
Drug-Induced Conformational Dynamics of P-Glycoprotein Underlies the Transport of Camptothecin Analogs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effect of Topotecan, Irinotecan, and SN-38 on Pgp-Coupled ATPase Activity
2.2. Determination of Drug-Pgp Binding Affinity by Protein Intrinsic Tryptophan Fluorescence Quenching
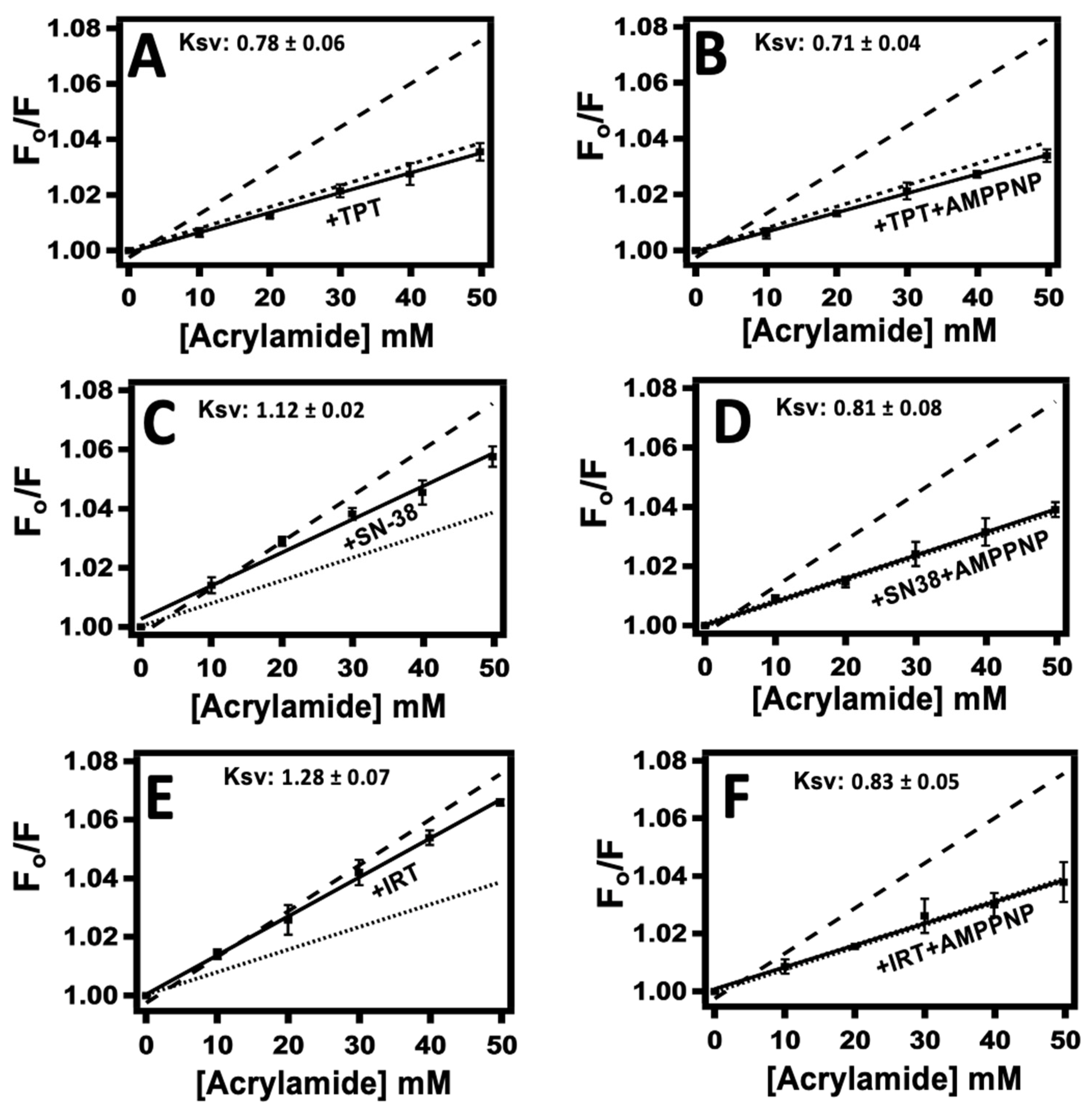
2.3. Probing TPT, SN-38, and IRT-Induced Pgp Conformational Changes Using Acrylamide Quenching of Protein Fluorescence
2.4. Ligand-Receptor Interactions Determined by STDD NMR
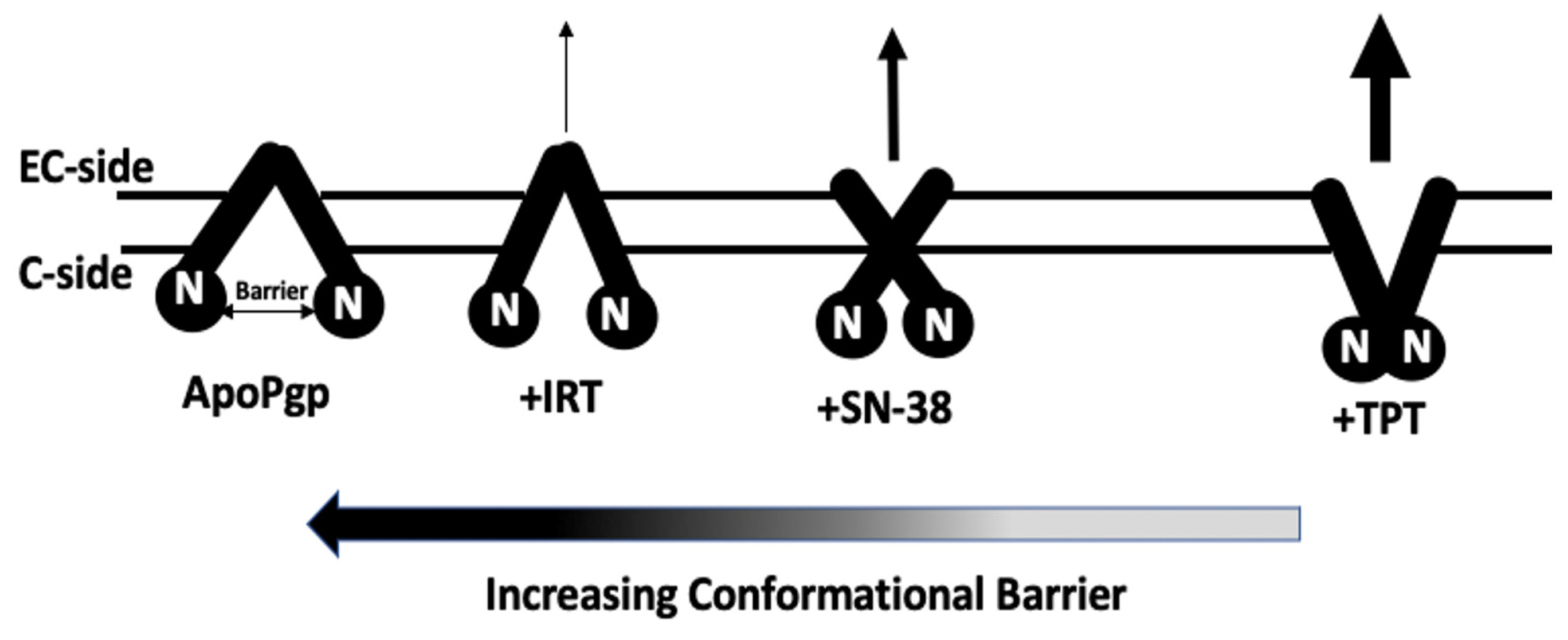
2.5. AFM Analysis of the Impact of Topotecan, SN-38, and Irinotecan on the Conformational Distribution of Pgp
3. Discussion
4. Methods
4.1. Chemical Reagents
4.2. Expression and Purification of P-Glycoprotein
4.3. Reconstitution of Pgp into Liposomes
4.4. Pgp-Mediated ATPase Activity Assay
4.5. Pgp-Drug Binding Affinity Determined by the Quenching of Protein Fluorescence
4.6. Drug-Induced Pgp Conformational Changes Probed Indirectly by Acrylamide Quenching of Protein Fluorescence
4.7. Atomic Force Microscopy Imaging
4.8. Saturation Transfer Double Difference (STDD) NMR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benadiba, M.; Maor, Y. Importance of ABC Transporters in Drug Development. Curr. Pharm. Des. 2016, 22, 5817–5829. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Xia, D.; Zhou, F.; Esser, L. Emerging consensus on the mechanism of polyspecific substrate recognition by the multidrug transporter P-glycoprotein. Cancer Drug Resist. 2019, 2, 471–489. [Google Scholar] [CrossRef]
- Abu-Qare, A.W.; Elmasry, E.; Abou-Donia, M.B. A role for P-glycoprotein in environmental toxicology. J. Toxicol. Environ. Health B Crit. Rev. 2003, 6, 279–288. [Google Scholar] [CrossRef]
- Georgantzopoulou, A.; Skoczyńska, E.; Van den Berg, J.H.J.; Brand, W.; Legay, S.; Klein, S.G.; Rietjens, I.M.; Murk, A.J. P-gp efflux pump inhibition potential of common environmental contaminants determined in vitro. Environ. Toxicol. Chem. 2014, 33, 804–813. [Google Scholar] [CrossRef]
- Durmus, S.; Hendrikx, J.J.M.A.; Schinkel, A.H. Chapter one—Apical ABC Transporters and Cancer Chemotherapeutic Drug Disposition. In Advances in Cancer Research; Schuetz, J.D., Ishikawa, T., Eds.; ABC Transporters and Cancer; Academic Press: Cambridge, MA, USA, 2015; Volume 125, pp. 1–41. [Google Scholar] [CrossRef]
- Fu, D. Where is it and how does it get there—Intracellular localization and traffic of P-glycoprotein. Front. Oncol. 2013, 3, 321. [Google Scholar] [CrossRef] [PubMed]
