
Increased IGFBP2 Levels by Placenta-Derived Mesenchymal Stem Cells Enhance Glucose Metabolism in a TAA-Injured Rat Model via AMPK Signaling Pathway

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results
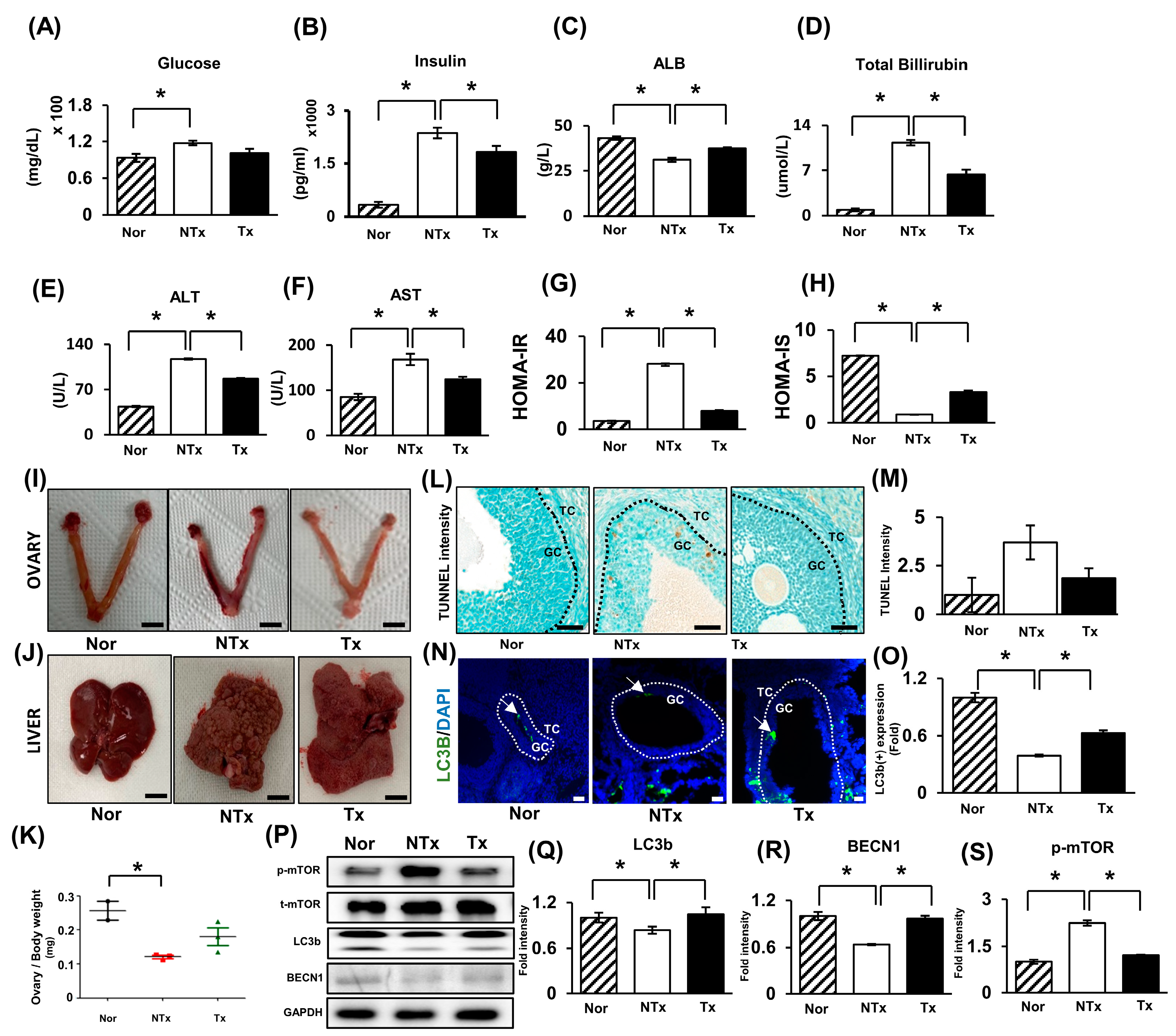
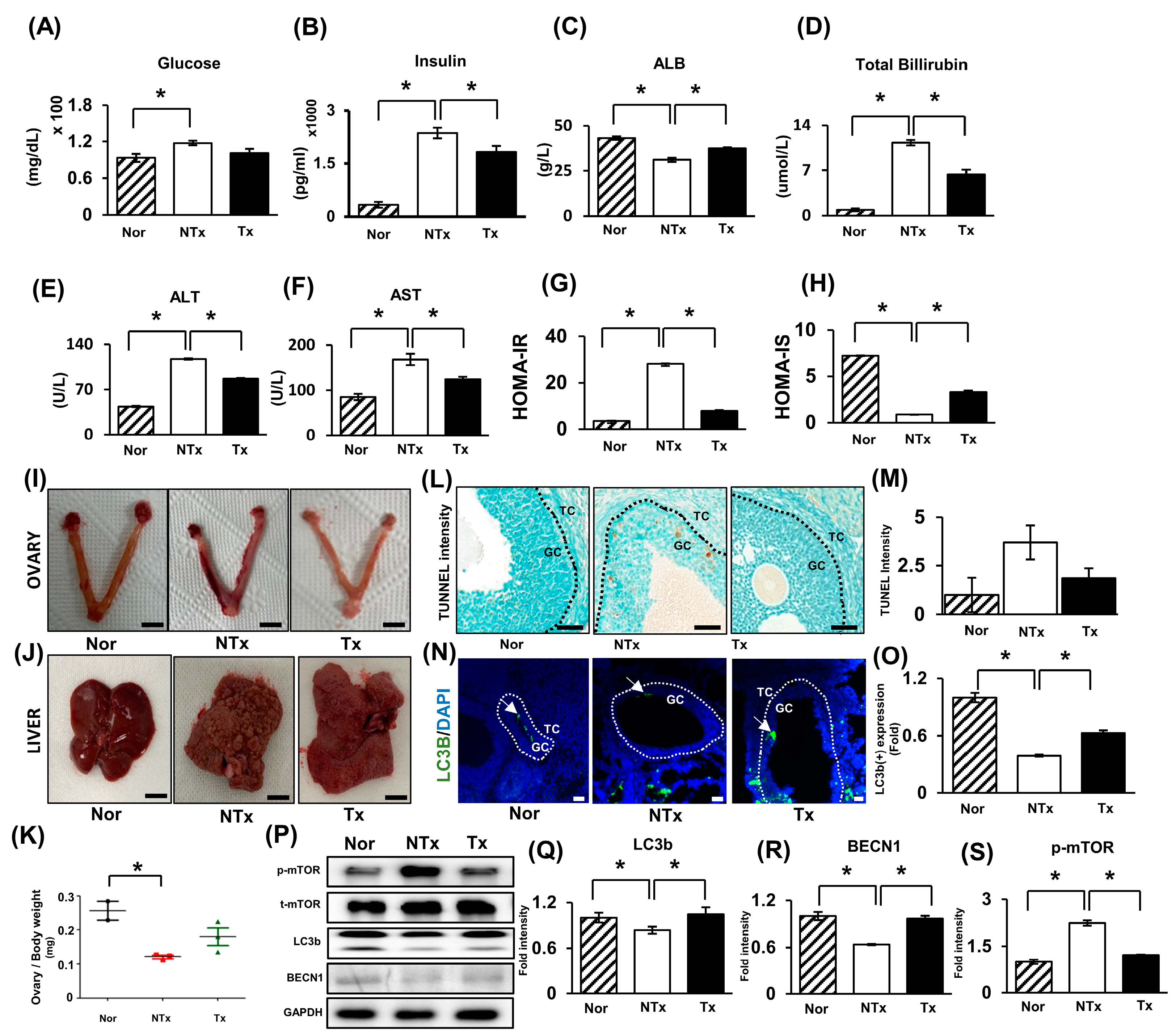
2.1. Thioacetamide Induces Ovarian Dysfunction and Insulin Resistance and Impairs Glucose Metabolism
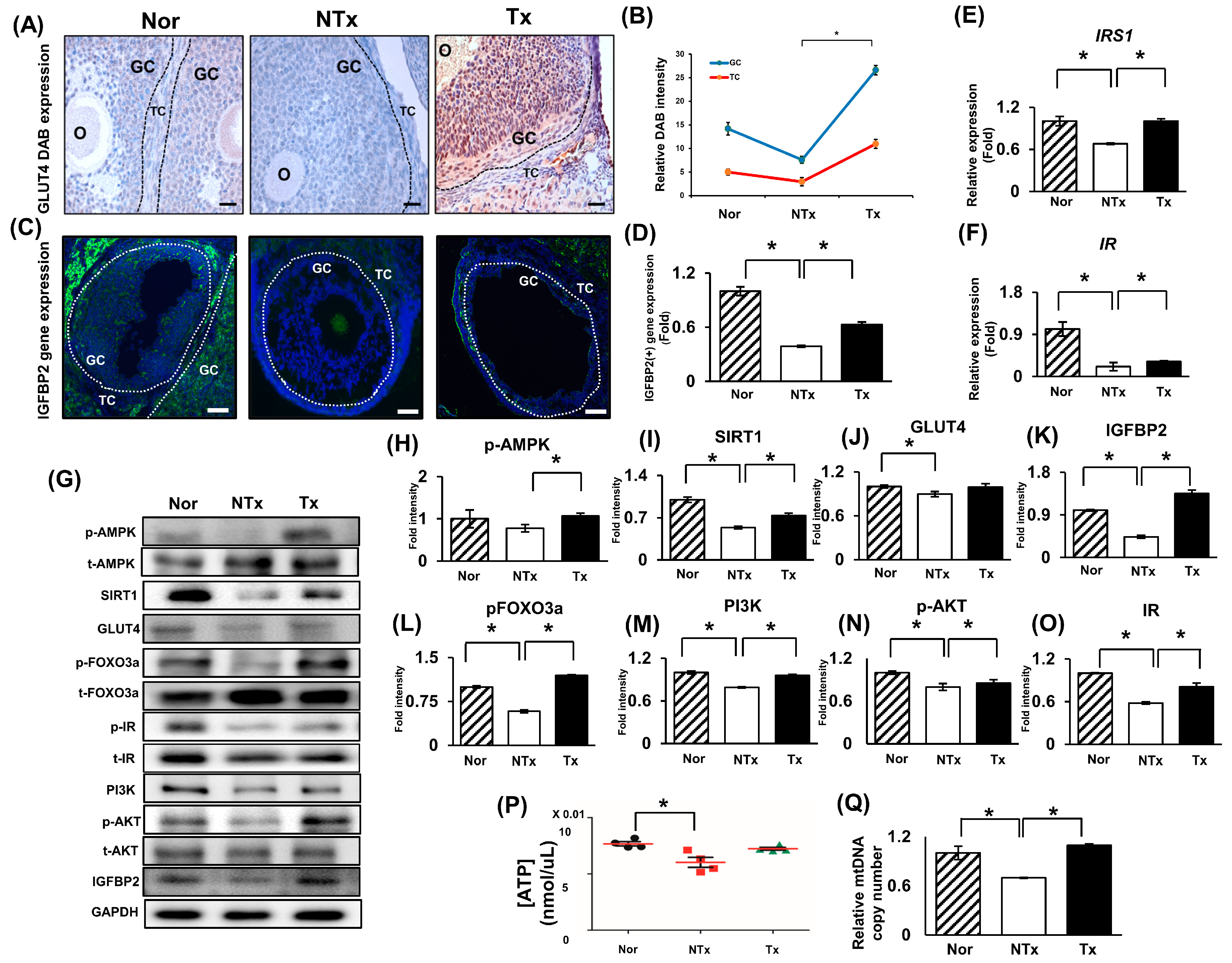
2.2. Glucose Metabolism and Insulin Pathway Signaling Activated by PD-MSCs through AMPK Signaling Pathway
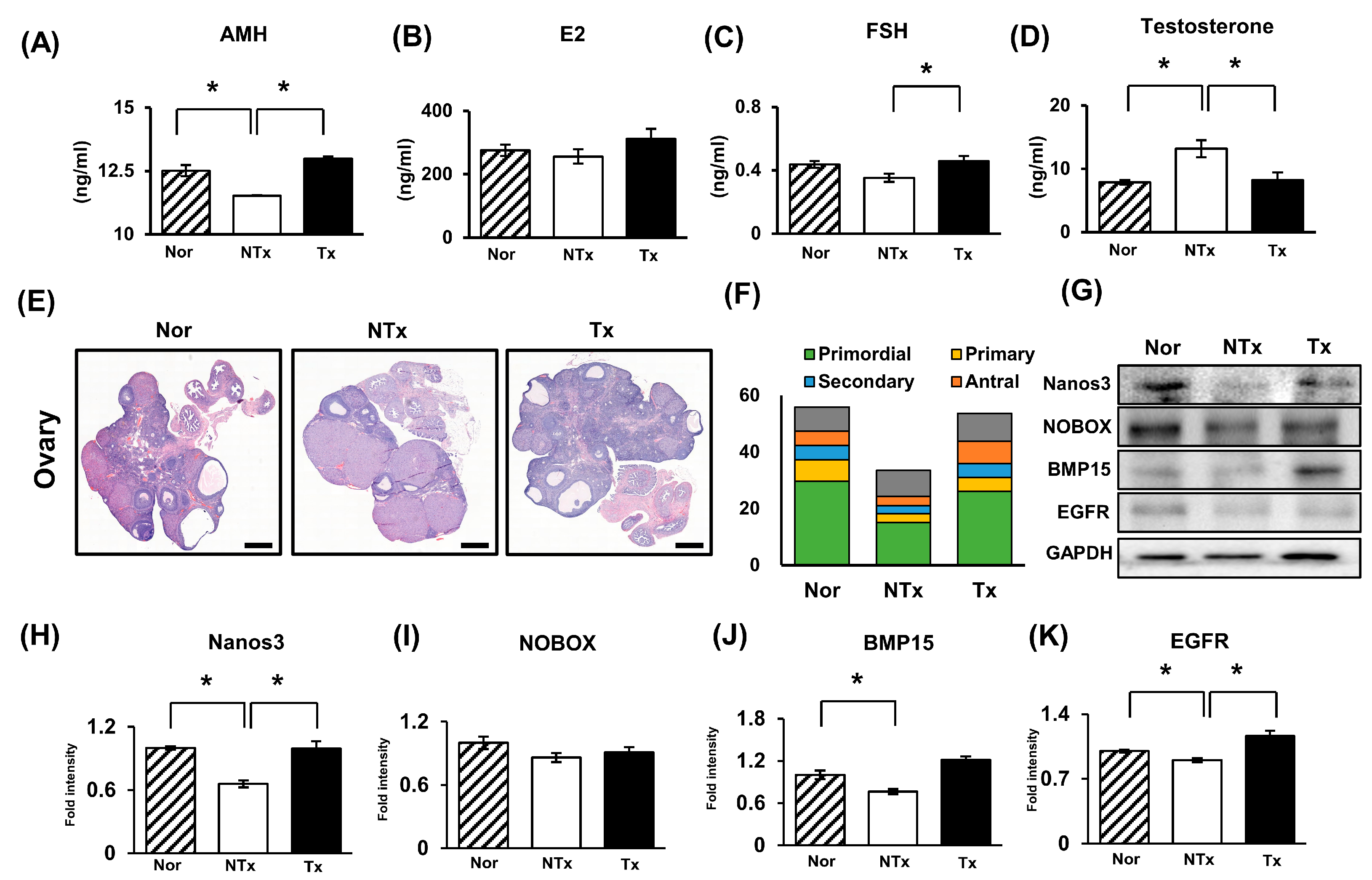
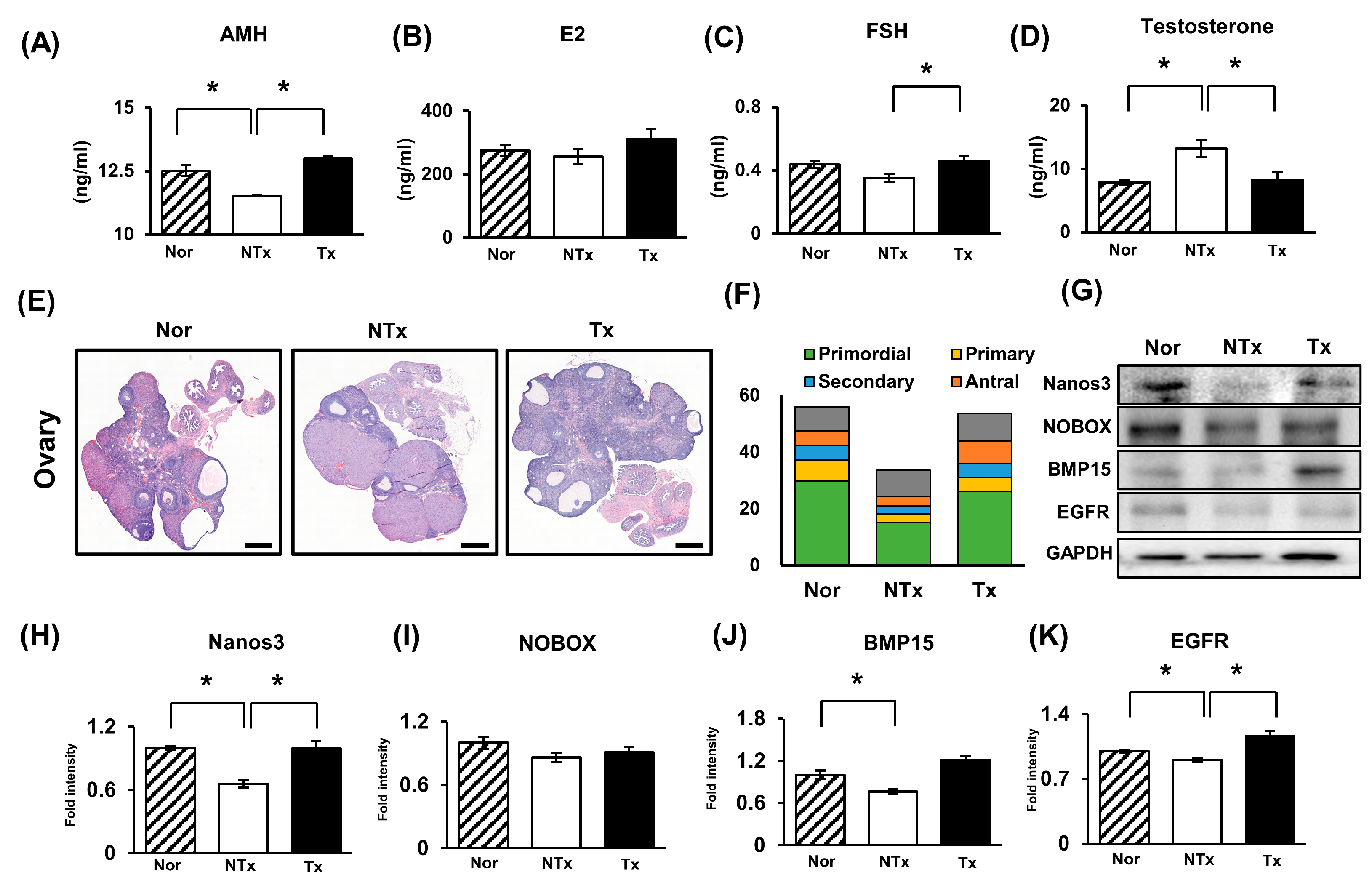
2.3. Effect of PD-MSCs on Ovarian Function in the Ovaries of the TAA-Injured Rats
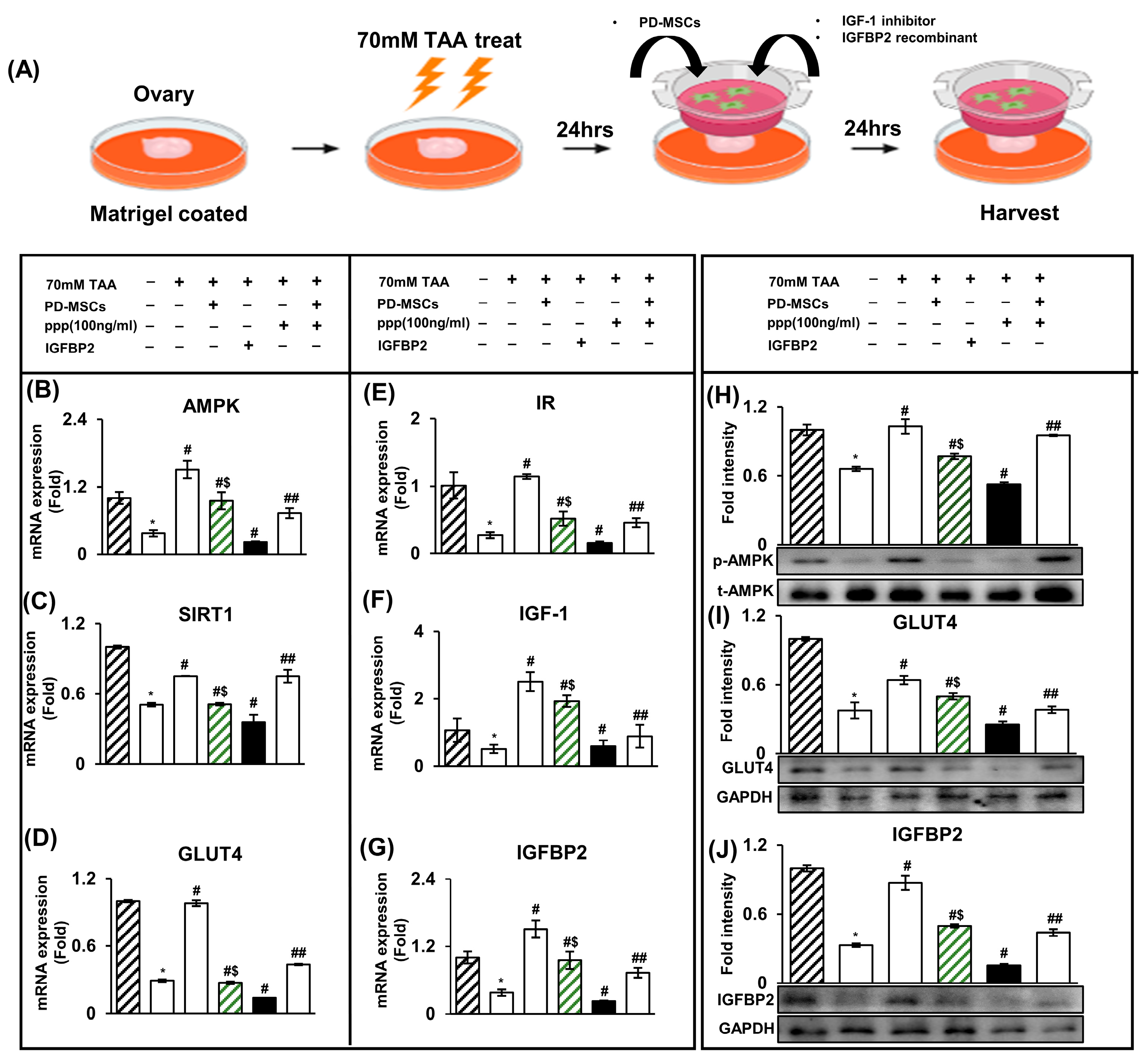
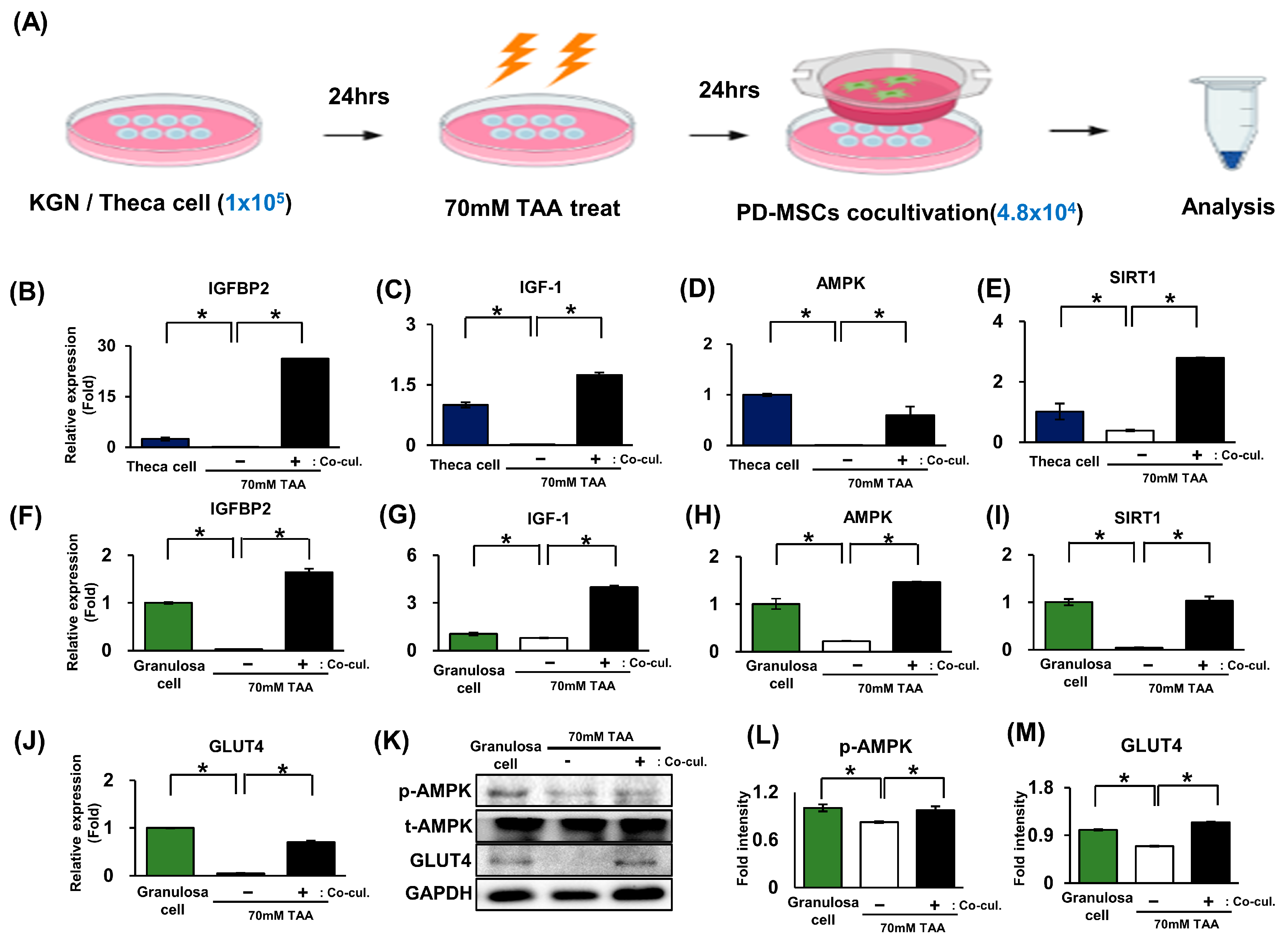
2.4. The Increase in IGFBP2 Secreted by PD-MSCs Triggers Glucose Metabolism in a TAA-Injured Rat Ovary Model
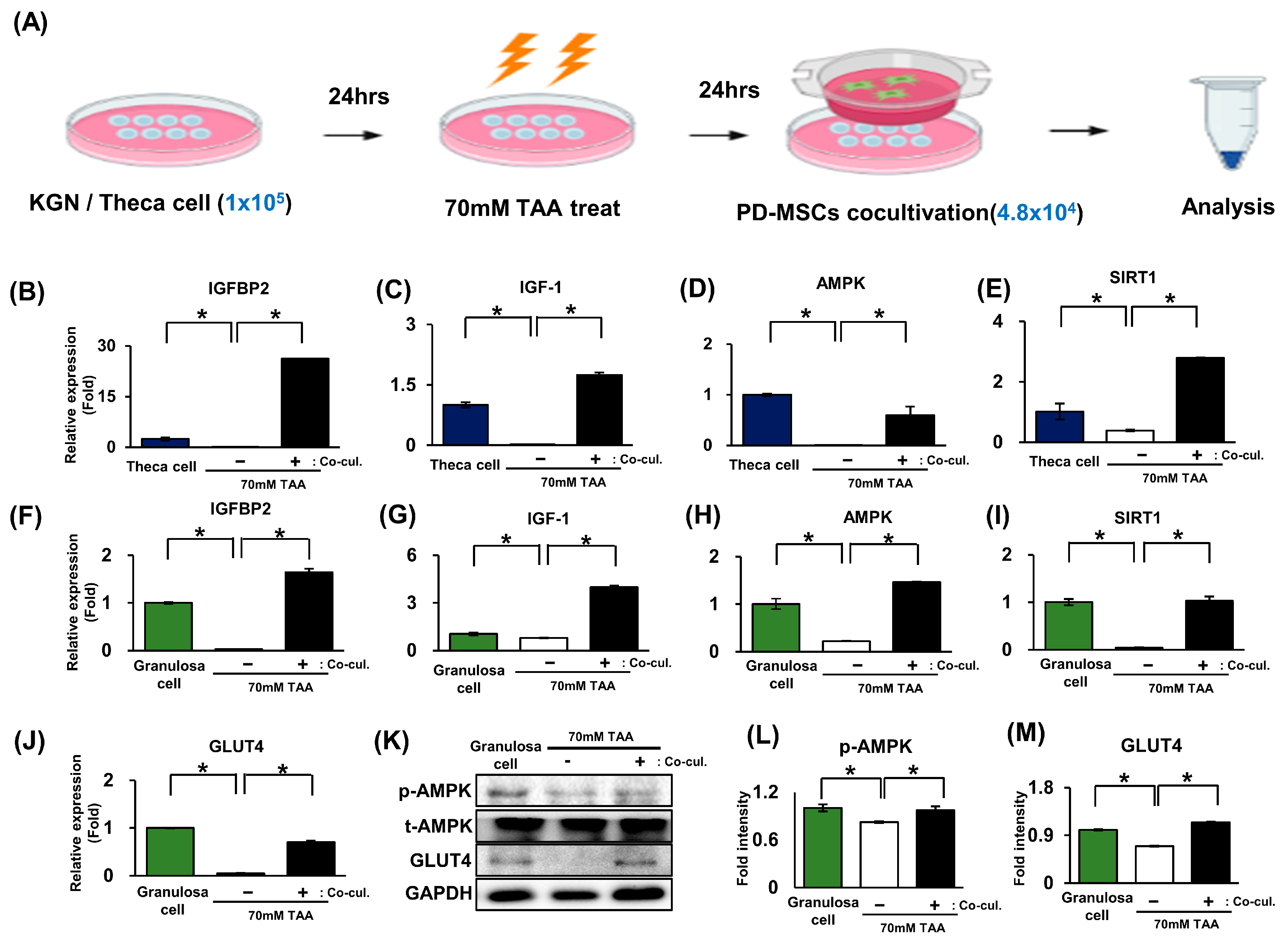
2.5. The Activation of Glucose Metabolism Markers Was Enhanced by the AMPK Signaling Pathway in Granulosa Cells (In Vitro)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. The Human GC-like Tumor Cell Line KGN
4.3. Primary Cell Culture
4.4. Ex Vivo Culture System
4.5. TAA Animal Modeling and PD-MSC Transplantation
4.6. Blood Chemistry Test
4.7. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
4.8. Protein Isolation and Western Blotting
4.9. The gDNA Extraction and Mitochondrial DNA Copy Number Analysis
4.10. Hematoxylin and Eosin (H&E) Staining for Follicle Count
4.11. Immunoflurescence Staining
4.12. Immunohistochemistry Staining
4.13. Enzyme-Linked Immunosorbent Assay
