

Epigenetic Dysregulation and Its Correlation with the Steroidogenic Machinery Impacting Breast Pathogenesis: Data Mining and Molecular Insights into Therapeutics



Abstract
1. Introduction
2. Results
2.1. Analyses of TCGA Pan-Cancer Normalized RNA-Seq Datasets for the Expression of Epigenetic Enzymes in Cancerous and Non-Cancerous Breast Tissues
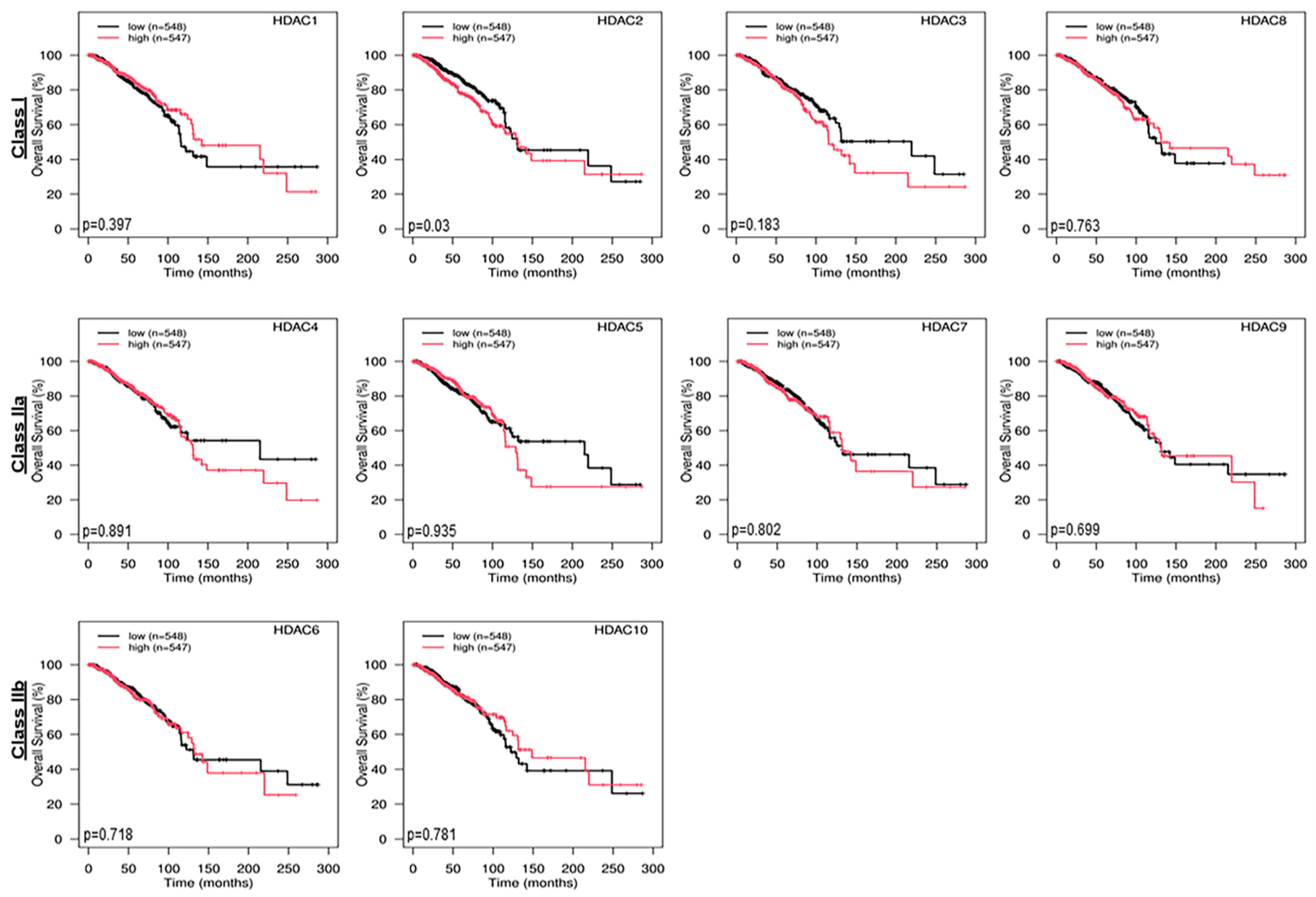
2.2. Genomic Expression Profiling of Epigenetic Enzymes in TCGA Cancerous and Non-Cancerous Breast Tissues and Their Correlation with Overall Survival
2.3. Analyses of a Variety of Steroidogenic Factors and Hormone Receptors Using TCGA Pan-Cancer Normalized BC RNA-Seq Datasets
2.4. Genomic Expression of Positive and Negative Regulatory Transcription Factors Using TCGA Cancerous and Non-Cancerous Breast RNA-Seq Datasets
2.5. Analyses of RNA-Seq Datasets for the Expression of Epigenetic Enzymes in a Variety of Human Cancerous and Non-Cancerous Breast Cell Lines
2.6. Genomic Expression of Key Steroidogenic Factors and Hormone Receptors in Cancerous and Non-Cancerous Breast Cell Lines
2.7. Ingenuity Pathway Analysis Using BC Microarray and RNA-Seq Datasets
3. Discussion
4. Materials and Methods
4.1. Analyses of TCGA Pan-Cancer Normalized RNA-Seq BC Datasets for the Expression of Various Epigenetic Enzymes
4.2. Generation of Kaplan–Meier Curves and Overall Survival Analyses
4.3. Analyses of RNA-Seq Datasets for the Expression of Various Steroidogenic Marker Genes, Hormone Receptors, and Transcription Factors in Cancerous and Non-Cancerous Breast Tissues
4.4. Generation of Heatmaps Using Transcriptome and RNA-Seq Data Pertaining to Various Breast Cell Lines and TCGA Tumors
4.5. Boxplot Analyses of Key Steroidogenic Markers in Normal and Cancerous Breast Tissues Using TCGA Transcriptome and RNA-Seq Datasets
4.6. Statistical Analysis
5. Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar]
- O’Sullivan, D.E.; Johnson, K.C.; Skinner, L.; Koestler, D.C.; Christensen, B.C. Epigenetic and genetic burden measures are associated with tumor characteristics in invasive breast carcinoma. Epigenetics 2016, 11, 344–353. [Google Scholar] [CrossRef]
- Buocikova, V.; Rios-Mondragon, I.; Pilalis, E.; Chatziioannou, A.; Miklikova, S.; Mego, M.; Pajuste, K.; Rucins, M.; Yamani, N.E.; Longhin, E.M.; et al. Epigenetics in Breast Cancer Therapy-New Strategies and Future Nanomedicine Perspectives. Cancers 2020, 12, 3622. [Google Scholar] [CrossRef]
- Ayipo, Y.O.; Ajiboye, A.T.; Osunniran, W.A.; Jimoh, A.A.; Mordi, M.N. Epigenetic oncogenesis, biomarkers and emerging chemotherapeutics for breast cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194873. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Patel, M.; Chandrasekaran, S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites 2021, 11, 792. [Google Scholar] [CrossRef]