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): Recent biochemical and structural studies. Adv. Cancer Res. 2015, 125, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Karthika, C.; Sureshkumar, R. P-Glycoprotein Efflux Transporters and Its Resistance Its Inhibitors and Therapeutic Aspects; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Robinson, K.; Tiriveedhi, V. Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front. Oncol. 2020, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-glycoprotein (P-gp) Inducers on Exposure of P-gp Substrates: Review of Clinical Drug–Drug Interaction Studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef] [PubMed]
- FDA. Drug Development and Drug Interactions|Table of Substrates, Inhibitors and Inducers. FDA; Published Online 26 May 2021. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 5 February 2022).
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar] [PubMed]
- Tesauro, C.; Simonsen, A.K.; Andersen, M.B.; Petersen, K.W.; Kristoffersen, E.L.; Algreen, L.; Hansen, N.Y.; Andersen, A.B.; Jakobsen, A.K.; Stougaard, M.; et al. Topoisomerase I activity and sensitivity to camptothecin in breast cancer-derived cells: A comparative study. BMC Cancer 2019, 19, 1158. [Google Scholar] [CrossRef] [PubMed]
- Laco, G.S. Evaluation of Two Models for Human Topoisomerase I Interaction with dsDNA and Camptothecin Derivatives. PLoS ONE 2011, 6, e24314. [Google Scholar] [CrossRef]
- Ando, K.; Shah, A.K.; Sachdev, V.; Kleinstiver, B.P.; Taylor-Parker, J.; Welch, M.M.; Hu, Y.; Salgia, R.; White, F.M.; Parvin, J.D.; et al. Camptothecin resistance is determined by the regulation of topoisomerase I degradation mediated by ubiquitin proteasome pathway. Oncotarget 2017, 8, 43733–43751. [Google Scholar] [CrossRef]
- Kostjukov, V.V. Theoretical analysis of lactone and carboxylate forms of camptothecin in aqueous solution: Electronic states, absorption spectra, and hydration. J. Mol. Liq. 2021, 344, 117804. [Google Scholar] [CrossRef]
- Kacprzak, K.M. Chemistry and Biology of Camptothecin and its Derivatives. In Natural Products; Ramawat, K.G., Mérillon, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 643–682. [Google Scholar] [CrossRef]
- Selvi, B.; Patel, S.; Savva, M. Physicochemical Characterization and Membrane Binding Properties of Camptothecin. J. Pharm. Sci. 2008, 97, 4379–4390. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan—Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef]
- Parvez, M.M.; Basit, A.; Jariwala, P.B.; Gáborik, Z.; Kis, E.; Heyward, S.; Redinbo, M.R.; Prasad, B. Quantitative investigation of irinotecan metabolism, transport and gut microbiome activation. Drug Metab. Dispos. 2021, 49, 683–693. [Google Scholar] [CrossRef]
