Abstract
Alzheimer’s disease (AD) is a complex multifactorial disorder that poses a substantial burden on patients, caregivers, and society. Considering the increased aging population and life expectancy, the incidence of AD will continue to rise in the following decades. However, the molecular pathogenesis of AD remains controversial, superior blood-based biomarker candidates for early diagnosis are still lacking, and effective therapeutics to halt or slow disease progression are urgently needed. As powerful genetic regulators, microRNAs (miRNAs) are receiving increasing attention due to their implications in the initiation, development, and theranostics of various diseases, including AD. In this review, we summarize miRNAs that directly target microtubule-associated protein tau (MAPT), amyloid precursor protein (APP), and β-site APP-cleaving enzyme 1 (BACE1) transcripts and regulate the alternative splicing of tau and APP. We also discuss related kinases, such as glycogen synthase kinase (GSK)-3β, cyclin-dependent kinase 5 (CDK5), and death-associated protein kinase 1 (DAPK1), as well as apolipoprotein E, that are directly targeted by miRNAs to control tau phosphorylation and amyloidogenic APP processing leading to Aβ pathologies. Moreover, there is evidence of miRNA-mediated modulation of inflammation. Furthermore, circulating miRNAs in the serum or plasma of AD patients as noninvasive biomarkers with diagnostic potential are reviewed. In addition, miRNA-based therapeutics optimized with nanocarriers or exosomes as potential options for AD treatment are discussed.
Keywords:
Alzheimer’s disease; microRNA; tau; amyloid precursor protein; beta-amyloid; APOE; neuroinflammation; diagnosis; therapy 1. Introduction
Characterized by a time-dependent decline in the function of organisms, aging is a weakening of the body’s defense and repair functions [1,2]. Owing to longer life expectancies in our modern society, a rapidly increasing aging human population has resulted in an elevated global burden of late-life diseases [3]. Notably, aging is associated with increased susceptibility to infections, tumors, including malignant tumors, and neurodegenerative diseases such as Alzheimer’s disease (AD) [4,5]. For individuals above 65 years of age, the odds of developing AD double every five years, and one out of three people aged 85 or older develop AD [6]. AD is the main cause of dementia, and its prevalence is projected to triple by 2050 according to a prevalence estimate of dementia from a study reporting it, reaching approximately 50 million worldwide in 2018 [7]. Thus, AD has emerged as a serious global public health threat as a result of an ever-increasing aging population.
AD is a subtle yet devastating age-related progressive neurodegenerative disorder defined by numerous cognitive or behavioral symptoms [7,8]. Currently, there are four hypotheses for the pathogenesis of sporadic or late-onset AD, including the amyloid cascade, and the inflammatory, vascular, and infectious factors [9]. The cardinal neuropathological hallmarks of AD are aggregation of neurofibrillary tangles (NFTs) due to hyperphosphorylated tau and the accumulation of beta-amyloid (Aβ) processed from amyloid precursor protein (APP) [10,11]. These two features have been recognized for more than one hundred years and they remain indispensable for AD diagnosis today [12]. Unfortunately, the pathogenesis of AD is still unknown. Moreover, early and accurate diagnosis of AD remains an enormous challenge. Furthermore, current disease-modifying therapeutics have been largely unsuccessful. Therefore, thoroughly understanding the molecular changes underlying the pathogenesis of AD may reveal potential targets for diagnosis and therapeutics.
The discovery of microRNAs (miRNAs) has revolutionized our comprehension of gene regulation as they have important target recognition and regulatory functions [13,14]. In human cells, at least 2300 mature miRNAs have been experimentally validated [15]. These miRNAs have been estimated to modulate approximately half of all protein-coding genes via posttranscriptional regulation [16]. The expression profiling of miRNAs has been found to be time- and tissue-dependent [17,18]. Dysregulation of miRNAs is regarded as a reflection of the state of cells in different tissues and a causal factor in various disorders [19,20], suggesting potential implications in the pathogenesis and theranostics of human diseases.
Up to 70% of all miRNAs are expressed in the human nervous system [21]. MiRNAs also play critical regulatory roles in the accumulation of toxic proteins that affect neuronal survival [22]. Importantly, altered expression of several miRNAs has been found in the early stages of AD, around two decades before the onset of clinical symptoms [23]. These findings suggest that miRNAs possess diagnostic and therapeutic value. In this review, we summarize the regulatory roles of miRNAs in tau pathologies and amyloidogenic APP processing resulting in Aβ pathologies in AD. Moreover, we explored the capacity of miRNAs as noninvasive biomarkers for AD diagnosis. Furthermore, miRNA-based therapeutics have been evaluated for potential AD treatment.
2. MiRNA-Mediated Regulation of Tau Pathologies in AD
2.1. Tau Pathologies in AD
In 1975, a protein named tau was found to be an essential regulator of microtubule assembly [24]. In humans, tau is encoded by microtubule-associated protein tau (MAPT) located on chromosome 17q21, generating six molecular isoforms due to alternative splicing [25]. Tau protein is expressed predominantly in neurons and is vital for the stabilization of the neuronal cytoskeleton [26]. Notably, tau abnormalities have been proven to result in various neurological diseases collectively known as tauopathies, including AD [27].
Among a series of posttranslational modifications such as methylation, acetylation, phosphorylation, and N-glycosylation, phosphorylation strongly correlates with pathological tau conditions [28]. Under normal physiological conditions, the phosphorylation of tau is developmentally modulated and promotes microtubule assembly [29,30]. However, in certain pathological situations, a substantial rise in the phosphorylation of tau has been observed, resulting in aberrant hyperphosphorylated tau [30]. Aberrant tau phosphorylation is regulated by protein kinases and phosphatases [26].
In AD, the abnormal tau phosphorylation not only breaks down neuronal microtubules but also prevents normal tau functioning by facilitating interactions with normal tau [31]. Notably, it is hyperphosphorylated tau instead of normal tau that form a component of NFTs called paired helical filaments (PHFs), thereby contributing to the generation of NFTs [32,33,34]. The resulting aggregation of NFTs correlates with the severity of the cognitive decline [35,36]. Interestingly, tau pathologies have recently been proposed to be essential initiating factors in the sporadic form of AD [37].
2.2. MiRNAs Regulate MAPT Expression
2.2.1. MiRNAs Directly Modulate MAPT Transcript Levels
Recently, through capture technology, miR-92a-3p, miR-320a, and miR-320b were found to directly bind to MAPT mRNA and inhibit tau protein expression in a human neuroblastoma cell line [38]. Overexpression of these miRNAs leads to a significant decrease in tau levels, while miRNA inhibitors dramatically upregulate the protein expression of tau [38]. Interestingly, the authors further identified that plasma miR-92a-3p levels were reduced in AD individuals when compared with healthy participants, which is consistent with several previous publications [39,40] but contradictory with others [41]. Downregulation of miR-320a has been shown in the serum [39] and cerebrospinal fluid (CSF) [42] of patients with AD versus controls, although contrary data exist [43]. Moreover, miR-132-3p, which is frequently downregulated in AD and other tauopathies, directly targets MAPT mRNA to inhibit its expression, and its deletion in AD model mice leads to tau aggregation [44]. In addition, miR-34c-5p [45], miR-186 [46], and miR-27a [47] can bind to the 3′UTR of MAPT in human cell lines or a rat model; however, these interactions have not been observed in AD. These miRNAs directly target the mRNA of tau, participating in the development of tau pathologies in AD (Figure 1).
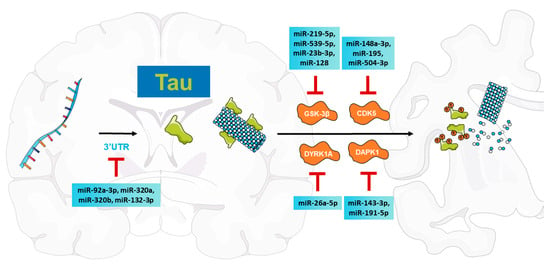
Figure 1.
A summary of miRNAs directly targeting MAPT transcripts and related kinases, including GSK-3β, CDK5, DYRK1A, and DAPK1.
2.2.2. MiRNAs Regulate Tau Alternative Splicing
In adult human brains, alternative splicing of exons 2, 3, and 10 of MAPT pre-mRNA generates six tau isoforms [25]. The exclusion or inclusion of exon 10, encoding the second microtubule-binding repeat of tau, results in tau isoforms containing either three (3R) or four (4R) microtubule-binding domain repeats [48]. Approximately equal 3R-tau and 4R-tau are physiologically expressed in adults, but tau exon 10 splicing becomes dysregulated in several tauopathies and alters the ratios between 4R-tau and 3R-tau isoforms, contributing to neurodegeneration and dementia [49]. Several miRNAs, such as miR-124, miR-9, miR-132, and miR-137, have been shown to be involved in the aberrant splicing of tau exon 10 and the modulation of 4R:3R-tau ratios in neuronal cells [50]. Specifically downregulated in the brains of patients with sporadic progressive supranuclear palsy, miR-132 directly targets polypyrimidine tract-binding protein, a neuronal splicing factor that is significantly upregulated in PSP brain regions [50]. Whether and how these miRNAs affect tau splicing in AD remains undetermined. Given that miR-132 [51,52], miR-124 [53,54], miR-9 [55,56], and miR-137 [57,58] are implicated in AD, it would be interesting to further investigate the potential roles of miRNAs in the alternative splicing of tau in AD.
2.3. MiRNAs Regulate Kinases That Phosphorylate Tau
The phosphorylation of tau is developmentally modulated, as phosphorylated tau levels are high in fetal brains and decline with age during development [30]. However, in AD brains, tau is aberrantly hyperphosphorylated [25]. Direct pathological events involved in tau hyperphosphorylation include the increase or abnormal activation of kinases [35], such as glycogen synthase kinase (GSK)-3β, cyclin-dependent kinase 5 (CDK5), dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A), and death-associated protein kinase 1 (DAPK1), which have been found to be directly regulated by miRNAs in tau pathologies (Figure 1).
2.3.1. MiRNAs Modulate GSK-3β to Inhibit Tau Phosphorylation
GSK-3β is a multifunctional serine/threonine kinase enriched in the brain and shows elevated levels with aging [59]. Notably, increased levels of active GSK-3β are observed in AD brains, which are considered an early event before NFT formation [60]. Several miRNAs have been demonstrated to modulate GSK-3β and participate in tau pathologies in AD. MiR-219-5p is downregulated in AD brains and inhibits tau phosphorylation at Ser198, Ser199, Ser201, and Ser422 via directly targeting GSK-3β in a human neuroblastoma cell line [61]. Moreover, in the CSF of AD patients, miR-539-5p levels are dramatically decreased compared with those in the CSF of healthy controls, showing a negative correlation with GSK-3β expression [62]. Upregulation of miR-539-5p through injection in AD model mice downregulates tau phosphorylation at Ser396 and Ser404 and improves memory ability [62]. Furthermore, in the plasma of AD patients, miR-23b-3p is remarkably downregulated compared to that in the plasma of healthy age-matched individuals [63]. MiR-23b-3p exhibits neuroprotection by inhibiting tau phosphorylation, alleviating AD-like symptoms in AD model mice, and interrupting GSK-3β-dependent tau phosphorylation at Ser396 and Ser404 in vivo [63]. In addition, the brain-specific miRNA miR-128 inhibits tau phosphorylation at Ser396, Ser404, and Thr217 by directly suppressing GSK-3β, and an increase in miR-128 levels in the hippocampus improves spatial learning and memory in 5×FAD mice [64].