- Hai, R.; Yang, D.; Zheng, F.; Wang, W.; Han, X.; Bode, A.M.; Luo, X. The emerging roles of HDACs and their therapeutic implications in cancer. Eur. J. Pharmacol. 2022, 931, 175216. [Google Scholar] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Manna, P.R.; Ahmed, A.U.; Yang, S.; Narasimhan, M.; Cohen-Tannoudji, J.; Slominski, A.T.; Pruitt, K. Genomic Profiling of the Steroidogenic Acute Regulatory Protein in Breast Cancer: In Silico Assessments and a Mechanistic Perspective. Cancers 2019, 11, 623. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [PubMed]
- Manna, P.R.; Ahmed, A.U.; Molehin, D.; Narasimhan, M.; Pruitt, K.; Reddy, P.H. Hormonal and Genetic Regulatory Events in Breast Cancer and Its Therapeutics: Importance of the Steroidogenic Acute Regulatory Protein. Biomedicines 2022, 10, 1313. [Google Scholar]
- Manna, P.R.; Ramachandran, S.; Pradeepkiran, J.A.; Molehin, D.; Castro-Piedras, I.; Pruitt, K.; Ganapathy, V.; Reddy, P.H. Expression and Function of StAR in Cancerous and Non-Cancerous Human and Mouse Breast Tissues: New Insights into Diagnosis and Treatment of Hormone-Sensitive Breast Cancer. Int. J. Mol. Sci. 2023, 24, 758. [Google Scholar] [PubMed]
- Castro-Piedras, I.; Sharma, M.; den Bakker, M.; Molehin, D.; Martinez, E.G.; Vartak, D.; Pruitt, W.M.; Deitrick, J.; Almodovar, S.; Pruitt, K. DVL1 and DVL3 differentially localize to CYP19A1 promoters and regulate aromatase mRNA in breast cancer cells. Oncotarget 2018, 9, 35639–35654. [Google Scholar]
- Sharma, M.; Molehin, D.; Castro-Piedras, I.; Martinez, E.G.; Pruitt, K. Acetylation of conserved DVL-1 lysines regulates its nuclear translocation and binding to gene promoters in triple-negative breast cancer. Sci. Rep. 2019, 9, 16257. [Google Scholar]
- Manna, P.R.; Ahmed, A.U.; Vartak, D.; Molehin, D.; Pruitt, K. Overexpression of the steroidogenic acute regulatory protein in breast cancer: Regulation by histone deacetylase inhibition. Biochem. Biophys. Res. Commun. 2019, 509, 476–482. [Google Scholar] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar]
- Kaypee, S.; Sudarshan, D.; Shanmugam, M.K.; Mukherjee, D.; Sethi, G.; Kundu, T.K. Aberrant lysine acetylation in tumorigenesis: Implications in the development of therapeutics. Pharmacol. Ther. 2016, 162, 98–119. [Google Scholar]
- Zhao, J.; Xie, C.; Edwards, H.; Wang, G.; Taub, J.W.; Ge, Y. Histone deacetylases 1 and 2 cooperate in regulating BRCA1, CHK1, and RAD51 expression in acute myeloid leukemia cells. Oncotarget 2017, 8, 6319–6329. [Google Scholar]
- Ruzic, D.; Djokovic, N.; Srdic-Rajic, T.; Echeverria, C.; Nikolic, K.; Santibanez, J.F. Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention. Pharmaceutics 2022, 14, 209. [Google Scholar]
- Cummins, C.L.; Volle, D.H.; Zhang, Y.; McDonald, J.G.; Sion, B.; Lefrancois-Martinez, A.M.; Caira, F.; Veyssiere, G.; Mangelsdorf, D.J.; Lobaccaro, J.M. Liver X receptors regulate adrenal cholesterol balance. J. Clin. Investig. 2006, 116, 1902–1912. [Google Scholar]