- Mao, Q.; Lai, Y.; Wang, J. Drug Transporters in Xenobiotic Disposition and Pharmacokinetic Prediction. Drug Metab. Dispos. 2018, 46, 561–566. [Google Scholar] [CrossRef]
- Kawahara, I.; Nishikawa, S.; Yamamoto, A.; Kono, Y.; Fujita, T. The Impact of Breast Cancer Resistance Protein (BCRP/ABCG2) on Drug Transport Across Caco-2 Cell Monolayers. Drug Metab. Dispos. 2020, 48, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kang, J.; Kwon, N.-Y.; Sivaraman, A.; Naik, R.; Jin, S.-Y.; Oh, A.R.; Shin, J.-H.; Na, Y.; Lee, K.; et al. Dual Inhibition of P-gp and BCRP Improves Oral Topotecan Bioavailability in Rodents. Pharmaceutics 2021, 13, 559. [Google Scholar] [CrossRef]
- Zhao, Y.; Wan, B.; Zhang, T.; Xu, Y.; Liu, H.; Lv, T.; Zhang, F.; Zhan, P.; Song, Y. Irinotecan, topotecan, paclitaxel or docetaxel for second-line treatment of small cell lung cancer: A single-center retrospective study of efficiency comparation and prognosis analysis. Transl. Lung Cancer Res. 2019, 8, 829–837. [Google Scholar] [CrossRef]
- Houghton, P.J.; Cheshire, P.J.; Hallman, J.D.; Lutz, L.; Friedman, H.S.; Danks, M.K.; Houghton, J.A. Efficacy of topoisomerase I inhibitors, topotecan and irinotecan, administered at low dose levels in protracted schedules to mice bearing xenografts of human tumors. Cancer Chemother. Pharmacol. 1995, 36, 393–403. [Google Scholar] [CrossRef]
- Joto, N.; Ishii, M.; Minami, M.; Kuga, H.; Mitsui, I.; Tohgo, A. DX-8951F, a water-soluble camptothecin analog, exhibits potent antitumor activity against a human lung cancer cell line and its SN-38-resistant variant. Int. J. Cancer 1997, 72, 680–686. [Google Scholar] [CrossRef]
- Hirose, K.; Kozu, C.; Yamashita, K.; Maruo, E.; Kitamura, M.; Hasegawa, J.; Omoda, K.; Murakami, T.; Maeda, Y. Correlation between plasma concentration ratios of SN-38 glucuronide and SN-38 and neutropenia induction in patients with colorectal cancer and wild-type UGT1A1 gene. Oncol. Lett. 2012, 3, 694–698. [Google Scholar] [CrossRef]
- Lin, X.; Skolnik, S.; Chen, X.; Wang, J. Attenuation of Intestinal Absorption by Major Efflux Transporters: Quantitative Tools and Strategies Using a Caco-2 Model. Drug Metab. Dispos. 2011, 39, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Salphati, L.; Plise, E.G.; Li, G. Expression and activity of the efflux transporters ABCB1, ABCC2 and ABCG2 in the human colorectal carcinoma cell line LS513. Eur. J. Pharm. Sci. 2009, 37, 463–468. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Ambudkar, S.V. About a switch: How P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 2007, 6, 13–23. [Google Scholar] [CrossRef]