2.3.2. MiRNAs Are Involved in CDK5 Regulation to Reduce Tau-Related Pathologies
CDK5 is an essential serine/threonine protein kinase and a unique member of the cyclin-dependent kinase family as the activity of CDK5 is restrictedly regulated by the neuronal-specific and membrane-localized activators p35 and p39 (or their respective truncated forms p25 and p29) instead of binding to cyclins in the central nervous system (CNS) [65,66]. In AD brain tissues, an increase in CDK5 immunoreactivity has been found in neurons bearing early-stage NFTs [67]. Increased activity of CDK5 contributes to the accumulation of aggregated tau and promotes neurofibrillary pathology development [68]. In the serum of AD patients, miR-148a-3p expression levels are lower [69]. MiR-148a-3p decreases tau hyperphosphorylation at Ser202/Thr205, Ser199, Ser396, and Ser404 by targeting cyclin-dependent kinase 5 regulatory subunit 1 (CDK5R1) mRNA, which encodes the p35 protein, and intracerebroventricular injections of miR-148a-3p ameliorate cognitive deficits in AD model mice [69]. Moreover, miR-195 overexpression prevents CDK5/p35 activity and tau hyperphosphorylation at Thr231, Ser262, and Ser422 in the hippocampus of model rats by targeting CDK5R1 [70]. Recently, our group found that miR-504-3p binds to the 3′UTR of p39 and downregulates p39 protein expression by modulating its mRNA level [71]. We further demonstrated that miR-504-3p attenuates tau hyperphosphorylation at CDK5-dependent phosphorylation sites, including Thr213 and Ser396, related to AD by targeting p39 [71].
2.3.3. MiRNAs Regulate DYRK1A to Suppress Tau Phosphorylation
DYRK1A is a serine/threonine kinase encoded on human chromosome 21 that plays an important role in early-onset neurodegeneration [72]. In the hippocampus of AD patients, DYRK1A mRNA levels are dramatically increased compared with those in the hippocampus of healthy controls [73]. DYRK1A is involved in the hyperphosphorylation of tau and its extra copy may lead to the early onset of AD [74]. MiR-26a-5p expression is downregulated in the brain tissues of AD model mice and negatively modulates DYRK1A by targeting its 3′UTR [75]. Overexpression of miR-26a-5p inhibits tau phosphorylation at Thr212, Ser202, and Ser404, and alleviates AD-like symptoms in mice [75].
2.3.4. MiRNAs Target DAPK1 to Attenuate Tau Pathologies
DAPK1 is a calcium/calmodulin-regulated serine/threonine kinase that plays essential roles in various types of cancers and neurodegenerative disorders including AD [76,77]. Genetic variations in DAPK1 have been found to show a significant association with late-onset AD [78,79,80,81]. Notably, DAPK1 is highly overexpressed in the hippocampal tissues of AD individuals compared with age-matched controls [82,83,84]. Mechanically, DAPK1 can phosphorylate and inhibit the activity of Pin1, a peptidyl-prolyl cis/trans isomerase that converts cis to trans p-tau to prevent a group of tau pathologies, including AD [8,85,86,87]. Recently, our group found that miR-143-3p inhibits aberrant tau phosphorylation at Thr231, Ser262, and Ser396 and promotes microtubule assembly via directly targeting DAPK1 in AD [88]. Moreover, miR-191-5p was also observed to suppress tau phosphorylation at Thr231, Ser262, and Ser396 and promote neurite outgrowth [89].
3. MiRNA-Mediated Modulation of Aβ Pathologies in AD
3.1. Amyloidogenic APP Processing in AD
Since the 1990s, the amyloid cascade hypothesis has dominated research and clinical trials of AD [90,91]. Accumulation of Aβ in the brain is hypothesized to drive AD pathogenesis and cause neurodegeneration [92]. Aβ is an aggregation-prone peptide consisting of 36 to 43 amino acids originating from APP proteolysis via the amyloidogenic pathway [6]. The human APP gene is mapped to chromosome 21 and encodes a type I transmembrane protein after the alternative splicing of 18 exons, primarily generating three isoforms, APP695, APP751, and APP770 [93,94,95]. Genomic duplications in the APP locus have been observed to cause early-onset AD [96,97].
APP is metabolized via two distinct proteolytic processes, the nonamyloidogenic pathway and the amyloidogenic pathway [98]. In the nonamyloidogenic processing pathway, APP is first cleaved by α-secretase within the Aβ sequence and further cleaved by γ-secretase, thus precluding intact Aβ generation [99]. However, in the amyloidogenic processing pathway, APP is cleaved by β-secretase (β-site APP-cleaving enzyme 1, BACE1) and subsequently cleaved by γ-secretase, inducing Aβ pathologies [99]. An upregulation in APP expression has also been found in AD brains [100,101]. The aberrant deposition of Aβ is associated with neurotoxicity and has emerged as a neuropathological hallmark of AD [92].
As a single transmembrane-spanning protein, APP possesses a variety of phosphorylation sites in both the short cytoplasmic domain and the long extracellular domain [102]. Within the cytoplasmic domain of APP, there are eight potential phosphorylation sites, seven of which are observed to be phosphorylated in AD brains [102,103]. Phosphorylation of APP is capable of modulating its processing and transport, exerting key regulatory roles in Aβ generation [104,105]. Thus, abnormal phosphorylation of APP and related kinases involved in amyloidogenic APP processing are critical for the development of Aβ pathologies.
3.2. MiRNAs Regulate Amyloidogenic APP Processing
3.2.1. MiRNAs Directly Modulate APP mRNA Expression
In attempts to address whether APP levels can be posttranscriptionally regulated, miR-106a and miR-520c were initially found to target the 3′UTR of APP mRNA and repress its protein expression in human cells [106]. MiR-20a, miR-17-5p, and miR-106b, as members of the same miRNA family, directly regulate APP expression in both human and mouse neuronal cells, while miR-106b is significantly downregulated in sporadic AD patients [107]. MiR-153 negatively regulates APP expression in human neuroblastoma cells, mouse models [108], and primary human fetal brain cultures [109]. MiR-153 levels are dramatically decreased in a subset of advanced AD postmortem brain specimens (Braak stage III-VI) compared with early-stage specimens (control, Braak stage I/II) [109]. In addition to miR-20a, miR-17, and miR-153, miRNAs miR-147, miR-655, miR-323-3p, and miR-644 can also regulate APP expression [110]. MiR-101 modulates APP levels in rat hippocampal neurons [111] and multiple human cell types [112]. MiR-16 directly inhibits APP expression in model mice [113,114] and rat hippocampal neurons [115]. MiR-200b and miR-429 downregulate APP mRNA and protein expression in primary mouse hippocampal neurons and human neuroblastoma cells [116]. MiR-200b levels are lower in the serum of patients with Alzheimer-type dementia than in healthy controls, and Aβ42 can downregulate miR-200b expression, leading to a vicious cycle resulting in the continuous accumulation of Aβ42 [116]. MiR-384 suppresses APP mRNA and protein levels, and APP levels are lower in the CSF and serum of patients with Alzheimer-type dementia [117]. MiR-193b inhibits APP mRNA and protein levels, and exosomal miR-193b levels are reduced in the CSF and blood of AD patients [118]. MiR-144-3p is a negative regulator of APP and inhibits its protein expression in human cells [119]. MiR-15b-5p has been shown to decrease APP mRNA and protein levels in a human AD cell model [120]. MiR-455-3p can decrease APP levels in a mouse neuroblastoma cell line [121]. MiR-31, previously found to be downregulated in AD patients, can reduce APP mRNA levels in human cells and an AD mouse model [122]. MiR-539-5p has also been observed to downregulate APP expression in an AD mouse model [62]. MiR-202 downregulates APP expression, and miR-202 levels are dramatically reduced in the serum of AD patients [123]. MiR-298 is a repressor of APP and downregulates APP expression in a primary human cell culture model [124]. MiR-130a-3p can downregulate the expression of APP in the primary hippocampal neurons of AD model mice [125]. MiR-185-5p can decrease APP transcript levels, and serum exosomal miR-185-5p levels are markedly reduced in the AD group versus the corresponding control groups [126]. All the above-mentioned miRNAs directly target the 3′UTR of APP mRNA to regulate its levels, playing vital roles in the development of Aβ pathologies (Figure 2). Although miR-373-3p can also target APP mRNA and inhibit its protein expression [127], the role of this interaction in AD pathogenesis has not been confirmed. Interestingly, APP 3’UTR polymorphisms located in miRNA target sites have been observed to influence the risk of AD occurrence [110,128]. In addition, miR-346 can target the 5′UTR of APP mRNA to promote APP translation and Aβ generation [129].
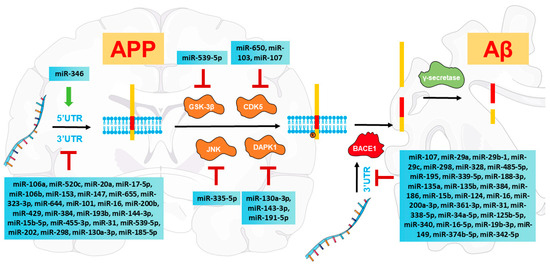
Figure 2.
A summary of miRNAs directly targeting APP transcripts, BACE1 transcripts, and related kinases, including GSK-3β, CDK5, JNK, and DAPK1.
3.2.2. MiRNAs Regulate the Alternative Splicing of APP
Apart from the direct regulation of APP by targeting the 5′UTR and 3′UTR in AD, miRNAs also participate in the alternative splicing of APP mRNA. APP695, APP751, and APP770 are the three primary isoforms generated after alternative splicing, and the APP695 isoform is predominantly expressed in neurons [95]. In AD brains, APP695 is generally downregulated, whereas APP751 and APP770 are upregulated [130]. Notably, in postmitotic neurons from the cortex of Dicer conditional knockout mice, the lack of miRNAs results in APP exon 7 and 8 inclusion [131]. MiR-124 is involved in this abnormal APP splicing, and miR-124 levels are decreased in AD brains [131].