4.14. The Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cakir Bicer, N.; Ermis, A.A.; Bas, D. The Role of Different Methods in Defining Cardiometabolic Risk and Metabolic Syndrome in Women with Polycystic Ovary Syndrome. Life 2023, 13, 1959. [Google Scholar] [CrossRef] [PubMed]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; International, P.N. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum. Reprod. 2018, 33, 1602–1618. [Google Scholar] [CrossRef] [PubMed]
- Joham, A.E.; Norman, R.J.; Stener-Victorin, E.; Legro, R.S.; Franks, S.; Moran, L.J.; Boyle, J.; Teede, H.J. Polycystic ovary syndrome. Lancet Diabetes Endocrinol. 2022, 10, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Sutton-McDowall, M.L.; Gilchrist, R.B.; Thompson, J.G. The pivotal role of glucose metabolism in determining oocyte developmental competence. Reproduction 2010, 139, 685–695. [Google Scholar] [CrossRef]
- Gawriluk, T.R.; Ko, C.; Hong, X.; Christenson, L.K.; Rucker, E.B., 3rd. Beclin-1 deficiency in the murine ovary results in the reduction of progesterone production to promote preterm labor. Proc. Natl. Acad. Sci. USA 2014, 111, E4194–E4203. [Google Scholar] [CrossRef]
- Zhang, J.; Bao, Y.; Zhou, X.; Zheng, L. Polycystic ovary syndrome and mitochondrial dysfunction. Reprod. Biol. Endocrinol. 2019, 17, 67. [Google Scholar] [CrossRef]
- Kirillova, A.; Smitz, J.E.J.; Sukhikh, G.T.; Mazunin, I. The Role of Mitochondria in Oocyte Maturation. Cells 2021, 10, 2484. [Google Scholar] [CrossRef]
- Das, M.; Sauceda, C.; Webster, N.J.G. Mitochondrial Dysfunction in Obesity and Reproduction. Endocrinology 2021, 162, bqaa158. [Google Scholar] [CrossRef]