- Patzlaff, J.S.; van der Heide, T.; Poolman, B. The ATP/Substrate Stoichiometry of the ATP-binding Cassette (ABC) Transporter OpuA. J. Biol. Chem. 2003, 278, 29546–29551. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Müller, M.; Peng, X.H.; Ambudkar, S.V. Importance of the Conserved Walker B Glutamate Residues, 556 and 1201, for the Completion of the Catalytic Cycle of ATP Hydrolysis by Human P-glycoprotein (ABCB1). Biochemistry 2002, 41, 13989–14000. [Google Scholar] [CrossRef]
- Litman, T.; Zeuthen, T.; Skovsgaard, T.; Stein, W.D. Structure-activity relationships of P-glycoprotein interacting drugs: Kinetic characterization of their effects on ATPase activity. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1997, 1361, 159–168. [Google Scholar] [CrossRef]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Cardarelli, C.O.; Pashinsky, I.; Stein, W.D. Relation Between the Turnover Number for Vinblastine Transport and for Vinblastine-stimulated ATP Hydrolysis by Human P-glycoprotein. J. Biol. Chem. 1997, 272, 21160–21166. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.M.; Sauna, Z.E.; Ambudkar, S.V. Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J. Biol. Chem. 2001, 276, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Li-Blatter, X.; Beck, A.; Seelig, A. P-Glycoprotein-ATPase Modulation: The Molecular Mechanisms. Biophys. J. 2012, 102, 1383–1393. [Google Scholar] [CrossRef]
- Borgnia, M.J.; Eytan, G.D.; Assaraf, Y.G. Competition of hydrophobic peptides, cytotoxic drugs, and chemosensitizers on a common P-glycoprotein pharmacophore as revealed by its ATPase activity. J. Biol. Chem. 1996, 271, 3163–3171. [Google Scholar] [CrossRef]
- Ritchie, T.K.; Grinkova, Y.V.; Bayburt, T.H.; Denisov, I.G.; Zolnerciks, J.K.; Atkins, W.M.; Sligar, S.G. Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009, 464, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Wilt, L.A.; Nguyen, D.; Roberts, A.G. Insights into the molecular mechanism of triptan transport by P-glycoprotein. J. Pharm. Sci. 2017, 106, 1670–1679. [Google Scholar] [CrossRef]
- Li, H.; Jin, H.-E.; Kim, W.; Han, Y.-H.; Kim, D.-D.; Chung, S.-J.; Shim, C.-K. Involvement of P-glycoprotein, multidrug resistance protein 2 and breast cancer resistance protein in the transport of belotecan and topotecan in Caco-2 and MDCKII cells. Pharm Res. 2008, 25, 2601–2612. [Google Scholar] [CrossRef]