3.2.3. MiRNAs Directly Regulate BACE1 Transcripts
APP is first cleaved by BACE1, which is the enzyme that initiates Aβ generation, and BACE1 cleavage of APP is the rate-limiting step for Aβ formation in the brain [132,133]. Initially, BACE1 was cloned and characterized in 1999, and its expression levels and activity have been found to be increased in the brains and body fluids of AD individuals [133]. To investigate the potential regulators contributing to BACE1 posttranscriptional regulation, miR-107 was found to be downregulated early in AD by miRNA expression microarrays, and miR-107 targeting of the 3′UTR of BACE1 mRNA was biochemically validated [134]. Moreover, miR-29a and miR-29b-1 can downregulate BACE1 expression in human neuroblastoma cells, and these miRNAs were found to be downregulated in a cohort of AD patients with aberrantly high BACE1 levels [55]. Belonging to the miR-29 family, miR-29c can also directly inhibit the expression of BACE1 in human cells and mouse models, and its downregulation correlates with the increase in BACE1 levels in sporadic AD patients [135,136,137]. MiR-298 and miR-328 negatively regulate BACE1 expression in cultured neuronal cells [124,138]. MiR-485-5p decreases in BACE1 protein levels in human cells [139]. MiR-195 downregulates BACE1 expression by suppressing its translation in human and mouse cells [140]. MiR-339-5p decreases BACE1 expression levels in human primary brain cultures, and miR-339-5p is downregulated in AD patient brain specimens [141]. MiR-188-3p can downregulate BACE1 expression, and the levels of miR-188-3p are decreased in AD brains [142]. MiR-135a inhibits BACE1 expression in human neuroblastoma cells and primary mouse hippocampal neurons and its levels are reduced in the serum of AD patients [116]. MiR-135b also decreases the protein expression levels of BACE1, and its levels are reduced in the blood of AD individuals [143]. MiR-384 can downregulate BACE1 expression in human neuroblastoma cells as well [117]. Moreover, miR-186 can downregulate BACE1 expression in mouse neuronal cells [144]. MiR-15b modulates BACE1 expression in human neuroblastoma cells, and its levels are decreased in sporadic AD brain tissues [145,146]. MiR-124 levels are downregulated in sporadic AD brain tissues, and miR-124 can inhibit BACE1 expression in human neuroblastoma cells [147] and AD mouse models [148]. MiR-16 downregulates BACE1 expression in an AD cell model, and its levels are decreased in AD brain tissues [149]. MiR-200a-3p inhibits BACE1 protein expression, and its levels are reduced in the blood plasma of AD individuals [150]. MiR-361-3p regulates the expression of BACE1, and its levels are decreased in AD brains [151]. MiR-31 downregulates BACE1 expression in an AD animal model [122]. MiR-338-5p can downregulate BACE1 expression in the hippocampus of AD model mice and is downregulated in the hippocampus of AD patients [152]. MiR-34a-5p and miR-125b-5p reduce BACE1 expression levels in primary mouse cortical neurons, and the levels of these miRNAs are decreased in the serum samples of AD patients [153]. MiR-340 can downregulate BACE1 expression in human neuroblastoma cells [154]. MiR-16-5p and miR-19b-3p can reduce BACE1 protein levels in human neuroblastoma cells [155]. MiR-149 inhibits BACE1 expression in an AD cell model, and its levels are reduced in the serum of AD individuals [156]. MiR-374b-5p has also been verified to interact with BACE1 [157]. MiR-342-5p downregulates BACE1 expression in mouse cells, and its levels are decreased in the circulating small extracellular vesicles from patients with AD [158]. All of these miRNAs directly bind to the 3′UTR of BACE1 mRNA, suggesting that they potentially play roles in modulating Aβ pathologies (Figure 2).
3.3. MiRNAs Regulate Kinases That Phosphorylate APP at Thr668
APP is a phosphoprotein with several phosphorylation sites that can be modulated by numerous kinases [104]. Phosphorylation of APP plays regulatory roles in APP processing and APP transport and is thus critical for the generation of Aβ [105]. Among various potential phosphorylation sites, Thr668 in the cytoplasmic region of APP has attracted considerable attention due to its profound impact on APP metabolism [102,103]. APP phosphorylation at Thr668 is increased in the brain samples of AD patients and promotes amyloidogenic APP processing to produce Aβ [103]. The abnormal phosphorylation of APP at Thr668 has been found to be regulated by kinases, such as GSK-3β, CDK5, c-Jun NH2-terminal kinase (JNK), and DAPK1, which have been reported to be directly modulated by miRNAs (Figure 2).
3.3.1. MiRNAs Are Involved in GSK-3β Regulation
GSK-3β is a kinase that is responsible for the Thr668 phosphorylation of APP [159,160]. The activity of GSK-3β is elevated in AD patients [60]. MiR-539-5p has been confirmed to directly target GSK-3β, decrease GSK-3β expression levels, and inhibit Aβ accumulation in APP/PS1 mice [62]. The expression levels of miR-539-5p and GSK-3β are negatively correlated in the CSF and brain tissues of AD patients [62].
3.3.2. MiRNAs Modulate CDK5 to Reduce Aβ Pathologies
CDK5 is also a well-known protein kinase that phosphorylates APP at Thr668 and plays a vital role in APP proteolytic cleavage [161,162]. Hyperactivity of CDK5 has been observed in human AD brains [67]. MiR-650 can target CDK5, and overexpression of miR-650 reduces CDK5 activity and attenuates AD pathologies, including plaque formation and Aβ production, in APP/PSEN1 mice [163]. MiR-103 and miR-107, both members of the miR-15/107 family, can influence CDK5 expression and activity and reduce APP phosphorylation at Thr668 [164]. A significant inverse correlation of expression also exists between these miRNAs and CDK5R1 in AD hippocampal tissues [164].
3.3.3. MiRNAs Target JNK to Inhibit Aβ Production
JNK also plays a fundamental role in the phosphorylation of APP at Thr668 [165,166]. The phosphorylation of JNK is elevated in the postmortem brain tissues of AD individuals [167]. MiR-335-5p can directly bind to the 3′UTR of JNK3 mRNA, decrease its protein levels, and alleviate Aβ accumulation in a human AD neuronal cell model [168]. The decrease in miR-335-5p levels is negatively correlated with the increased levels of JNK3 in AD brain tissues [168].
3.3.4. MiRNAs Regulate DAPK1 to Alleviate Amyloidogenic APP Processing
DAPK1, without its kinase-deficient mutant, has been shown to promote APP phosphorylation at Thr668, initiate APP amyloidogenic processing, and increase Aβ secretion [83]. MiR-130a-3p can target the 3′UTR of DAPK1 and improve the cognitive function of APP/PS1 mice [169]. Recently, our studies indicated that miR-143-3p decreases APP phosphorylation at Thr668 and reduces Aβ40 and Aβ42 production by directly targeting DAPK1 [88]. Moreover, miR-191-5p was also found to inhibit DAPK1, decrease APP phosphorylation levels, and reduce Aβ secretion [89].
3.4. MiRNAs Regulate Apolipoprotein E (APOE)-Mediated Aβ Pathologies
Polymorphism in the APOE gene is the strongest genetic risk factor for late-onset AD [170]. Mounting evidence suggests that genetic variation in the APOE gene increases the risk of AD by driving Aβ pathologies [171]. Intriguingly, by evaluating a sample of female patients aged 55 years or older carrying the ε4 allele of APOE, a median 3-fold decrease in miR-9-5p levels was observed when compared with controls, which may be implicated in accelerating amyloidogenic processing [172]. In the plasma of AD patients, miR-1908 is upregulated and is negatively associated with APOE levels [173]. MiR-1908 can directly target the 3′-UTR of APOE mRNA, modulating APOE-mediated Aβ clearance [173].
4. MiRNA-Mediated Modulation of Inflammation in AD
Among patients with AD, upregulated levels of inflammatory markers and AD risk genes in association with innate immune functions have been unveiled, suggesting a prominent role of inflammation in the pathogenesis of AD [174]. The inflammatory process includes the activation of microglia and the production of pro-inflammatory cytokines [175]. Notably, microglial activation has been proven to be a vital factor linking the deleterious effects of Aβ to tau spread [176]. The concomitant presence of Aβ, tau, and microglial activation abnormalities emerges as the strongest predictor of cognitive impairment [176]. Several miRNAs have been found to be strongly associated with inflammation in AD.
4.1. MiRNAs Induce Pro-Inflammatory Responses
MiR-155 acts as a central pro-inflammatory mediator of neuroinflammation of the CNS [177]. Expressions of a number of pro-inflammatory cytokines, including interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and interferon regulatory factor 3 (IRF3), can be upregulated by miR-155 [178]. MiR-155 is involved in the innate immune response pathways and proamyloidogenic pathways leading to Aβ [179]. In microglia, pro-inflammatory miR-155 overexpression can downregulate fibrillar Aβ1-42 catabolism [180].
4.2. MiRNAs Promote Anti-Inflammatory Responses
MiR-146 is another key regulator of innate immune responses by targeting several inflammation-related mRNAs [181]. As a negative regulator, miR-146 can bind to the mRNA 3′UTRs of the TNF receptor-associated factor 6 and IL-1 receptor-associated kinase 1, controlling the Toll-like receptor and cytokine signaling [182]. Pro-inflammatory cytokines such as IL-6 and TNF-α are also demonstrated to be negatively modulated by miR-146 [183]. Intriguingly, the interactions between miR-155 and miR-146 may result in microglia activation [183].
5. Diagnostic Potential of miRNAs in AD
The occurrence of AD doubles every five years for individuals older than 65 years of age [6]. Currently, few effective disease-modifying drugs can prevent or reverse this devastating disorder [184]. One explanation for numerous failed clinical trials might be that the neuropathological alterations of AD start 10–20 years before detectable clinical onset [184,185]. Hence, it is crucial to identify potential biomarker candidates for early diagnosis and disease progression monitoring. A biological rather than a syndromal definition of AD has been defined by the intravital diagnosis of AD, which may better characterize and understand the disease [186]. Currently, CSF and positron emission tomography (PET) biomarkers, including Aβ, tau, and neurofilament light chain (NfL), are employed for the prediction of dementia onset and progression from mild cognitive impairment (MCI) to AD [187,188]. However, these costly, inconvenient, and invasive strategies limit their use as first-line diagnostic tools.
Notably, blood-based biomarkers have attracted considerable attention since they can offer noninvasive, easily accessible, cost-effective, fast, real-time, and repeatable approaches to provide valuable early diagnostic and prognostic insights into multifarious diseases, including cancers and AD [189]. In AD, blood-based biomarkers hold great potential for both primary care screening and personalized precision medicine [189]. Interestingly, miRNAs in blood are stable, providing invaluable diagnostic, prognostic, and predictive information [190,191]. Circulating miRNAs associated with extracellular vesicles such as exosomes present remarkable stability even at room temperature [192]. Importantly, ten miRNAs deregulated both in the bloodstream and in the brain of Braak stage III AD patients have been reported to have diagnostic value nearly two decades prior to clinical symptom onset, offering attractive early miRNA biomarkers for AD [23].
To determine the potential of miRNAs as early noninvasive markers for the diagnosis of AD, various screening strategies have been carried out. After assessing 654 human miRNAs, a unique circulating 7-miRNA signature was validated to be downregulated in the plasma of AD individuals to distinguish AD patients from normal controls [193]. Employing next-generation sequencing to miRNAs from blood samples, a 12-miRNA signature was observed to separate AD patients from healthy controls [194]. Six previously reported miRNAs dysregulated in AD were further examined, and serum miR-125b alone was able to distinguish AD patients from healthy controls [195]. Moreover, via genome-wide serum miRNA expression analysis, a serum miRNA panel consisting of six miRNAs or miR-342-3p alone was shown to be able to distinguish AD patients from healthy controls [196]. By determining the expression of 84 miRNAs, the levels of miR-125b, miR-23a, and miR-26b in serum were validated to be decreased in AD patients, and serum miR-125 levels can distinguish AD patients from healthy controls [197]. Based on Solexa sequencing analysis, the levels of miR-31, miR-93, miR-143, and miR-146a were confirmed to be reduced in the serum of AD patients compared with healthy controls, and this panel can be used to discriminate AD patients from healthy controls [198]. By using omiRas, a total of 27 miRNAs with differential expression between AD patients and healthy controls were identified, and this panel can separate several AD subgroups from healthy controls [199]. Using next-generation deep sequencing, 16 dysregulated miRNAs in exosomes isolated from serum were selected for predicting AD [200]. Among 20 plasma exosomal miRNAs that presented differential expression in AD resulting from initial screening after deep sequencing, a panel of seven miRNAs was reported to predict AD status [201]. In a parallel whole-blood-based study on the seven miRNAs, a decrease in the levels of miR-9-5p, miR-106a-5p, miR-106b-5p, and miR-107 was found to be correlated with a higher risk of AD [202]. By high-throughput next-generation sequencing, serum miR-501-3p levels were observed to be decreased in AD patients and correlated with Mini-Mental State Examination (MMSE) scores [203]. Among 179 miRNAs assessed in the plasma, six miRNAs were selected to differentiate AD from healthy controls [43]. Among 10 mature miRNAs dysregulated in the plasma of AD patients, miR-34a-5p and miR-545-3p were observed to present diagnostic accuracy distinguishing AD patients from control subjects [204]. Using microarray analysis, miR-455-3p was validated to be increased in the serum of AD patients and was able to distinguish individuals with or without AD [205]. Through next-generation sequencing, a 9-miRNA signature in serum was utilized for detecting AD [206]. By examining miRNAs related to synaptic proteins in the plasma of AD subjects, the levels of miR-92a-3p, miR-181c-5p, and miR-210-3p were found to be increased and were able to distinguish AD patients from healthy controls [41]. Based on miRNA profiling in patient plasma, miR-206 levels were found to be elevated in AD patients and predict cognitive decline [207]. In a machine learning approach, a serum 12-miRNA signature was constructed to discriminate AD patients from healthy controls [208]. Among 853 miRNAs in the blood samples, a panel of six dysregulated exosomal miRNAs was selected to detect preclinical AD [209]. Using capture technology, plasma miR-92a-3p and miR-320a among miRNAs directly binding to MAPT mRNA were validated to discriminate AD patients from healthy controls [38].