- Manna, P.R.; Dyson, M.T.; Stocco, D.M. Role of basic leucine zipper proteins in transcriptional regulation of the steroidogenic acute regulatory protein gene. Mol. Cell Endocrinol. 2009, 302, 1–11. [Google Scholar] [PubMed]
- Manna, P.R.; Cohen-Tannoudji, J.; Counis, R.; Garner, C.W.; Huhtaniemi, I.; Kraemer, F.B.; Stocco, D.M. Mechanisms of action of hormone-sensitive lipase in mouse Leydig cells: Its role in the regulation of the steroidogenic acute regulatory protein. J. Biol. Chem. 2013, 288, 8505–8518. [Google Scholar]
- Casper, J.; Zweig, A.S.; Villarreal, C.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Karolchik, D.; et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018, 46, D762–D769. [Google Scholar] [PubMed]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [PubMed]
- Heiser, L.M.; Sadanandam, A.; Kuo, W.L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Gibb, W.J.; Wang, N.J.; Ziyad, S.; Tong, F.; et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729. [Google Scholar]
- Munir, M.T.; Ponce, C.; Powell, C.A.; Tarafdar, K.; Yanagita, T.; Choudhury, M.; Gollahon, L.S.; Rahman, S.M. The contribution of cholesterol and epigenetic changes to the pathophysiology of breast cancer. J. Steroid Biochem. Mol. Biol. 2018, 183, 1–9. [Google Scholar]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. [Google Scholar]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar]
- Clem, B.F.; Clark, B.J. Association of the mSin3A-histone deacetylase 1/2 corepressor complex with the mouse steroidogenic acute regulatory protein gene. Mol. Endocrinol. 2006, 20, 100–113. [Google Scholar]
- Manna, P.R.; Stetson, C.L.; Slominski, A.T.; Pruitt, K. Role of the steroidogenic acute regulatory protein in health and disease. Endocrine 2016, 51, 7–21. [Google Scholar] [PubMed]
- Manna, P.R.; Wang, X.J.; Stocco, D.M. Involvement of multiple transcription factors in the regulation of steroidogenic acute regulatory protein gene expression. Steroids 2003, 68, 1125–1134. [Google Scholar] [PubMed]
- Clem, B.F.; Hudson, E.A.; Clark, B.J. Cyclic adenosine 3′,5′-monophosphate (cAMP) enhances cAMP-responsive element binding (CREB) protein phosphorylation and phospho-CREB interaction with the mouse steroidogenic acute regulatory protein gene promoter. Endocrinology 2005, 146, 1348–1356. [Google Scholar] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar]
- Pramanik, S.D.; Kumar Halder, A.; Mukherjee, U.; Kumar, D.; Dey, Y.N.; R, M. Potential of histone deacetylase inhibitors in the control and regulation of prostate, breast and ovarian cancer. Front. Chem. 2022, 10, 948217. [Google Scholar]
- Pruitt, K. Molecular and Cellular Changes During Cancer Progression Resulting From Genetic and Epigenetic Alterations. Prog. Mol. Biol. Transl. Sci. 2016, 144, 3–47. [Google Scholar]
