pereira, J.M.; Rondon, A.G.; Barroso, S.; Andujar, E.; Perez-Alegre, M.; Garcia-Muse, T.; Aguilera, A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, L.; Wang, Q.; Hu, Y.; Zhao, W.; Gautam, M.; Li, L. H3K9 demethylation-induced R-loop accumulation is linked to disorganized nucleoli. Front. Genet. 2020, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Fazzio, T.G. Regulation of chromatin structure and cell fate by R-loops. Transcription 2016, 7, 121–126. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chedin, F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016, 32, 828–838. [Google Scholar] [CrossRef]
- Drolet, M.; Bi, X.; Liu, L.F. Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J. Biol. Chem. 1994, 269, 2068–2074. [Google Scholar] [CrossRef]
- Drolet, M. Growth inhibition mediated by excess negative supercoiling: The interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 2006, 59, 723–730. [Google Scholar] [CrossRef]
- Stolz, R.; Sulthana, S.; Hartono, S.R.; Malig, M.; Benham, C.J.; Chedin, F. Interplay between DNA sequence and negative superhelicity drives R-loop structures. Proc. Natl. Acad. Sci. USA 2019, 116, 6260–6269. [Google Scholar] [CrossRef]
- Phoenix, P.; Raymond, M.A.; Masse, E.; Drolet, M. Roles of DNA topoisomerases in the regulation of R-loop formation in vitro. J. Biol. Chem. 1997, 272, 1473–1479. [Google Scholar] [CrossRef]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Manzo, S.G.; Hartono, S.R.; Sanz, L.A.; Marinello, J.; De Biasi, S.; Cossarizza, A.; Capranico, G.; Chedin, F. DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 2018, 19, 100. [Google Scholar] [CrossRef]
- Masse, E.; Drolet, M. Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J. Biol. Chem. 1999, 274, 16659–16664. [Google Scholar] [CrossRef]
- Wahba, L.; Gore, S.K.; Koshland, D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife 2013, 2, e00505. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef]
- Wimberly, H.; Shee, C.; Thornton, P.C.; Sivaramakrishnan, P.; Rosenberg, S.M.; Hastings, P.J. R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat. Commun. 2013, 4, 2115. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Hall, A.N.; Merrikh, C.N.; Ragheb, M.; Tabakh, H.; Pollock, A.J.; Woodward, J.J.; Dreifus, J.E.; Merrikh, H. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell 2017, 170, 787–799.e718. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786.e719. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Zhang, Z.; Lu, Z.; Hsieh, C.L.; Lieber, M.R. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: A nick can serve as a strong R-loop initiation site. Mol. Cell. Biol. 2010, 30, 146–159. [Google Scholar] [CrossRef] [PubMed]
- San Martin-Alonso, M.; Soler-Oliva, M.E.; Garcia-Rubio, M.; Garcia-Muse, T.; Aguilera, A. Harmful R-loops are prevented via different cell cycle-specific mechanisms. Nat. Commun. 2021, 12, 4451. [Google Scholar] [CrossRef]
- Gonzalez-Aguilera, C.; Tous, C.; Gomez-Gonzalez, B.; Huertas, P.; Luna, R.; Aguilera, A. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol. Biol. Cell 2008, 19, 4310–4318. [Google Scholar] [CrossRef]
- Moreira, M.C.; Klur, S.; Watanabe, M.; Nemeth, A.H.; Le Ber, I.; Moniz, J.C.; Tranchant, C.; Aubourg, P.; Tazir, M.; Schols, L.; et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004, 36, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Puget, N.; Lin, Y.L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Tran, P.L.T.; Pohl, T.J.; Chen, C.F.; Chan, A.; Pott, S.; Zakian, V.A. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat. Commun. 2017, 8, 15025. [Google Scholar] [CrossRef]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef]
- Yuan, W.; Al-Hadid, Q.; Wang, Z.; Shen, L.; Cho, H.; Wu, X.; Yang, Y. TDRD3 promotes DHX9 chromatin recruitment and R-loop resolution. Nucleic Acids Res. 2021, 49, 8573–8591. [Google Scholar] [CrossRef]
- Yang, S.; Winstone, L.; Mondal, S.; Wu, Y. Helicases in R-loop formation and resolution. J. Biol. Chem. 2023, 299, 105307. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 19110. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Abe, M.; Itaya, A.; Tomida, J.; Ishiai, M.; Takaori-Kondo, A.; Taoka, M.; Isobe, T.; Takata, M. FANCD2 protects genome stability by recruiting RNA processing enzymes to resolve R-loops during mild replication stress. FEBS J. 2019, 286, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- San Martin Alonso, M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021, 49, 4848–4863. [Google Scholar] [CrossRef]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, B.; Liu, X.; Gao, G.; Che, Z.; Fan, M.; Meng, S.; Zhao, X.; Sugimura, R.; Cao, H.; et al. ZFP281-BRCA2 prevents R-loop accumulation during DNA replication. Nat. Commun. 2022, 13, 3493. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef]