The detailed miRNAs described in the above-mentioned screening processes are listed in Table 1. In addition, miR-137, miR-181c, miR-9, miR-29a, and miR-29b in the serum [210], miR-34a and miR-146a in the plasma [211], miR-34c in the plasma [212], miR-210 in the serum [213], miR-223 in the serum [214], miR-29c-3p and miR-19b-3p in the serum [215], miR-135a, miR-193b, and miR-384 in the serum [216], miR-133b in the serum [217], miR-103 and miR-107 in the plasma [218], miR-28-3p in the serum [219], miR-483-5p in the plasma [220], miR-331-3p in the serum [221], miR-128 in the serum [222], and miR-106b in the serum [223] have also been observed to be dysregulated in AD patients compared with healthy controls and serve as potential biomarkers to predict AD. In addition, the miR-132 family (miR-128, miR-132, and miR-874) normalized per miR-491-5p and the miR-134 family (miR-134, miR-323-3p, and miR-382) normalized per miR-370 in the plasma [224,225], miR-107 in the plasma [226], miR-206 in the serum and plasma [207,227], miR-132 in the plasma [228], and miR-1185-2-3p, miR-1909-3p, miR-22-5p, miR-134-3p, and miR-107 in the plasma [229] have been shown to play predictive roles in MCI diagnosis and progression from MCI to AD.

Table 1.
Identification of miRNAs as potential blood-based biomarkers for AD diagnosis through screening processes.
Nevertheless, a series of limitations need to be addressed. Gender differences and variations among different human populations may exist for some circulating miRNAs as peripheral biomarkers in AD [230,231]. A standardized and reliable method or equipment with high specificity and sensitivity is needed for miRNA detection. Further investigations are also needed to validate the correlations of these blood-based biomarkers with established AD biomarkers in a larger cohort of participants.
6. Therapeutic Potential of miRNAs in AD
In recent decades, AD has rapidly become one of the most hindering and costly disorders affecting the elderly with a high mortality rate, emerging as a worldwide burden for both patients and caregivers [7]. Unfortunately, effective disease-modifying therapeutics are still lacking [184]. Since advanced age is considered the most influential risk factor for AD, antioxidants, including melatonin and resveratrol, have been regarded as potential candidates for neuroprotection against aging. Recently, we observed that melatonin alleviates tau pathologies by upregulating miR-504-3p expression [71], suggesting that miRNAs play critical roles in AD treatment. Resveratrol can also rescue aberrant expression of miRNAs to exert neuroprotective effects [232]. Currently, the majority of drug development programs are targeting Aβ and tau [7]. Nevertheless, the resulting clinical trials have been disappointing despite the positive therapeutic outcomes in cell and animal models [233]. Thus, more therapeutic approaches are needed to confront this complex multifactorial disorder.
Compared with conventional therapeutic methods that target proteins instead of underlying causes resulting in transient effects, employing nucleic acids as therapeutics may trigger long-lasting or curative effects due to the powerful capacities of gene inhibition, addition, replacement, or editing [234]. Due to their considerable merits of specificity, safety, and suitability for targets that remain undruggable, nucleic acid therapeutics possess the potential to emerge as the third pillar of drug development in addition to small molecule inhibitors and antibodies [235]. As one of the most advanced and efficacious therapeutic options for rare genetic disorders and debilitating diseases, nucleic acid therapeutics can achieve precise targeting with sequence-specific nucleic acid recognition, opening new avenues for CNS disease treatment [236]. Over the last quarter century, a total of 18 nucleic acid therapeutics have received clinical approval for treating various diseases [237]. Notably, among approximately 20,000 human proteins, only approximately 3000 proteins are druggable, and fewer than 700 proteins can be targeted by approved drugs [238,239]. Therefore, nucleic acid therapeutics with abundant candidate targets and relatively simple preparation processes may provide novel insights into targeting previously undruggable proteins to treat diseases that are difficult to cure, such as AD.
Recently, miRNA-based therapeutics that replenish or inhibit miRNA function via delivery of synthetic small RNA molecules have drawn considerable interest and shown promise in preclinical studies with multiple advantages [240]. First, miRNAs are natural molecules that originally exist in human cells and are thus involved in biological processes in vivo [241]. Second, in contrast to conventional therapeutics, miRNAs can directly bind to downstream targets to modulate their expression. Moreover, the size of miRNAs can facilitate the development of reliable and effective delivery systems. Furthermore, miRNAs may simultaneously modulate several genes within one specific pathway, leading to a more robust yet specific regulation [241]. As synthetic double-stranded oligonucleotides, miRNA mimics play roles in restoring lost miRNA function among different types of diseases [240,241]. To date, two miRNA mimics, MRX34 (miR-34 mimics) and MesomiR-1 (miR-16 mimics), have been tested in clinical trials for cancer treatment [241]. The potential clinical applications of miRNA-based therapeutics for AD treatment remain to be exploited.
Notably, despite the appealing therapeutic use of miRNAs, numerous challenges need to be overcome, including poor stability, low cell membrane permeability, the existence of the blood-brain barrier (BBB), and the limited targeting of specific tissues [18]. Naked RNA molecules are prone to degradation in circulation, and their negative charges as well as large size make it difficult for them to cross the cell membrane [242]. The presence of the BBB further hinders RNA uptake and delivery into brain tissues [239]. The rapid advancements in nanotechnology provide new possibilities for therapies for brain disorders with promising delivery systems. To address the issue of BBB penetration, we previously developed a BBB-permeable nanocapsule with 2-methacryloyloxyethyl phosphorylcholine (MPC) on the surface [243]. The nanocapsule is able to cross the BBB and release its cargo in the brain [243]. To date, a wide variety of nanocarriers have been fabricated with improved RNA delivery efficiency and minimized toxicity [242]. Both conventional and advanced nanocarriers are being extensively explored for effective drug delivery in the treatment of AD [244,245,246]. As naturally secreted nanosized extracellular vesicles, exosomes have sparked great interest as vehicles for the delivery of drugs due to their unique characteristics, including increased blood stability, reduced cytotoxicity, and limited immunogenicity [247]. Recently, we functionalized exosomes with the peptide angiopep-2, resulting in efficient nanocarriers with enhanced BBB permeability and improved biosafety [248]. In AD, exosomes have been demonstrated to restore cognitive function in animal models, offering an attractive therapeutic tool for treatment [249,250,251]. Thus, well-designed artificial and natural nanocarriers may facilitate therapeutic miRNA delivery in AD.
7. Concluding Remarks
In conclusion, miRNAs can directly regulate MAPT transcripts, the alternative splicing of tau, and related kinases to modulate tau, playing key roles in tau pathologies in AD. Moreover, miRNAs can directly modulate APP transcripts, the alternative splicing of APP, BACE1 transcripts, and related kinases to control amyloidogenic APP processing, exerting regulatory effects on Aβ pathologies in AD. Circulating miRNAs in blood have emerged as potential noninvasive biomarkers for AD diagnosis. The appealing potential of miRNA-based therapeutics holds promise, exploiting advanced delivery systems such as nanocarriers and exosomes. While miRNAs play a crucial role in regulating gene expression in a sequence-specific manner, the dual nature of their ability to modulate multiple targets poses a complex scenario. The question arises of whether miRNAs function as precise and specific regulators or if their impact is more potent within a particular pathway compared to a combination of siRNAs. Further investigation is required to clarify this aspect. With more insights into the role of miRNAs in AD pathogenesis and growing interest in developing novel technologies, miRNAs are expected to have diagnostic and therapeutic applications in the coming decades.