- Hohos, N.M.; Skaznik-Wikiel, M.E. High-Fat Diet and Female Fertility. Endocrinology 2017, 158, 2407–2419. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Froment, P.; Plotton, I.; Giulivi, C.; Fabre, S.; Khoueiry, R.; Mourad, N.I.; Horman, S.; Rame, C.; Rouillon, C.; Grandhaye, J.; et al. At the crossroads of fertility and metabolism: The importance of AMPK-dependent signaling in female infertility associated with hyperandrogenism. Hum. Reprod. 2022, 37, 1207–1228. [Google Scholar] [CrossRef] [PubMed]
- Aghanoori, M.R.; Smith, D.R.; Shariati-Ievari, S.; Ajisebutu, A.; Nguyen, A.; Desmond, F.; Jesus, C.H.A.; Zhou, X.; Calcutt, N.A.; Aliani, M.; et al. Insulin-like growth factor-1 activates AMPK to augment mitochondrial function and correct neuronal metabolism in sensory neurons in type 1 diabetes. Mol. Metab. 2019, 20, 149–165. [Google Scholar] [CrossRef]
- Spitschak, M.; Hoeflich, A. Potential Functions of IGFBP-2 for Ovarian Folliculogenesis and Steroidogenesis. Front. Endocrinol. 2018, 9, 119. [Google Scholar] [CrossRef]
- Kamangar, B.B.; Gabillard, J.C.; Bobe, J. Insulin-like growth factor-binding protein (IGFBP)-1, -2, -3, -4, -5, and -6 and IGFBP-related protein 1 during rainbow trout postvitellogenesis and oocyte maturation: Molecular characterization, expression profiles, and hormonal regulation. Endocrinology 2006, 147, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Webber, L.; Anderson, R.A.; Davies, M.; Janse, F.; Vermeulen, N. HRT for women with premature ovarian insufficiency: A comprehensive review. Hum. Reprod. Open 2017, 2017, hox007. [Google Scholar] [CrossRef] [PubMed]
- Esfandyari, S.; Chugh, R.M.; Park, H.S.; Hobeika, E.; Ulin, M.; Al-Hendy, A. Mesenchymal Stem Cells as a Bio Organ for Treatment of Female Infertility. Cells 2020, 9, 2253. [Google Scholar] [CrossRef] [PubMed]