- Anglicheau, D.; Pallet, N.; Rabant, M.; Marquet, P.; Cassinat, B.; Méria, P.; Beaune, P.; Legendre, C.; Thervet, E. Role of P-glycoprotein in cyclosporine cytotoxicity in the cyclosporine–sirolimus interaction. Kidney Int. 2006, 70, 1019–1025. [Google Scholar] [CrossRef]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef]
- Srivalli, K.M.R.; Lakshmi, P.K. Overview of P-glycoprotein inhibitors: A rational outlook. Braz. J. Pharm. Sci. 2012, 48, 353–367. [Google Scholar] [CrossRef]
- Gayer, A.V.; Yakimov, B.P.; Budylin, G.S.; Shirshin, E.A. Evaluating the Number of Ligand Binding Sites on Protein from Tryptophan Fluorescence Quenching under Typical Experimental Conditions. J. Biomed. Photonics Eng. 2020, 6, 020303. [Google Scholar] [CrossRef]
- Yammine, A.; Gao, J.; Kwan, A.H. Tryptophan Fluorescence Quenching Assays for Measuring Protein-ligand Binding Affinities: Principles and a Practical Guide. Bio-Protocol 2019, 9, e3253. [Google Scholar] [CrossRef]
- Campbell, K.; Zappas, A.; Bunz, U.; Thio, Y.S.; Bucknall, D.G. Fluorescence quenching of a poly(para-phenylene ethynylenes) by C60 fullerenes. J. Photochem. Photobiol. A Chem. 2012, 249, 41–46. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2011. [Google Scholar]
- Ward, A.; Reyes, C.L.; Yu, J.; Roth, C.B.; Chang, G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA 2007, 104, 19005–19010. [Google Scholar] [CrossRef]
- Sonveaux, N.; Vigano, C.; Shapiro, A.B.; Ling, V.; Ruysschaert, J.M. Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. A tryptophan fluorescence quenching analysis. J. Biol. Chem. 1999, 274, 17649–17654. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, M.E.; Mok, L.; Swartz, D.J.; Singh, A.; Fendley, G.A.; Urbatsch, I.L.; Altenberg, G.A. Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer-associated P-glycoprotein during ATP hydrolysis. J. Biol. Chem. 2017, 292, 20412–20424. [Google Scholar] [CrossRef]
- Akbar, S.M.D.; Sreeramulu, K.; Sharma, H.C. Tryptophan fluorescence quenching as a binding assay to monitor protein conformation changes in the membrane of intact mitochondria. J. Bioenerg. Biomembr. 2016, 48, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E.; Wilt, L.A.; Ledwitch, K.V.; Roberts, A.G. A conformationally-gated model of methadone and loperamide transport by P-glycoprotein. J. Pharm. Sci. 2018, 107, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Jazaj, D.; Ghadami, S.A.; Bemporad, F.; Chiti, F. Probing conformational changes of monomeric transthyretin with second derivative fluorescence. Sci. Rep. 2019, 9, 10988. [Google Scholar] [CrossRef]
- Liu, R.; Siemiarczuk, A.; Sharom, F.J. Intrinsic fluorescence of the P-glycoprotein multidrug transporter: Sensitivity of tryptophan residues to binding of drugs and nucleotides. Biochemistry 2000, 39, 14927–14938. [Google Scholar] [CrossRef] [PubMed]