Author Contributions
Conceptualization, L.W., X.S., Y.Z. and T.H.L.; investigation, L.W., X.S., Y.D. and D.C.; writing—original draft preparation, L.W., X.S. and Y.Z.; writing—review and editing, Y.Z. and T.H.L.; supervision, Y.Z. and T.H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81970993 and 82271449), the Natural Science Foundation of Fujian Province (2022J01203 and 2022J01666), the Research Foundation for Advanced Talents of Fujian Medical University (XRCZX2017019), and the Startup Fund for Scientific Research, Fujian Medical University (2022QH2005).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Pietrocola, F.; Roiz-Valle, D.; Galluzzi, L.; Kroemer, G. Meta-hallmarks of aging and cancer. Cell Metab. 2023, 35, 12–35. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Y.; Chen, D.; Lee, T.H. Peptidyl-Prolyl Cis/Trans Isomerase Pin1 and Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 8, 355. [Google Scholar] [CrossRef]
- Grobler, C.; van Tongeren, M.; Gettemans, J.; Kell, D.B.; Pretorius, E. Alzheimer’s Disease: A Systems View Provides a Unifying Explanation of Its Development. J. Alzheimers Dis. 2023, 91, 43–70. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grasser, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stahler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Diener, C.; Keller, A.; Meese, E. Emerging concepts of miRNA therapeutics: From cells to clinic. Trends Genet. 2022, 38, 613–626. [Google Scholar] [CrossRef]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Iacomino, G. miRNAs: The Road from Bench to Bedside. Genes 2023, 14, 314. [Google Scholar] [CrossRef]
- Nowak, J.S.; Michlewski, G. miRNAs in development and pathogenesis of the nervous system. Biochem. Soc. Trans. 2013, 41, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Eacker, S.M.; Dawson, T.M.; Dawson, V.L. Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 2009, 10, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6156–6167. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Almansoub, H.; Tang, H.; Wu, Y.; Wang, D.Q.; Mahaman, Y.A.R.; Wei, N.; Almansob, Y.A.M.; He, W.; Liu, D. Tau Abnormalities and the Potential Therapy in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 67, 13–33. [Google Scholar] [CrossRef]
- Montalto, G.; Ricciarelli, R. Tau, tau kinases, and tauopathies: An updated overview. Biofactors 2023, 49, 502–511. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef]
- Lee, V.M.; Balin, B.J.; Otvos, L., Jr.; Trojanowski, J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Cairns, N.J.; Crowther, R.A. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Matsuo, E.S.; Shin, R.W.; Billingsley, M.L.; Van deVoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Piscopo, P.; Grasso, M.; Manzini, V.; Zeni, A.; Castelluzzo, M.; Fontana, F.; Talarico, G.; Castellano, A.E.; Rivabene, R.; Crestini, A.; et al. Identification of miRNAs regulating MAPT expression and their analysis in plasma of patients with dementia. Front. Mol. Neurosci. 2023, 16, 1127163. [Google Scholar] [CrossRef] [PubMed]
- Denk, J.; Oberhauser, F.; Kornhuber, J.; Wiltfang, J.; Fassbender, K.; Schroeter, M.L.; Volk, A.E.; Diehl-Schmid, J.; Prudlo, J.; Danek, A.; et al. Specific serum and CSF microRNA profiles distinguish sporadic behavioural variant of frontotemporal dementia compared with Alzheimer patients and cognitively healthy controls. PLoS ONE 2018, 13, e0197329. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Tarazona-Sanchez, A.; Braza-Boils, A.; Balaguer, A.; Ferre-Gonzalez, L.; Canada-Martinez, A.J.; Baquero, M.; Chafer-Pericas, C. Plasma microRNAs as potential biomarkers in early Alzheimer disease expression. Sci. Rep. 2022, 12, 15589. [Google Scholar] [CrossRef]
- Siedlecki-Wullich, D.; Catala-Solsona, J.; Fabregas, C.; Hernandez, I.; Clarimon, J.; Lleo, A.; Boada, M.; Saura, C.A.; Rodriguez-Alvarez, J.; Minano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.J.; Wong, B.Y.X.; Vaidyanathan, R.; Sreejith, S.; Chia, S.Y.; Kandiah, N.; Ng, A.S.L.; Zeng, L. Altered Cerebrospinal Fluid Exosomal microRNA Levels in Young-Onset Alzheimer’s Disease and Frontotemporal Dementia. J. Alzheimers Dis. Rep. 2021, 5, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Laskowska-Kaszub, K.; Debski, K.J.; Wojsiat, J.; Dabrowski, M.; Gabryelewicz, T.; Kuznicki, J.; Wojda, U. Profile of 6 microRNA in blood plasma distinguish early stage Alzheimer’s disease patients from non-demented subjects. Oncotarget 2017, 8, 16122–16143. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef]
- Wu, H.; Huang, M.; Lu, M.; Zhu, W.; Shu, Y.; Cao, P.; Liu, P. Regulation of microtubule-associated protein tau (MAPT) by miR-34c-5p determines the chemosensitivity of gastric cancer to paclitaxel. Cancer Chemother. Pharmacol. 2013, 71, 1159–1171. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Z.; Sun, L.; Fang, Y.; Xu, X.; Zhou, G. miR-186 regulates chemo-sensitivity to paclitaxel via targeting MAPT in non-small cell lung cancer (NSCLC). Mol. Biosyst. 2016, 12, 3417–3424. [Google Scholar] [CrossRef]
- Li, J.W.; Ren, S.H.; Ren, J.R.; Zhen, Z.G.; Li, L.R.; Hao, X.D.; Ji, H.M. Nimodipine Improves Cognitive Impairment After Subarachnoid Hemorrhage in Rats Through IncRNA NEAT1/miR-27a/MAPT Axis. Drug Des. Devel. Ther. 2020, 14, 2295–2306. [Google Scholar] [CrossRef]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef]
- Liu, F.; Gong, C.X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 8. [Google Scholar] [CrossRef]
- Smith, P.Y.; Delay, C.; Girard, J.; Papon, M.A.; Planel, E.; Sergeant, N.; Buee, L.; Hebert, S.S. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 2011, 20, 4016–4024. [Google Scholar] [CrossRef]
- Salta, E.; Sierksma, A.; Vanden Eynden, E.; De Strooper, B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol. Med. 2016, 8, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- El Fatimy, R.; Li, S.; Chen, Z.; Mushannen, T.; Gongala, S.; Wei, Z.; Balu, D.T.; Rabinovsky, R.; Cantlon, A.; Elkhal, A.; et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018, 136, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, D.; Huang, H.Z.; Wang, Z.H.; Hou, T.Y.; Yang, X.; Pang, P.; Wei, N.; Zhou, Y.F.; Dupras, M.J.; et al. A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 395–405. [Google Scholar] [CrossRef]
- Hou, T.Y.; Zhou, Y.; Zhu, L.S.; Wang, X.; Pang, P.; Wang, D.Q.; Liuyang, Z.Y.; Man, H.; Lu, Y.; Zhu, L.Q.; et al. Correcting abnormalities in miR-124/PTPN1 signaling rescues tau pathology in Alzheimer’s disease. J. Neurochem. 2020, 154, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef]
- Subramanian, M.; Hyeon, S.J.; Das, T.; Suh, Y.S.; Kim, Y.K.; Lee, J.S.; Song, E.J.; Ryu, H.; Yu, K. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat. Commun. 2021, 12, 3291. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid beta, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, B.; Chen, J.; Sui, Y.; Ren, L.; Li, J.; Zhang, H.; Guo, L.; Sun, X. Micro-RNA-137 Inhibits Tau Hyperphosphorylation in Alzheimer’s Disease and Targets the CACNA1C Gene in Transgenic Mice and Human Neuroblastoma SH-SY5Y Cells. Med. Sci. Monit. 2018, 24, 5635–5644. [Google Scholar] [CrossRef]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Li, J.; Chen, W.; Yi, Y.; Tong, Q. miR-219-5p inhibits tau phosphorylation by targeting TTBK1 and GSK-3beta in Alzheimer’s disease. J. Cell. Biochem. 2019, 120, 9936–9946. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, Y.; Su, L. MiR-539-5p Decreases amyloid beta-protein production, hyperphosphorylation of Tau and Memory Impairment by Regulating PI3K/Akt/GSK-3beta Pathways in APP/PS1 Double Transgenic Mice. Neurotox. Res. 2020, 38, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Liu, J.; Guo, S.; Zeng, L.; Cai, Z.; Zhang, J.; Wang, L.; Li, Z.; Liu, R. miR-23b-3p rescues cognition in Alzheimer’s disease by reducing tau phosphorylation and apoptosis via GSK-3beta signaling pathways. Mol. Ther. Nucleic Acids 2022, 28, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Poon, C.H.; Zhang, Z.; Yue, M.; Chen, R.; Zhang, Y.; Hossain, M.F.; Pan, Y.; Zhao, J.; Rong, L.; et al. MicroRNA-128 suppresses tau phosphorylation and reduces amyloid-beta accumulation by inhibiting the expression of GSK3beta, APPBP2, and mTOR in Alzheimer’s disease. CNS Neurosci. Ther. 2023, 29, 1848–1864. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Maitra, S.; Vincent, B. Cdk5-p25 as a key element linking amyloid and tau pathologies in Alzheimer’s disease: Mechanisms and possible therapeutic interventions. Life Sci. 2022, 308, 120986. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef]
- Noble, W.; Olm, V.; Takata, K.; Casey, E.; Mary, O.; Meyerson, J.; Gaynor, K.; LaFrancois, J.; Wang, L.; Kondo, T.; et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 2003, 38, 555–565. [Google Scholar] [CrossRef]
- Zeng, L.; Jiang, H.; Ashraf, G.M.; Liu, J.; Wang, L.; Zhao, K.; Liu, M.; Li, Z.; Liu, R. Implications of miR-148a-3p/p35/PTEN signaling in tau hyperphosphorylation and autoregulatory feedforward of Akt/CREB in Alzheimer’s disease. Mol. Ther. Nucleic Acids 2022, 27, 256–275. [Google Scholar] [CrossRef]
- Sun, L.H.; Ban, T.; Liu, C.D.; Chen, Q.X.; Wang, X.; Yan, M.L.; Hu, X.L.; Su, X.L.; Bao, Y.N.; Sun, L.L.; et al. Activation of Cdk5/p25 and tau phosphorylation following chronic brain hypoperfusion in rats involves microRNA-195 down-regulation. J. Neurochem. 2015, 134, 1139–1151. [Google Scholar] [CrossRef]
- Chen, D.; Lan, G.; Li, R.; Mei, Y.; Shui, X.; Gu, X.; Wang, L.; Zhang, T.; Gan, C.L.; Xia, Y.; et al. Melatonin ameliorates tau-related pathology via the miR-504-3p and CDK5 axis in Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Gong, C.X.; Hwang, Y.W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Xie, F.; Wang, X.; Hou, Y.; Wang, X.; Liu, J. Overexpression of miR-26a-5p Suppresses Tau Phosphorylation and Abeta Accumulation in the Alzheimer’s Disease Mice by Targeting DYRK1A. Curr. Neurovasc Res. 2020, 17, 241–248. [Google Scholar] [CrossRef]
- Chen, D.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 as a Promising Drug Target in Cancer and Alzheimer’s Disease. Recent. Pat. Anticancer. Drug Discov. 2019, 14, 144–157. [Google Scholar] [CrossRef]
- Kim, N.; Chen, D.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease. Int. J. Mol. Sci. 2019, 20, 3131. [Google Scholar] [CrossRef]
- Li, Y.; Grupe, A.; Rowland, C.; Nowotny, P.; Kauwe, J.S.; Smemo, S.; Hinrichs, A.; Tacey, K.; Toombs, T.A.; Kwok, S.; et al. DAPK1 variants are associated with Alzheimer’s disease and allele-specific expression. Hum. Mol. Genet. 2006, 15, 2560–2568. [Google Scholar] [CrossRef]