- Qiu, T.; Zhou, L.; Zhu, W.; Wang, T.; Wang, J.; Shu, Y.; Liu, P. Effects of treatment with histone deacetylase inhibitors in solid tumors: A review based on 30 clinical trials. Future Oncol. 2013, 9, 255–269. [Google Scholar] [CrossRef]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cancer. Cholesterol and cancer, in the balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef]
- Goldstein, T.C.; Paull, E.O.; Ellis, M.J.; Stuart, J.M. Molecular pathways: Extracting medical knowledge from high-throughput genomic data. Clin. Cancer Res. 2013, 19, 3114–3120. [Google Scholar] [CrossRef] [PubMed]
- Schech, A.J.; Shah, P.; Yu, S.; Sabnis, G.J.; Goloubeva, O.; Rosenblatt, P.; Kazi, A.; Chumsri, S.; Brodie, A. Histone deacetylase inhibitor entinostat in combination with a retinoid downregulates HER2 and reduces the tumor initiating cell population in aromatase inhibitor-resistant breast cancer. Breast Cancer Res. Treat. 2015, 152, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, D.; Rampias, T. HDAC Inhibitors: Dissecting Mechanisms of Action to Counter Tumor Heterogeneity. Cancers 2021, 13, 3575. [Google Scholar] [CrossRef] [PubMed]
- Parveen, R.; Harihar, D.; Chatterji, B.P. Recent histone deacetylase inhibitors in cancer therapy. Cancer 2023, 129, 3372–3380. [Google Scholar] [CrossRef]
- Molehin, D.; Castro-Piedras, I.; Sharma, M.; Sennoune, S.R.; Arena, D.; Manna, P.R.; Pruitt, K. Aromatase Acetylation Patterns and Altered Activity in Response to Sirtuin Inhibition. Mol. Cancer Res. 2018, 16, 1530–1542. [Google Scholar] [CrossRef]
- Yi, X.; Wei, W.; Wang, S.Y.; Du, Z.Y.; Xu, Y.J.; Yu, X.D. Histone deacetylase inhibitor SAHA induces ERalpha degradation in breast cancer MCF-7 cells by CHIP-mediated ubiquitin pathway and inhibits survival signaling. Biochem. Pharmacol. 2008, 75, 1697–1705. [Google Scholar] [CrossRef]
- Wawruszak, A.; Luszczki, J.J.; Grabarska, A.; Gumbarewicz, E.; Dmoszynska-Graniczka, M.; Polberg, K.; Stepulak, A. Assessment of Interactions between Cisplatin and Two Histone Deacetylase Inhibitors in MCF7, T47D and MDA-MB-231 Human Breast Cancer Cell Lines—An Isobolographic Analysis. PLoS ONE 2015, 10, e0143013. [Google Scholar] [CrossRef]
- Xu, P.; Xiong, W.; Lin, Y.; Fan, L.; Pan, H.; Li, Y. Histone deacetylase 2 knockout suppresses immune escape of triple-negative breast cancer cells via downregulating PD-L1 expression. Cell Death Dis. 2021, 12, 779. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y. Trichostatin A and Tamoxifen inhibit breast cancer cell growth by miR-204 and ERalpha reducing AKT/mTOR pathway. Biochem. Biophys. Res. Commun. 2015, 467, 242–247. [Google Scholar] [CrossRef]
- Legare, S.; Basik, M. Minireview: The Link Between ERalpha Corepressors and Histone Deacetylases in Tamoxifen Resistance in Breast Cancer. Mol. Endocrinol. 2016, 30, 965–976. [Google Scholar] [CrossRef]