- Shareghi-Oskoue, O.; Aghebati-Maleki, L.; Yousefi, M. Transplantation of human umbilical cord mesenchymal stem cells to treat premature ovarian failure. Stem Cell Res. Ther. 2021, 12, 454. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Seok, J.; Lim, S.M.; Kim, T.H.; Kim, G.J. Microenvironmental changes induced by placenta-derived mesenchymal stem cells restore ovarian function in ovariectomized rats via activation of the PI3K-FOXO3 pathway. Stem Cell Res. Ther. 2020, 11, 486. [Google Scholar] [CrossRef]
- Cho, J.; Kim, T.H.; Seok, J.; Jun, J.H.; Park, H.; Kweon, M.; Lim, J.Y.; Kim, G.J. Vascular remodeling by placenta-derived mesenchymal stem cells restores ovarian function in ovariectomized rat model via the VEGF pathway. Lab. Investig. 2021, 101, 304–317. [Google Scholar] [CrossRef]
- Park, H.J.; Jun, J.H.; Kim, J.Y.; Jang, H.J.; Lim, J.Y.; Bae, S.H.; Kim, G.J. Phosphatase of Regenerating Liver-1 (PRL-1)-Overexpressing Placenta-Derived Mesenchymal Stem Cells Enhance Antioxidant Effects via Peroxiredoxin 3 in TAA-Injured Rat Livers. Antioxidants 2022, 12, 46. [Google Scholar] [CrossRef]
- Kim, J.J.; Hwang, K.R.; Oh, S.H.; Chae, S.J.; Yoon, S.H.; Choi, Y.M. Prevalence of insulin resistance in Korean women with polycystic ovary syndrome according to various homeostasis model assessment for insulin resistance cutoff values. Fertil. Steril. 2019, 112, 959–966.e951. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Song, J.; Kim, H.H.; Seok, J.; Kim, J.Y.; Jun, J.H.; Kim, G.J. Human placenta-derived mesenchymal stem cells trigger repair system in TAA-injured rat model via antioxidant effect. Aging 2020, 13, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Franks, S.; Stark, J.; Hardy, K. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum. Reprod. Update 2008, 14, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Silvestris, E.; de Pergola, G.; Rosania, R.; Loverro, G. Obesity as disruptor of the female fertility. Reprod. Biol. Endocrinol. 2018, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Chen, L.; Cai, L.; Ge, S.; Deng, X. Regulatory effects of the AMPKalpha-SIRT1 molecular pathway on insulin resistance in PCOS mice: An in vitro and in vivo study. Biochem. Biophys. Res. Commun. 2017, 494, 615–620. [Google Scholar] [CrossRef]
- Boughanem, H.; Yubero-Serrano, E.M.; Lopez-Miranda, J.; Tinahones, F.J.; Macias-Gonzalez, M. Potential Role of Insulin Growth-Factor-Binding Protein 2 as Therapeutic Target for Obesity-Related Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 1133. [Google Scholar] [CrossRef]
- Nishi, Y.; Yanase, T.; Mu, Y.; Oba, K.; Ichino, I.; Saito, M.; Nomura, M.; Mukasa, C.; Okabe, T.; Goto, K.; et al. Establishment and characterization of a steroidogenic human granulosa-like tumor cell line, KGN, that expresses functional follicle-stimulating hormone receptor. Endocrinology 2001, 142, 437–445. [Google Scholar] [CrossRef]
- Cao, M.; Pan, Q.; Dong, H.; Yuan, X.; Li, Y.; Sun, Z.; Dong, X.; Wang, H. Adipose-derived mesenchymal stem cells improve glucose homeostasis in high-fat diet-induced obese mice. Stem Cell Res. Ther. 2015, 6, 208. [Google Scholar] [CrossRef]
- Davinelli, S.; Nicolosi, D.; Di Cesare, C.; Scapagnini, G.; Di Marco, R. Targeting Metabolic Consequences of Insulin Resistance in Polycystic Ovary Syndrome by D-chiro-inositol and Emerging Nutraceuticals: A Focused Review. J. Clin. Med. 2020, 9, 987. [Google Scholar] [CrossRef]
- Seok, J.; Park, H.; Choi, J.H.; Lim, J.Y.; Kim, K.G.; Kim, G.J. Placenta-Derived Mesenchymal Stem Cells Restore the Ovary Function in an Ovariectomized Rat Model via an Antioxidant Effect. Antioxidants 2020, 9, 591. [Google Scholar] [CrossRef]