- Li, H.; Wetten, S.; Li, L.; St Jean, P.L.; Upmanyu, R.; Surh, L.; Hosford, D.; Barnes, M.R.; Briley, J.D.; Borrie, M.; et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 2008, 65, 45–53. [Google Scholar] [CrossRef]
- Laumet, G.; Chouraki, V.; Grenier-Boley, B.; Legry, V.; Heath, S.; Zelenika, D.; Fievet, N.; Hannequin, D.; Delepine, M.; Pasquier, F.; et al. Systematic analysis of candidate genes for Alzheimer’s disease in a French, genome-wide association study. J. Alzheimers Dis. 2010, 20, 1181–1188. [Google Scholar] [CrossRef]
- Gaj, P.; Paziewska, A.; Bik, W.; Dabrowska, M.; Baranowska-Bik, A.; Styczynska, M.; Chodakowska-Zebrowska, M.; Pfeffer-Baczuk, A.; Barcikowska, M.; Baranowska, B.; et al. Identification of a late onset Alzheimer’s disease candidate risk variant at 9q21.33 in Polish patients. J. Alzheimers Dis. 2012, 32, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; You, M.H.; Chen, C.H.; Lee, S.; Hong, Y.; Hong, Y.; Kimchi, A.; Zhou, X.Z.; Lee, T.H. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis. 2014, 5, e1237. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; You, M.H.; Chen, C.H.; Suh, J.; Tanzi, R.E.; Ho Lee, T. Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum. Mol. Genet. 2016, 25, 2498–2513. [Google Scholar] [CrossRef]
- You, M.H.; Kim, B.M.; Chen, C.H.; Begley, M.J.; Cantley, L.C.; Lee, T.H. Death-associated protein kinase 1 phosphorylates NDRG2 and induces neuronal cell death. Cell Death Differ. 2017, 24, 238–250. [Google Scholar] [CrossRef]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.H.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.H.; Yao, Y.; Lin, Y.M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef]
- Wang, R.; Lu, K.P.; Zhou, X.Z. Function and regulation of cis P-tau in the pathogenesis and treatment of conventional and nonconventional tauopathies. J. Neurochem. 2023, 166, 904–914. [Google Scholar] [CrossRef]
- Wang, L.; Shui, X.; Mei, Y.; Xia, Y.; Lan, G.; Hu, L.; Zhang, M.; Gan, C.L.; Li, R.; Tian, Y.; et al. miR-143-3p Inhibits Aberrant Tau Phosphorylation and Amyloidogenic Processing of APP by Directly Targeting DAPK1 in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 7992. [Google Scholar] [CrossRef]
- Wang, L.; Shui, X.; Zhang, M.; Mei, Y.; Xia, Y.; Lan, G.; Hu, L.; Gan, C.L.; Tian, Y.; Li, R.; et al. MiR-191-5p Attenuates Tau Phosphorylation, Abeta Generation, and Neuronal Cell Death by Regulating Death-Associated Protein Kinase 1. ACS Chem. Neurosci. 2022, 13, 3554–3566. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Goldgaber, D.; Lerman, M.I.; McBride, O.W.; Saffiotti, U.; Gajdusek, D.C. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 1987, 235, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Gusella, J.F.; Watkins, P.C.; Bruns, G.A.; St George-Hyslop, P.; Van Keuren, M.L.; Patterson, D.; Pagan, S.; Kurnit, D.M.; Neve, R.L. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 1987, 235, 880–884. [Google Scholar] [CrossRef]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef][Green Version]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerriere, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef]
- Sleegers, K.; Brouwers, N.; Gijselinck, I.; Theuns, J.; Goossens, D.; Wauters, J.; Del-Favero, J.; Cruts, M.; van Duijn, C.M.; Van Broeckhoven, C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 2006, 129, 2977–2983. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Thinakaran, G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 2006, 66, S69–S73. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Cohen, M.L.; Golde, T.E.; Usiak, M.F.; Younkin, L.H.; Younkin, S.G. In situ hybridization of nucleus basalis neurons shows increased beta-amyloid mRNA in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 1227–1231. [Google Scholar] [CrossRef]
- Higgins, G.A.; Lewis, D.A.; Bahmanyar, S.; Goldgaber, D.; Gajdusek, D.C.; Young, W.G.; Morrison, J.H.; Wilson, M.C. Differential regulation of amyloid-beta-protein mRNA expression within hippocampal neuronal subpopulations in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 1297–1301. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, D.; Lee, T.H. Phosphorylation Signaling in APP Processing in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 21, 209. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kao, S.C.; Lemere, C.A.; Xia, W.; Tseng, H.C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.H. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef]
- Suzuki, T.; Nakaya, T. Regulation of amyloid beta-protein precursor by phosphorylation and protein interactions. J. Biol. Chem. 2008, 283, 29633–29637. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; Zhou, D.; D’Adamio, L. The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 2009, 4, 28. [Google Scholar] [CrossRef]
- Patel, N.; Hoang, D.; Miller, N.; Ansaloni, S.; Huang, Q.; Rogers, J.T.; Lee, J.C.; Saunders, A.J. MicroRNAs can regulate human APP levels. Mol. Neurodegener. 2008, 3, 10. [Google Scholar] [CrossRef]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef]
- Delay, C.; Calon, F.; Mathews, P.; Hebert, S.S. Alzheimer-specific variants in the 3’UTR of Amyloid precursor protein affect microRNA function. Mol. Neurodegener. 2011, 6, 70. [Google Scholar] [CrossRef]
- Vilardo, E.; Barbato, C.; Ciotti, M.; Cogoni, C.; Ruberti, F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010, 285, 18344–18351. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Lahiri, D.K. MicroRNA-101 downregulates Alzheimer’s amyloid-beta precursor protein levels in human cell cultures and is differentially expressed. Biochem. Biophys. Res. Commun. 2011, 404, 889–895. [Google Scholar] [CrossRef]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Parsi, S.; Smith, P.Y.; Goupil, C.; Dorval, V.; Hebert, S.S. Preclinical Evaluation of miR-15/107 Family Members as Multifactorial Drug Targets for Alzheimer’s Disease. Mol. Ther. Nucleic Acids 2015, 4, e256. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, C.F.; Wang, A.H.; Lin, Q.F. MiR-16 regulates cell death in Alzheimer’s disease by targeting amyloid precursor protein. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4020–4027. [Google Scholar]
- Liu, C.G.; Wang, J.L.; Li, L.; Xue, L.X.; Zhang, Y.Q.; Wang, P.C. MicroRNA-135a and -200b, potential Biomarkers for Alzheimer׳s disease, regulate beta secretase and amyloid precursor protein. Brain Res. 2014, 1583, 55–64. [Google Scholar] [CrossRef]
- Liu, C.G.; Wang, J.L.; Li, L.; Wang, P.C. MicroRNA-384 regulates both amyloid precursor protein and beta-secretase expression and is a potential biomarker for Alzheimer’s disease. Int. J. Mol. Med. 2014, 34, 160–166. [Google Scholar] [CrossRef]
- Liu, C.G.; Song, J.; Zhang, Y.Q.; Wang, P.C. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer’s disease. Mol. Med. Rep. 2014, 10, 2395–2400. [Google Scholar] [CrossRef]
- Li, K.; Zhang, J.; Ji, C.; Wang, L. MiR-144-3p and Its Target Gene beta-Amyloid Precursor Protein Regulate 1-Methyl-4-Phenyl-1,2-3,6-Tetrahydropyridine-Induced Mitochondrial Dysfunction. Mol. Cells 2016, 39, 543–549. [Google Scholar] [CrossRef]
- Liu, H.Y.; Fu, X.; Li, Y.F.; Li, X.L.; Ma, Z.Y.; Zhang, Y.; Gao, Q.C. miR-15b-5p targeting amyloid precursor protein is involved in the anti-amyloid eflect of curcumin in swAPP695-HEK293 cells. Neural Regen. Res. 2019, 14, 1603–1609. [Google Scholar]
- Kumar, S.; Reddy, A.P.; Yin, X.; Reddy, P.H. Novel MicroRNA-455-3p and its protective effects against abnormal APP processing and amyloid beta toxicity in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2428–2440. [Google Scholar] [CrossRef] [PubMed]
- Barros-Viegas, A.T.; Carmona, V.; Ferreiro, E.; Guedes, J.; Cardoso, A.M.; Cunha, P.; Pereira de Almeida, L.; Resende de Oliveira, C.; Pedro de Magalhaes, J.; Peca, J.; et al. miRNA-31 Improves Cognition and Abolishes Amyloid-beta Pathology by Targeting APP and BACE1 in an Animal Model of Alzheimer’s Disease. Mol. Ther. Nucleic Acids 2020, 19, 1219–1236. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.H.; Sun, L.; Zhang, W.J.; Wang, X.Y.; Li, J.M. Reduced serum miR-202 may promote the progression of Alzheimer’s disease patients via targeting amyloid precursor protein. Kaohsiung J. Med. Sci. 2021, 37, 730–738. [Google Scholar] [CrossRef]
- Chopra, N.; Wang, R.; Maloney, B.; Nho, K.; Beck, J.S.; Pourshafie, N.; Niculescu, A.; Saykin, A.J.; Rinaldi, C.; Counts, S.E.; et al. MicroRNA-298 reduces levels of human amyloid-beta precursor protein (APP), beta-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol. Psychiatry 2021, 26, 5636–5657. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhu, L.; Tan, J.; Chen, K.; Yu, B. Suppression of miR-130a-3p Attenuates Oxygen-Glucose Deprivation/Reoxygenation-Induced Dendritic Spine Loss by Promoting APP. Front. Neurosci. 2021, 15, 601850. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, X.; Xia, X.; Li, Y.; Wang, Y.; Li, C.; Sun, Y.; Gao, G.; Zhao, S.; Sheng, S.; et al. Exosomes Mediate APP Dysregulation via APP-miR-185-5p Axis. Front. Cell Dev. Biol. 2022, 10, 793388. [Google Scholar] [CrossRef]
- Fan, X.; Xu, S.; Yang, C. miR-373-3p promotes lung adenocarcinoma cell proliferation via APP. Oncol. Lett. 2018, 15, 1046–1050. [Google Scholar] [CrossRef]
- Zhou, Q.; Luo, L.; Wang, X.; Li, X. Relationship between single nucleotide polymorphisms in the 3’UTR of amyloid precursor protein and risk of Alzheimer’s disease and its mechanism. Biosci. Rep. 2019, 39, BSR20182485. [Google Scholar] [CrossRef]
- Long, J.M.; Maloney, B.; Rogers, J.T.; Lahiri, D.K. Novel upregulation of amyloid-beta precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5’-untranslated region: Implications in Alzheimer’s disease. Mol. Psychiatry 2019, 24, 345–363. [Google Scholar] [CrossRef]
- Nikom, D.; Zheng, S. Alternative splicing in neurodegenerative disease and the promise of RNA therapies. Nat. Rev. Neurosci. 2023, 24, 457–473. [Google Scholar] [CrossRef]
- Smith, P.; Al Hashimi, A.; Girard, J.; Delay, C.; Hebert, S.S. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J. Neurochem. 2011, 116, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Vassar, R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The beta-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Zong, Y.; Wang, H.; Dong, W.; Quan, X.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Qin, C. miR-29c regulates BACE1 protein expression. Brain Res. 2011, 1395, 108–115. [Google Scholar] [CrossRef]
- Lei, X.; Lei, L.; Zhang, Z.; Zhang, Z.; Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565–1574. [Google Scholar]
- Yang, G.; Song, Y.; Zhou, X.; Deng, Y.; Liu, T.; Weng, G.; Yu, D.; Pan, S. MicroRNA-29c targets beta-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol. Med. Rep. 2015, 12, 3081–3088. [Google Scholar] [CrossRef]