- Park, M.J.; Sohrabji, F. The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. J. Neuroinflammation 2016, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, U.; Kamran, M.; Manai, M.; Cristofanilli, M.; Ince, T.A. Cell-of-Origin Targeted Drug Repurposing for Triple-Negative and Inflammatory Breast Carcinoma with HDAC and HSP90 Inhibitors Combined with Niclosamide. Cancers 2023, 15, 332. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef]
- Santen, R.J.; Brodie, H.; Simpson, E.R.; Siiteri, P.K.; Brodie, A. History of aromatase: Saga of an important biological mediator and therapeutic target. Endocr. Rev. 2009, 30, 343–375. [Google Scholar] [CrossRef]
- Revilla, G.; Pons, M.P.; Baila-Rueda, L.; Garcia-Leon, A.; Santos, D.; Cenarro, A.; Magalhaes, M.; Blanco, R.M.; Moral, A.; Ignacio Perez, J.; et al. Cholesterol and 27-hydroxycholesterol promote thyroid carcinoma aggressiveness. Sci. Rep. 2019, 9, 10260. [Google Scholar] [CrossRef]
- Inasu, M.; Bendahl, P.O.; Ferno, M.; Malmstrom, P.; Borgquist, S.; Kimbung, S. High CYP27A1 expression is a biomarker of favorable prognosis in premenopausal patients with estrogen receptor positive primary breast cancer. npj Breast Cancer 2021, 7, 127. [Google Scholar] [CrossRef]
- Manna, P.R.; Reddy, A.P.; Pradeepkiran, J.A.; Kshirsagar, S.; Reddy, P.H. Regulation of retinoid mediated StAR transcription and steroidogenesis in hippocampal neuronal cells: Implications for StAR in protecting Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166596. [Google Scholar] [CrossRef]
- Vassilev, B.; Sihto, H.; Li, S.; Holtta-Vuori, M.; Ilola, J.; Lundin, J.; Isola, J.; Kellokumpu-Lehtinen, P.L.; Joensuu, H.; Ikonen, E. Elevated levels of StAR-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: Potential mechanisms contributing to progression of HER2-positive breast cancers. Am. J. Pathol. 2015, 185, 987–1000. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhang, Z.Y.; Du, W.D.; Ye, L.; Xu, S.; Zuo, X.B.; Zhou, F.S.; Chen, G.; Ma, X.L.; Schneider, M.E.; et al. Association analysis of ERBB2 amplicon genetic polymorphisms and STARD3 expression with risk of gastric cancer in the Chinese population. Gene 2014, 535, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Kim, R.N.; Song, K.; Jeon, S.; Jeong, H.M.; Kim, J.S.; Han, J.; Hong, S.; Oh, E.; Choi, J.S.; et al. Genes co-amplified with ERBB2 or MET as novel potential cancer-promoting genes in gastric cancer. Oncotarget 2017, 8, 92209–92226. [Google Scholar] [CrossRef]
- Fararjeh, A.F.S.; Al Khader, A.; Kaddumi, E.; Obeidat, M.; Al-Fawares, O. Differential Expression and Prognostic Significance of STARD3 Gene in Breast Carcinoma. Int. J. Mol. Cell Med. 2021, 10, 34–41. [Google Scholar] [PubMed]
- Fararjeh, A.; Kaddumi, E.; Al-Khader, A.; Aburayyan, W. The significance of StAR-related lipid transfer protein-3 expression in breast cancer. Pol. J. Pathol. 2022, 73, 215–222. [Google Scholar] [CrossRef]