- Wu, X.K.; Zhou, S.Y.; Liu, J.X.; Pollanen, P.; Sallinen, K.; Makinen, M.; Erkkola, R. Selective ovary resistance to insulin signaling in women with polycystic ovary syndrome. Fertil. Steril. 2003, 80, 954–965. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, L.; Cheng, F.; Jiang, Q.; Ye, Y.; Ren, Y.; He, Y.; Su, D.; Cheng, L.; Shi, G.; et al. PPARgamma-dependent hepatic macrophage switching acts as a central hub for hUCMSC-mediated alleviation of decompensated liver cirrhosis in rats. Stem Cell Res. Ther. 2023, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Carmina, E.; Rosato, F.; Janni, A.; Rizzo, M.; Longo, R.A. Extensive clinical experience: Relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J. Clin. Endocrinol. Metab. 2006, 91, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.H.; Poulsen, G.J.; Videbech, P.; Wohlfahrt, J.; Melbye, M. Endocrine disease history and the risk of postpartum depression. Br. J. Psychiatry 2023, 222, 119–124. [Google Scholar] [CrossRef]
- Chugh, R.M.; Park, H.S.; El Andaloussi, A.; Elsharoud, A.; Esfandyari, S.; Ulin, M.; Bakir, L.; Aboalsoud, A.; Ali, M.; Ashour, D.; et al. Mesenchymal stem cell therapy ameliorates metabolic dysfunction and restores fertility in a PCOS mouse model through interleukin-10. Stem Cell Res. Ther. 2021, 12, 388. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Shen, R.; Song, L.; Lu, M.; Wang, J.; Zhao, S.; Tang, Y.; Meng, X.; Li, Z.; He, Z.X. Bone Marrow Mesenchymal Stem Cells (BM-MSCs) Improve Heart Function in Swine Myocardial Infarction Model through Paracrine Effects. Sci. Rep. 2016, 6, 28250. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, S.; Lee, H.J.; Lew, H.; Kim, G.J. Functionally enhanced placenta-derived mesenchymal stem cells inhibit adipogenesis in orbital fibroblasts with Graves’ ophthalmopathy. Stem Cell Res. Ther. 2020, 11, 469. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.; Wang, Z.; Zheng, N.; Yuan, F.; Li, B.; Li, X.; Deng, L.; Lin, M.; Chen, X.; et al. Enhanced glycolysis in granulosa cells promotes the activation of primordial follicles through mTOR signaling. Cell Death Dis. 2022, 13, 87. [Google Scholar] [CrossRef]
- Hoque, S.A.M.; Kawai, T.; Zhu, Z.; Shimada, M. Mitochondrial Protein Turnover Is Critical for Granulosa Cell Proliferation and Differentiation in Antral Follicles. J. Endocr. Soc. 2019, 3, 324–339. [Google Scholar] [CrossRef]
- Myers, M.; Britt, K.L.; Wreford, N.G.; Ebling, F.J.; Kerr, J.B. Methods for quantifying follicular numbers within the mouse ovary. Reproduction 2004, 127, 569–580. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Annealing Temperature (°C) | NM Number |
|---|---|---|---|
| hIGFBP2 | F: 5′-ACA TCC CCA ACT GTG ACA AG-3′ | 56.8 | NM_001313993.2 |
| R: 5′-ATC AGC TTC CCG GTG TTG A-3′ | |||
| hAMPK | F: 5′- CCA TGA AGA GGG CCA CAA TC-3′ | 58 | NM_001355035.2 |
| R: 5′-TGC CAA AGG ATC CTG GTG AT -3′ | |||
| hGLUT4 | F: 5’-ATC CTT GGA CGA TTC CTC ATT GG-3’ | 59.4 | NM_175697.3 |
| R: 5’-CAG GTG AGT GGG AGC AAT CT -3’ | |||
| hGAPDH | F: 5’-CTC CTC TTC GGC AGC ACA-3’ | 58.2 | NM_001357943.2 |
| R: 5’-AAC GCT TCA CCT AAT TTG CGT-3’ | |||
| rIGFBP2 | F: 5’-GCA AAG GTG CCA AAC ACC TC-3’ | 59.3 | NM_001310659.1 |
| R: 5’-TTC CAG AGG ACC CCG ATC AT -3’ | |||
| rGLUT4 | F: 5’-TCA TCT TCA CCT TCC TAA-3’ | 49.1 | NM_001039163.2 |
| R: 5’-CCT CAG TCA TTC TCA TCT -3’ | |||