- Boissonneault, V.; Plante, I.; Rivest, S.; Provost, P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J. Biol. Chem. 2009, 284, 1971–1981. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-beta production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-339-5p down-regulates protein expression of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J. Biol. Chem. 2014, 289, 5184–5198. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.; Teng, Z.; Tang, Y.P.; Chen, C. Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188-3p in a mouse model of Alzheimer’s disease. J. Neurosci. 2014, 34, 14919–14933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xing, H.; Guo, S.; Zheng, Z.; Wang, H.; Xu, D. MicroRNA-135b has a neuroprotective role via targeting of beta-site APP-cleaving enzyme 1. Exp. Ther. Med. 2016, 12, 809–814. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, H.; Chung, D.E.; Brown, J.L.; Belmonte, K.C.; Kim, J. miR-186 is decreased in aged brain and suppresses BACE1 expression. J. Neurochem. 2016, 137, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; An, F.; Wang, Y.; Bian, M.; Yu, L.J.; Wei, C. miR-15b represses BACE1 expression in sporadic Alzheimer’s disease. Oncotarget 2017, 8, 91551–91557. [Google Scholar] [CrossRef]
- Li, J.; Wang, H. miR-15b reduces amyloid-beta accumulation in SH-SY5Y cell line through targetting NF-kappaB signaling and BACE1. Biosci. Rep. 2018, 38, BSR20180051. [Google Scholar]
- An, F.; Gong, G.; Wang, Y.; Bian, M.; Yu, L.; Wei, C. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 2017, 8, 114065–114071. [Google Scholar] [CrossRef]
- Du, X.; Huo, X.; Yang, Y.; Hu, Z.; Botchway, B.O.A.; Jiang, Y.; Fang, M. miR-124 downregulates BACE 1 and alters autophagy in APP/PS1 transgenic mice. Toxicol. Lett. 2017, 280, 195–205. [Google Scholar] [CrossRef]
- Zhong, Z.; Yuan, K.; Tong, X.; Hu, J.; Song, Z.; Zhang, G.; Fang, X.; Zhang, W. MiR-16 attenuates beta-amyloid-induced neurotoxicity through targeting beta-site amyloid precursor protein-cleaving enzyme 1 in an Alzheimer’s disease cell model. Neuroreport 2018, 29, 1365–1372. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Wang, Q.; Jiang, H.; Zeng, L.; Li, Z.; Liu, R. MicroRNA-200a-3p Mediates Neuroprotection in Alzheimer-Related Deficits and Attenuates Amyloid-Beta Overproduction and Tau Hyperphosphorylation via Coregulating BACE1 and PRKACB. Front. Pharmacol. 2019, 10, 806. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, D.; Zhang, B.; Lu, H. MiR-361-3p inhibits beta-amyloid accumulation and attenuates cognitive deficits through targeting BACE1 in Alzheimer’s disease. J. Integr. Neurosci. 2019, 18, 285–291. [Google Scholar]
- Qian, Q.; Zhang, J.; He, F.P.; Bao, W.X.; Zheng, T.T.; Zhou, D.M.; Pan, H.Y.; Zhang, H.; Zhang, X.Q.; He, X.; et al. Down-regulated expression of microRNA-338-5p contributes to neuropathology in Alzheimer’s disease. FASEB J. 2019, 33, 4404–4417. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xu, Y.; Wang, B.; Huang, J.; Li, Q. miR-34a-5p and miR-125b-5p attenuate Abeta-induced neurotoxicity through targeting BACE1. J. Neurol. Sci. 2020, 413, 116793. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Luo, Y.; Pi, D.; Xia, L.; Li, Z.; Tu, Q. MiR-340 Reduces the Accumulation of Amyloid-beta Through Targeting BACE1 (beta-site Amyloid Precursor Protein Cleaving Enzyme 1) in Alzheimer’s Disease. Curr. Neurovasc Res. 2020, 17, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, W.W.; Lv, C.M.; Gao, Y.W.; Liu, X.L.; Zhao, L. miR-16-5p and miR-19b-3p prevent amyloid beta-induced injury by targeting BACE1 in SH-SY5Y cells. Neuroreport 2020, 31, 205–212. [Google Scholar] [CrossRef]
- Du, W.; Lei, C.; Dong, Y. MicroRNA-149 is downregulated in Alzheimer’s disease and inhibits beta-amyloid accumulation and ameliorates neuronal viability through targeting BACE1. Genet. Mol. Biol. 2021, 44, e20200064. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, R. Deregulated lncRNA MAGI2-AS3 in Alzheimer’s disease attenuates amyloid-beta induced neurotoxicity and neuroinflammation by sponging miR-374b-5p. Exp. Gerontol. 2021, 144, 111180. [Google Scholar] [CrossRef]
- Dong, Z.; Gu, H.; Guo, Q.; Liu, X.; Li, F.; Liu, H.; Sun, L.; Ma, H.; Zhao, K. Circulating Small Extracellular Vesicle-Derived miR-342-5p Ameliorates Beta-Amyloid Formation via Targeting Beta-site APP Cleaving Enzyme 1 in Alzheimer’s Disease. Cells 2022, 11, 3830. [Google Scholar] [CrossRef]
- Aplin, A.E.; Gibb, G.M.; Jacobsen, J.S.; Gallo, J.M.; Anderton, B.H. In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J. Neurochem. 1996, 67, 699–707. [Google Scholar] [CrossRef]
- Acevedo, K.M.; Opazo, C.M.; Norrish, D.; Challis, L.M.; Li, Q.X.; White, A.R.; Bush, A.I.; Camakaris, J. Phosphorylation of amyloid precursor protein at threonine 668 is essential for its copper-responsive trafficking in SH-SY5Y neuroblastoma cells. J. Biol. Chem. 2014, 289, 11007–11019. [Google Scholar] [CrossRef]
- Iijima, K.; Ando, K.; Takeda, S.; Satoh, Y.; Seki, T.; Itohara, S.; Greengard, P.; Kirino, Y.; Nairn, A.C.; Suzuki, T. Neuron-specific phosphorylation of Alzheimer’s beta-amyloid precursor protein by cyclin-dependent kinase 5. J. Neurochem. 2000, 75, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Su, Y.; Li, B.; Zhou, Y.; Ryder, J.; Gonzalez-DeWhitt, P.; May, P.C.; Ni, B. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003, 547, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, X.; Cheng, X.; Li, Y.; Gearing, M.; Levey, A.; Huang, X.; Li, Y.; Jin, P.; Li, X. MicroRNA-650 Regulates the Pathogenesis of Alzheimer’s Disease Through Targeting Cyclin-Dependent Kinase 5. Mol. Neurobiol. 2023, 60, 2426–2441. [Google Scholar] [CrossRef] [PubMed]
- Moncini, S.; Lunghi, M.; Valmadre, A.; Grasso, M.; Del Vescovo, V.; Riva, P.; Denti, M.A.; Venturin, M. The miR-15/107 Family of microRNA Genes Regulates CDK5R1/p35 with Implications for Alzheimer’s Disease Pathogenesis. Mol. Neurobiol. 2017, 54, 4329–4342. [Google Scholar] [CrossRef] [PubMed]
- Standen, C.L.; Brownlees, J.; Grierson, A.J.; Kesavapany, S.; Lau, K.F.; McLoughlin, D.M.; Miller, C.C. Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer’s disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3). J. Neurochem. 2001, 76, 316–320. [Google Scholar] [CrossRef]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Wang, D.; Fei, Z.; Luo, S.; Wang, H. MiR-335-5p Inhibits beta-Amyloid (Abeta) Accumulation to Attenuate Cognitive Deficits Through Targeting c-jun-N-terminal Kinase 3 in Alzheimer’s Disease. Curr. Neurovasc Res. 2020, 17, 93–101. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, M.; Hong, Z.; Kang, J.; Pan, H.; Yan, C. MiR-130a-3p Has Protective Effects in Alzheimer’s Disease via Targeting DAPK1. Am. J. Alzheimers Dis. Other Demen 2021, 36, 15333175211020572. [Google Scholar] [CrossRef]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Souza, V.C.; Morais, G.S., Jr.; Henriques, A.D.; Machado-Silva, W.; Perez, D.I.V.; Brito, C.J.; Camargos, E.F.; Moraes, C.F.; Nobrega, O.T. Whole-Blood Levels of MicroRNA-9 Are Decreased in Patients With Late-Onset Alzheimer Disease. Am. J. Alzheimers Dis. Other Demen 2020, 35, 1533317520911573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qin, W.; Wei, C.B.; Tang, Y.; Zhao, L.N.; Jin, H.M.; Li, Y.; Wang, Q.; Luan, X.Q.; He, J.C.; et al. The microRNA-1908 up-regulation in the peripheral blood cells impairs amyloid clearance by targeting ApoE. Int. J. Geriatr. Psychiatry 2018, 33, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Poli, G.; Cecchetti, R.; Bastiani, P.; Scamosci, M.; Febo, M.; Mazzon, E.; Bruscoli, S.; Brancorsini, S.; Mecocci, P. miRNAs and Alzheimer’s Disease: Exploring the Role of Inflammation and Vitamin E in an Old-Age Population. Nutrients 2023, 15, 634. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Long, Y.F.; Xu, P.; Guo, H.D.; Cui, G.H. Pathogenesis of miR-155 on nonmodifiable and modifiable risk factors in Alzheimer’s disease. Alzheimers Res. Ther. 2023, 15, 122. [Google Scholar] [CrossRef]
- Rastegar-Moghaddam, S.H.; Ebrahimzadeh-Bideskan, A.; Shahba, S.; Malvandi, A.M.; Mohammadipour, A. Roles of the miR-155 in Neuroinflammation and Neurological Disorders: A Potent Biological and Therapeutic Target. Cell Mol. Neurobiol. 2023, 43, 455–467. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.V.; Mastroeni, D.; Audrain, M.; Fanutza, T.; Kim, S.H.; Blitzer, R.D.; Gandy, S.; Dudley, J.T.; Ehrlich, M.E. miR155 regulation of behavior, neuropathology, and cortical transcriptomics in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 295–315. [Google Scholar] [CrossRef]
- Aloi, M.S.; Prater, K.E.; Sopher, B.; Davidson, S.; Jayadev, S.; Garden, G.A. The pro-inflammatory microRNA miR-155 influences fibrillar beta-Amyloid(1)(-42) catabolism by microglia. Glia 2021, 69, 1736–1748. [Google Scholar] [CrossRef]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Su, W.; Aloi, M.S.; Garden, G.A. MicroRNAs mediating CNS inflammation: Small regulators with powerful potential. Brain Behav. Immun. 2016, 52, 1–8. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Zetterberg, H.; Skillback, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K.; Alzheimer’s Disease Neuroimaging, I. Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol. 2016, 73, 60–67. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Glinge, C.; Clauss, S.; Boddum, K.; Jabbari, R.; Jabbari, J.; Risgaard, B.; Tomsits, P.; Hildebrand, B.; Kaab, S.; Wakili, R.; et al. Stability of Circulating Blood-Based MicroRNAs—Pre-Analytic Methodological Considerations. PLoS ONE 2017, 12, e0167969. [Google Scholar] [CrossRef]
- Coenen-Stass, A.M.L.; Pauwels, M.J.; Hanson, B.; Martin Perez, C.; Conceicao, M.; Wood, M.J.A.; Mager, I.; Roberts, T.C. Extracellular microRNAs exhibit sequence-dependent stability and cellular release kinetics. RNA Biol. 2019, 16, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Dezso, Z.; MacKenzie, C.; Oestreicher, J.; Agoulnik, S.; Byrne, M.; Bernier, F.; Yanagimachi, M.; Aoshima, K.; Oda, Y. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e69807. [Google Scholar] [CrossRef] [PubMed]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.C.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stahler, C.; et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14, R78. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Liu, Q.Y.; Tan, M.S.; Zhang, W.; Hu, N.; Wang, Y.L.; Sun, L.; Jiang, T.; Tan, L. Circulating miR-125b as a biomarker of Alzheimer’s disease. J. Neurol. Sci. 2014, 336, 52–56. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.T.; Tan, M.S.; Liu, Q.Y.; Wang, H.F.; Zhang, W.; Jiang, T.; Tan, L. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 1017–1027. [Google Scholar] [CrossRef]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as potential biomarkers in Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Li, J.; Huang, L.; Chen, X.; Li, D.; Wang, T.; Hu, C.; Xu, J.; Zhang, C.; Zen, K.; et al. Serum MicroRNA Profiles Serve as Novel Biomarkers for the Diagnosis of Alzheimer’s Disease. Dis. Markers 2015, 2015, 625659. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Niida, S. MicroRNA-Seq Data Analysis Pipeline to Identify Blood Biomarkers for Alzheimer’s Disease from Public Data. Biomark. Insights 2015, 10, 21–31. [Google Scholar] [CrossRef]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.N.; Rowe, C.C.; Macaulay, S.L.; Masters, C.L.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry 2015, 20, 1188–1196. [Google Scholar] [CrossRef]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef]