- Lodi, M.; Voilquin, L.; Alpy, F.; Moliere, S.; Reix, N.; Mathelin, C.; Chenard, M.P.; Tomasetto, C.L. STARD3: A New Biomarker in HER2-Positive Breast Cancer. Cancers 2023, 15, 362. [Google Scholar] [CrossRef]
- Bulun, S.E.; Lin, Z.; Zhao, H.; Lu, M.; Amin, S.; Reierstad, S.; Chen, D. Regulation of aromatase expression in breast cancer tissue. Ann. N. Y. Acad. Sci. 2009, 1155, 121–131. [Google Scholar] [CrossRef]
- Vriend, J.; Reiter, R.J. Breast cancer cells: Modulation by melatonin and the ubiquitin-proteasome system—A review. Mol. Cell Endocrinol. 2015, 417, 1–9. [Google Scholar] [CrossRef]
- Park, W.C. Role of Ovarian Function Suppression in Premenopausal Women with Early Breast Cancer. J. Breast Cancer 2016, 19, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Nooshinfar, E.; Safaroghli-Azar, A.; Bashash, D.; Akbari, M.E. Melatonin, an inhibitory agent in breast cancer. Breast Cancer 2017, 24, 42–51. [Google Scholar] [CrossRef]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Chien, T.J. A review of the endocrine resistance in hormone-positive breast cancer. Am. J. Cancer Res. 2021, 11, 3813–3831. [Google Scholar]
- Perez Pinero, C.; Giulianelli, S.; Lamb, C.A.; Lanari, C. New Insights in the Interaction of FGF/FGFR and Steroid Receptor Signaling in Breast Cancer. Endocrinology 2022, 163, bqab265. [Google Scholar] [CrossRef]
- Clusan, L.; Ferriere, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Glickman, M.E.; Rao, S.R.; Schultz, M.R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol. 2014, 67, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Korthauer, K.; Kimes, P.K.; Duvallet, C.; Reyes, A.; Subramanian, A.; Teng, M.; Shukla, C.; Alm, E.J.; Hicks, S.C. A practical guide to methods controlling false discoveries in computational biology. Genome Biol. 2019, 20, 118. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Molehin, D.; Ahmed, A.U. Dysregulation of Aromatase in Breast, Endometrial, and Ovarian Cancers: An Overview of Therapeutic Strategies. Prog. Mol. Biol. Transl. Sci. 2016, 144, 487–537. [Google Scholar]
- Smith, K.T.; Workman, J.L. Histone deacetylase inhibitors: Anticancer compounds. Int. J. Biochem. Cell Biol. 2009, 41, 21–25. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDAC Members | HDAC Classes | Expression in Normal Breast Tissues | Expression in Cancerous Breast Tissues | Fold Changes (95% Confident Interval) | p-Values |
|---|---|---|---|---|---|
| HDAC1 | Class I | 0.697 | 0.969 | 1.390 (1.298, 1.488) | <0.001, *** |
| HDAC2 | 0.913 | 1.169 | 1.281 (1.173, 1.399) | <0.001, *** | |
| HDAC3 | 0.972 | 1.016 | 1.046 (0.996, 1.098) | 0.071 | |
| HDAC8 | 0.833 | 1.037 | 1.245 (1.176, 1.317) | <0.001, *** | |
| HDAC4 | Class IIa | 1.269 | 0.637 | 0.502 (0.457, 0.552) | <0.001, *** |
| HDAC5 | 1.260 | 0.883 | 0.701 (0.645, 0.762) | <0.001, *** | |
| HDAC7 | 1.170 | 0.992 | 0.848 (0.789, 0.911) | <0.001, *** | |
| HDAC9 | 1.115 | 0.681 | 0.611 (0.506, 0.738) | <0.001, *** | |