| rAMPK | F: 5’-AGA TTG CAA AGG GCA TGA ACT AC-3’ | 57.8 | NM_023991.1 |
| R: 5’-ACA TTC cTG CT GCC AAG TC-3’ | |||
| rIR | F: 5’-TTT TTG TCC CCA GGC CAT CC -3’ | 59.6 | NM_017071.2 |
| R: 5’-CCT GTG CTC CTC CTG ACT TG-3’ | |||
| rGAPDH | F: 5’-TCT CTG CTC CTC CCT GTT CTA-3’ | 59.4 | NM_017008.4 |
| R: 5’-ATG AAG GGG TCG TTG ATG GC -3’ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-H.; Park, H.; You, J.-H.; Seok, J.; Kwon, D.-W.; Kim, Y.-R.; Kim, G.-J. Increased IGFBP2 Levels by Placenta-Derived Mesenchymal Stem Cells Enhance Glucose Metabolism in a TAA-Injured Rat Model via AMPK Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 16531. https://doi.org/10.3390/ijms242216531
Lee D-H, Park H, You J-H, Seok J, Kwon D-W, Kim Y-R, Kim G-J. Increased IGFBP2 Levels by Placenta-Derived Mesenchymal Stem Cells Enhance Glucose Metabolism in a TAA-Injured Rat Model via AMPK Signaling Pathway. International Journal of Molecular Sciences. 2023; 24(22):16531. https://doi.org/10.3390/ijms242216531
Chicago/Turabian StyleLee, Dae-Hyun, Hyeri Park, Jun-Hyeong You, Jin Seok, Dong-Wook Kwon, Young-Ran Kim, and Gi-Jin Kim. 2023. "Increased IGFBP2 Levels by Placenta-Derived Mesenchymal Stem Cells Enhance Glucose Metabolism in a TAA-Injured Rat Model via AMPK Signaling Pathway" International Journal of Molecular Sciences 24, no. 22: 16531. https://doi.org/10.3390/ijms242216531
APA StyleLee, D.-H., Park, H., You, J.-H., Seok, J., Kwon, D.-W., Kim, Y.-R., & Kim, G.-J. (2023). Increased IGFBP2 Levels by Placenta-Derived Mesenchymal Stem Cells Enhance Glucose Metabolism in a TAA-Injured Rat Model via AMPK Signaling Pathway. International Journal of Molecular Sciences, 24(22), 16531. https://doi.org/10.3390/ijms242216531