- Yilmaz, S.G.; Erdal, M.E.; Ozge, A.A.; Sungur, M.A. Can Peripheral MicroRNA Expression Data Serve as Epigenomic (Upstream) Biomarkers of Alzheimer’s Disease? OMICS 2016, 20, 456–461. [Google Scholar] [CrossRef]
- Hara, N.; Kikuchi, M.; Miyashita, A.; Hatsuta, H.; Saito, Y.; Kasuga, K.; Murayama, S.; Ikeuchi, T.; Kuwano, R. Serum microRNA miR-501-3p as a potential biomarker related to the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 10. [Google Scholar] [CrossRef]
- Cosin-Tomas, M.; Antonell, A.; Llado, A.; Alcolea, D.; Fortea, J.; Ezquerra, M.; Lleo, A.; Marti, M.J.; Pallas, M.; Sanchez-Valle, R.; et al. Plasma miR-34a-5p and miR-545-3p as Early Biomarkers of Alzheimer’s Disease: Potential and Limitations. Mol. Neurobiol. 2017, 54, 5550–5562. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Vijayan, M.; Reddy, P.H. MicroRNA-455-3p as a potential peripheral biomarker for Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 3808–3822. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Fan, G.; Zhang, J.; Wu, C.; Du, Y.; Ye, H.; Li, Z.; Wang, L.; Zhang, Z.; Zhang, L.; et al. A 9-microRNA Signature in Serum Serves as a Noninvasive Biomarker in Early Diagnosis of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1365–1377. [Google Scholar] [CrossRef]
- Kenny, A.; McArdle, H.; Calero, M.; Rabano, A.; Madden, S.F.; Adamson, K.; Forster, R.; Spain, E.; Prehn, J.H.M.; Henshall, D.C.; et al. Elevated Plasma microRNA-206 Levels Predict Cognitive Decline and Progression to Dementia from Mild Cognitive Impairment. Biomolecules 2019, 9, 734. [Google Scholar] [CrossRef]
- Zhao, X.; Kang, J.; Svetnik, V.; Warden, D.; Wilcock, G.; David Smith, A.; Savage, M.J.; Laterza, O.F. A Machine Learning Approach to Identify a Circulating MicroRNA Signature for Alzheimer Disease. J. Appl. Lab. Med. 2020, 5, 15–28. [Google Scholar] [CrossRef]
- Jia, L.; Zhu, M.; Yang, J.; Pang, Y.; Wang, Q.; Li, T.; Li, F.; Wang, Q.; Li, Y.; Wei, Y. Exosomal MicroRNA-Based Predictive Model for Preclinical Alzheimer’s Disease: A Multicenter Study. Biol. Psychiatry 2022, 92, 44–53. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, C.; Sun, A.; Wang, Y.; Zhou, S. Quantification of microRNA-210 in the cerebrospinal fluid and serum: Implications for Alzheimer’s disease. Exp. Ther. Med. 2015, 9, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.H.; Liu, Y.N. Downregulated serum miR-223 servers as biomarker in Alzheimer’s disease. Cell Biochem. Funct. 2016, 34, 233–237. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Xu, J.; Cheng, J.; Jiao, D.; Zhou, C.; Dai, Y.; Chen, Q. Lower Serum Levels of miR-29c-3p and miR-19b-3p as Biomarkers for Alzheimer’s Disease. Tohoku J. Exp. Med. 2017, 242, 129–136. [Google Scholar] [CrossRef]
- Yang, T.T.; Liu, C.G.; Gao, S.C.; Zhang, Y.; Wang, P.C. The Serum Exosome Derived MicroRNA-135a, -193b, and -384 Were Potential Alzheimer’s Disease Biomarkers. Biomed. Environ. Sci. 2018, 31, 87–96. [Google Scholar]
- Yang, Q.; Zhao, Q.; Yin, Y. miR-133b is a potential diagnostic biomarker for Alzheimer’s disease and has a neuroprotective role. Exp. Ther. Med. 2019, 18, 2711–2718. [Google Scholar] [CrossRef]
- Wang, J.; Chen, C.; Zhang, Y. An investigation of microRNA-103 and microRNA-107 as potential blood-based biomarkers for disease risk and progression of Alzheimer’s disease. J. Clin. Lab. Anal. 2020, 34, e23006. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, S.; Sun, W. Expression of miR-28-3p in patients with Alzheimer’s disease before and after treatment and its clinical value. Exp. Ther. Med. 2020, 20, 2218–2226. [Google Scholar] [CrossRef]
- Sabry, R.; El Sharkawy, R.E.; Gad, N.M. MiRNA -483-5p as a Potential Noninvasive Biomarker for Early Detection of Alzheimer’s Disease. Egypt. J. Immunol. 2020, 27, 59–72. [Google Scholar]
- Liu, Q.; Lei, C. Neuroprotective effects of miR-331-3p through improved cell viability and inflammatory marker expression: Correlation of serum miR-331-3p levels with diagnosis and severity of Alzheimer’s disease. Exp. Gerontol. 2021, 144, 111187. [Google Scholar] [CrossRef]
- Zhang, M.; Han, W.; Xu, Y.; Li, D.; Xue, Q. Serum miR-128 Serves as a Potential Diagnostic Biomarker for Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2021, 17, 269–275. [Google Scholar] [CrossRef]
- Madadi, S.; Saidijam, M.; Yavari, B.; Soleimani, M. Downregulation of serum miR-106b: A potential biomarker for Alzheimer disease. Arch. Physiol. Biochem. 2022, 128, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Tsivinsky, V.G.; Crawford, F.; Mullan, M.J.; Abdullah, L.; Umansky, S.R. Plasma microRNA biomarkers for detection of mild cognitive impairment. Aging 2012, 4, 590–605. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Tsivinsky, V.G.; Abdullah, L.; Crawford, F.; Umansky, S.R. Plasma microRNA biomarkers for detection of mild cognitive impairment: Biomarker validation study. Aging 2013, 5, 925–938. [Google Scholar] [CrossRef]
- Wang, T.; Chen, K.; Li, H.; Dong, S.; Su, N.; Liu, Y.; Cheng, Y.; Dai, J.; Yang, C.; Xiao, S. The feasibility of utilizing plasma MiRNA107 and BACE1 messenger RNA gene expression for clinical diagnosis of amnestic mild cognitive impairment. J. Clin. Psychiatry 2015, 76, 135–141. [Google Scholar] [CrossRef]
- Xie, B.; Liu, Z.; Jiang, L.; Liu, W.; Song, M.; Zhang, Q.; Zhang, R.; Cui, D.; Wang, X.; Xu, S. Increased Serum miR-206 Level Predicts Conversion from Amnestic Mild Cognitive Impairment to Alzheimer’s Disease: A 5-Year Follow-up Study. J. Alzheimers Dis. 2017, 55, 509–520. [Google Scholar] [CrossRef]
- Salama, I.I.; Salama, S.I.; Elmosalami, D.M.; Saleh, R.M.; Rasmy, H.; Ibrahim, M.H.; Kamel, S.A.; Ganem, M.M.F.; Raslan, H.M. Risk Factors Associated with Mild Cognitive Impairment among Apparently Healthy People and the Role of MicroRNAs. Open Access Maced. J. Med. Sci. 2019, 7, 3253–3261. [Google Scholar] [CrossRef]
- He, H.; Liu, A.; Zhang, W.; Yang, H.; Zhang, M.; Xu, H.; Liu, Y.; Hong, B.; Yan, F.; Yue, L.; et al. Novel Plasma miRNAs as Biomarkers and Therapeutic Targets of Alzheimer’s Disease at the Prodromal Stage. J. Alzheimers Dis. 2021, 83, 779–790. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, P.H. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta 2016, 1862, 1617–1627. [Google Scholar] [CrossRef]
- Piscopo, P.; Bellenghi, M.; Manzini, V.; Crestini, A.; Pontecorvi, G.; Corbo, M.; Ortona, E.; Care, A.; Confaloni, A. A Sex Perspective in Neurodegenerative Diseases: microRNAs as Possible Peripheral Biomarkers. Int. J. Mol. Sci. 2021, 22, 4423. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Chen, N. Resveratrol as a Natural Autophagy Regulator for Prevention and Treatment of Alzheimer’s Disease. Nutrients 2017, 9, 927. [Google Scholar] [CrossRef]
- Chakravarthy, M.; Chen, S.; Dodd, P.R.; Veedu, R.N. Nucleic Acid-Based Theranostics for Tackling Alzheimer’s Disease. Theranostics 2017, 7, 3933–3947. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Taniguchi, H.; Suzuki, Y.; Imai, K.; Adachi, Y. Antitumoral RNA-targeted oligonucleotide therapeutics: The third pillar after small molecule inhibitors and antibodies. Cancer Sci. 2022, 113, 2952–2961. [Google Scholar] [CrossRef]
- Padmakumar, S.; D’Souza, A.; Parayath, N.N.; Bleier, B.S.; Amiji, M.M. Nucleic acid therapies for CNS diseases: Pathophysiology, targets, barriers, and delivery strategies. J. Control Release 2022, 352, 121–145. [Google Scholar] [CrossRef]
- Egli, M.; Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [Google Scholar] [CrossRef]
- Rodgers, G.; Austin, C.; Anderson, J.; Pawlyk, A.; Colvis, C.; Margolis, R.; Baker, J. Glimmers in illuminating the druggable genome. Nat. Rev. Drug Discov. 2018, 17, 301–302. [Google Scholar] [CrossRef]
- Dammes, N.; Peer, D. Paving the Road for RNA Therapeutics. Trends Pharmacol. Sci. 2020, 41, 755–775. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Choi, Y.H.; Tu, M.J. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Guo, Y.; Chen, L.; Zhang, X.; Wu, W.; Yang, Z.; Li, X.; Wang, Y.; Hu, Z.; Wang, Z. Co-delivery of phagocytosis checkpoint and STING agonist by a Trojan horse nanocapsule for orthotopic glioma immunotherapy. Theranostics 2022, 12, 5488–5503. [Google Scholar] [CrossRef]
- Ouyang, Q.; Meng, Y.; Zhou, W.; Tong, J.; Cheng, Z.; Zhu, Q. New advances in brain-targeting nano-drug delivery systems for Alzheimer’s disease. J. Drug Target. 2022, 30, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Taliyan, R.; Kakoty, V.; Sarathlal, K.C.; Kharavtekar, S.S.; Karennanavar, C.R.; Choudhary, Y.K.; Singhvi, G.; Riadi, Y.; Dubey, S.K.; Kesharwani, P. Nanocarrier mediated drug delivery as an impeccable therapeutic approach against Alzheimer’s disease. J. Control Release 2022, 343, 528–550. [Google Scholar] [CrossRef]
- Hu, L.; Tao, Y.; Jiang, Y.; Qin, F. Recent progress of nanomedicine in the treatment of Alzheimer’s disease. Front. Cell Dev. Biol. 2023, 11, 1228679. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Chen, L.F.; Wu, W.; Yang, Z.M.; Wang, Y.Z.; Wang, A.Q.; Jiang, S.J.; Qin, X.Z.; Ye, Z.C.; et al. Glioblastoma cell-derived exosomes functionalized with peptides as efficient nanocarriers for synergistic chemotherapy of glioblastoma with improved biosafety. Nano Res. 2023, 1–11. [Google Scholar] [CrossRef]
- Soares Martins, T.; Trindade, D.; Vaz, M.; Campelo, I.; Almeida, M.; Trigo, G.; da Cruz, E.S.O.A.B.; Henriques, A.G. Diagnostic and therapeutic potential of exosomes in Alzheimer’s disease. J. Neurochem. 2021, 156, 162–181. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, F.; Wang, K.; Zhong, Y.; Wei, X.; Wang, Q.; Zhang, H. Engineered Exosomes: A Promising Drug Delivery Strategy for Brain Diseases. Curr. Med. Chem. 2022, 29, 3111–3124. [Google Scholar] [CrossRef]
- Joo, H.S.; Jeon, H.Y.; Hong, E.B.; Kim, H.Y.; Lee, J.M. Exosomes for the diagnosis and treatment of dementia. Curr. Opin. Psychiatry 2023, 36, 119–125. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).