| HDAC6 | Class IIb | 0.918 | 0.858 | 0.935 (0.883, 0.989) | 0.022, * |
| HDAC10 | 0.476 | 0.683 | 1.434 (1.289, 1.596) | <0.001, *** | |
| SIRT1 | Class III | 1.899 | 1.294 | 0.682 (0.630, 0.737) | <0.001, *** |
| SIRT2 | 0.967 | 0.731 | 0.755 (0.706, 0.808) | <0.001, *** | |
| SIRT3 | 1.162 | 1.055 | 0.908 (0.838, 0.984) | 0.022, * | |
| SIRT4 | 1.329 | 0.951 | 0.716 (0.650, 0.788) | <0.001, *** | |
| SIRT5 | 1.004 | 0.940 | 0.937 (0.875, 1.003) | 0.063 | |
| SIRT6 | 0.389 | 0.717 | 1.846 (1.680, 2.028) | <0.001, *** | |
| SIRT7 | 0.484 | 0.912 | 1.885 (1.720, 2.066) | <0.001, *** | |
| HDAC11 | Class IV | 0.807 | 1.443 | 1.789 (1.570, 2.037) | <0.001, *** |
| Transcription Factors | Expression in Normal Breast Tissues | Expression in Cancerous Breast Tissues | Fold Changes (95% Confident Interval) | p-Values |
|---|---|---|---|---|
| CREB1 | 5.237 | 4.050 | 0.773 (0.728, 0.821) | <0.001, *** |
| CREM | 0.902 | 0.903 | 1.001 (0.935, 1.072) | 0.974 |
| SF1 | 1.830 | 1.691 | 0.924 (0.892, 0.958) | <0.001, *** |
| NR4A1 | 2.224 | 0.524 | 0.235 (0.192, 0.289) | <0.001, *** |
| CEBPB | 1.116 | 0.846 | 0.757 (0.661, 0.868) | <0.001, *** |
| GATA1 | 0.956 | 0.906 | 0.948 (0.837, 1.074) | 0.427 |
| SREBF1 | 3.391 | 6.327 | 1.866 (1.616, 2.155) | <0.001, *** |
| SREBF2 | 3.064 | 3.373 | 1.101 (1.004, 1.207) | 0.050, * |
| SP1 | 2.047 | 1.620 | 0.791 (0.746, 0.838) | <0.001, *** |
| FOS | 5.959 | 0.745 | 0.125 (0.099, 0.158) | <0.001, *** |
| JUN | 4.286 | 1.431 | 0.334 (0.290, 0.385) | <0.001, *** |
| NR0B1 | 0.834 | 0.305 | 0.366 (0.301, 0.445) | <0.001, *** |
| YY1 | 2.032 | 2.295 | 1.129 (1.073, 1.189) | <0.001, *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manna, P.R.; Yang, S.; Reddy, P.H. Epigenetic Dysregulation and Its Correlation with the Steroidogenic Machinery Impacting Breast Pathogenesis: Data Mining and Molecular Insights into Therapeutics. Int. J. Mol. Sci. 2023, 24, 16488. https://doi.org/10.3390/ijms242216488
Manna PR, Yang S, Reddy PH. Epigenetic Dysregulation and Its Correlation with the Steroidogenic Machinery Impacting Breast Pathogenesis: Data Mining and Molecular Insights into Therapeutics. International Journal of Molecular Sciences. 2023; 24(22):16488. https://doi.org/10.3390/ijms242216488
Chicago/Turabian StyleManna, Pulak R., Shengping Yang, and P. Hemachandra Reddy. 2023. "Epigenetic Dysregulation and Its Correlation with the Steroidogenic Machinery Impacting Breast Pathogenesis: Data Mining and Molecular Insights into Therapeutics" International Journal of Molecular Sciences 24, no. 22: 16488. https://doi.org/10.3390/ijms242216488
APA StyleManna, P. R., Yang, S., & Reddy, P. H. (2023). Epigenetic Dysregulation and Its Correlation with the Steroidogenic Machinery Impacting Breast Pathogenesis: Data Mining and Molecular Insights into Therapeutics. International Journal of Molecular Sciences, 24(22), 16488. https://doi.org/10.3390/ijms242216488