- Lima, W.F.; Murray, H.M.; Damle, S.S.; Hart, C.E.; Hung, G.; De Hoyos, C.L.; Liang, X.H.; Crooke, S.T. Viable RNaseH1 knockout mice show RNaseH1 is essential for R loop processing, mitochondrial and liver function. Nucleic Acids Res. 2016, 44, 5299–5312. [Google Scholar] [CrossRef]
- Cornelio, D.A.; Sedam, H.N.; Ferrarezi, J.A.; Sampaio, N.M.; Argueso, J.L. Both R-loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair 2017, 52, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Cristini, A.; Tellier, M.; Constantinescu, F.; Accalai, C.; Albulescu, L.O.; Heiringhoff, R.; Bery, N.; Sordet, O.; Murphy, S.; Gromak, N. RNase H2, mutated in Aicardi-Goutieres syndrome, resolves co-transcriptional R-loops to prevent DNA breaks and inflammation. Nat. Commun. 2022, 13, 2961. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell 2017, 65, 832–847.e834. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Melchionda, L.; Nasca, A.; Carrara, F.; Lamantea, E.; Zanolini, A.; Lamperti, C.; Fang, M.; Zhang, J.; Ronchi, D.; et al. RNASEH1 mutations Impair mtDNA replication and cause adult-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2015, 97, 186–193. [Google Scholar] [CrossRef]
- Hromas, R.; Srinivasan, G.; Yang, M.; Jaiswal, A.; Totterdale, T.A.; Phillips, L.; Kirby, A.; Khodayari, N.; Brantley, M.; Williamson, E.A.; et al. BRCA1 mediates protein homeostasis through the ubiquitination of PERK and IRE1. iScience 2022, 25, 105626. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.Q. The UPR transducer IRE1 promotes breast cancer malignancy by degrading tumor suppressor microRNAs. iScience 2020, 23, 101503. [Google Scholar] [CrossRef]
- Lee, K.Y.; Cheon, S.H.; Kim, D.G.; Lee, S.J.; Lee, B.J. A structural study of TatD from Staphylococcus aureus elucidates a putative DNA-binding mode of a Mg2+-dependent nuclease. IUCrJ 2020, 7, 509–521. [Google Scholar] [CrossRef]
- Chen, Y.C.; Li, C.L.; Hsiao, Y.Y.; Duh, Y.; Yuan, H.S. Structure and function of TatD exonuclease in DNA repair. Nucleic Acids Res. 2014, 42, 10776–10785. [Google Scholar] [CrossRef]
- Singh, D.; Rahi, A.; Kumari, R.; Gupta, V.; Gautam, G.; Aggarwal, S.; Rehan, M.; Bhatnagar, R. Computational and mutational analysis of TatD DNase of Bacillus anthracis. J. Cell. Biochem. 2019, 120, 11318–11330. [Google Scholar] [CrossRef]
- Dorival, J.; Eichman, B.F. Human and bacterial TatD enzymes exhibit apurinic/apyrimidinic (AP) endonuclease activity. Nucleic Acids Res. 2023, 51, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A. Targeting replication stress response pathways to enhance genotoxic chemo- and radiotherapy. Molecules 2022, 27, 4736. [Google Scholar] [CrossRef]
- Jackson, S.P.; Helleday, T. Drugging DNA repair. Science 2016, 352, 1178–1179. [Google Scholar] [CrossRef]
- Carrassa, L.; Damia, G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat. Rev. 2017, 60, 139–151. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Lee, S.H.; Hromas, R. Metnase and EEPD1: DNA repair functions and potential targets in cancer therapy. Front. Oncol. 2022, 12, 808757. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Weng, J. A pan-cancer bioinformatic analysis of RAD51 regarding the values for diagnosis, prognosis, and therapeutic prediction. Front. Oncol. 2022, 12, 858756. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickoloff, J.A.; Jaiswal, A.S.; Sharma, N.; Williamson, E.A.; Tran, M.T.; Arris, D.; Yang, M.; Hromas, R. Cellular Responses to Widespread DNA Replication Stress. Int. J. Mol. Sci. 2023, 24, 16903. https://doi.org/10.3390/ijms242316903
Nickoloff JA, Jaiswal AS, Sharma N, Williamson EA, Tran MT, Arris D, Yang M, Hromas R. Cellular Responses to Widespread DNA Replication Stress. International Journal of Molecular Sciences. 2023; 24(23):16903. https://doi.org/10.3390/ijms242316903
Chicago/Turabian StyleNickoloff, Jac A., Aruna S. Jaiswal, Neelam Sharma, Elizabeth A. Williamson, Manh T. Tran, Dominic Arris, Ming Yang, and Robert Hromas. 2023. "Cellular Responses to Widespread DNA Replication Stress" International Journal of Molecular Sciences 24, no. 23: 16903. https://doi.org/10.3390/ijms242316903
APA StyleNickoloff, J. A., Jaiswal, A. S., Sharma, N., Williamson, E. A., Tran, M. T., Arris, D., Yang, M., & Hromas, R. (2023). Cellular Responses to Widespread DNA Replication Stress. International Journal of Molecular Sciences, 24(23), 16903. https://doi.org/10.3390/ijms242316903