- Moller, M.; Denicola, A. Protein tryptophan accessibility studied by fluorescence quenching. Biochem. Mol. Biol. Edu. 2002, 30, 175–178. [Google Scholar] [CrossRef]
- Ledwitch, K.V.; Barnes, R.W.; Roberts, A.G. Unravelling the complex drug-drug interactions of the cardiovascular drugs, verapamil and digoxin, with P-glycoprotein. Biosci. Rep. 2016, 36, e00309. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Mukhopadhyay, C.; Chakrabarti, A. Probing Conformational Stability and Dynamics of Erythroid and Nonerythroid Spectrin: Effects of Urea and Guanidine Hydrochloride. PLoS ONE 2015, 10, e0116991. [Google Scholar] [CrossRef]
- Strambini, G.B.; Gonnelli, M. Fluorescence Quenching of Buried Trp Residues by Acrylamide Does Not Require Penetration of the Protein Fold. J. Phys. Chem. B 2010, 114, 1089–1093. [Google Scholar] [CrossRef]
- Russell, P.L.; Sharom, F.J. Conformational and functional characterization of trapped complexes of the P-glycoprotein multidrug transporter. Biochem. J. 2006, 399, 315–323. [Google Scholar] [CrossRef]
- Siarheyeva, A.; Liu, R.; Sharom, F.J. Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: Implications for catalysis. J. Biol. Chem. 2010, 285, 7575–7586. [Google Scholar] [CrossRef]
- Barbieri, A.; Thonghin, N.; Shafi, T.; Prince, S.M.; Collins, R.F.; Ford, R.C. Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor. Membranes 2021, 11, 923. [Google Scholar] [CrossRef]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.-K.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. Publ. Protein Soc. 2014, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Lee, S.C.; Tao, H.; Speir, J.A.; Chang, G.; Urbatsch, I.L.; Potter, C.S.; Carragher, B.; Zhang, Q. Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 2015, 23, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Cryst. D 2015, 71, 732–741. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef]
- Monaco, S.; Tailford, L.E.; Juge, N.; Angulo, J. Differential Epitope Mapping by STD NMR Spectroscopy To Reveal the Nature of Protein–Ligand Contacts. Angew. Chem. Int. Ed. Engl. 2017, 56, 15289–15293. [Google Scholar] [CrossRef]
- Xu, Y.D.; Lai, R.Y.; Procházková, E.; Stenzel, M.H. Saturation Transfer Difference NMR Spectroscopy for the Elucidation of Supramolecular Albumin–Polymer Interactions. ACS Macro Lett. 2021, 10, 819–824. [Google Scholar] [CrossRef]
- D’Amelio, N.; Aroulmoji, V.; Toraldo, A.; Sundaraganesan, N.; Anbarasan, P.M. Aggregation properties and structural studies of anticancer drug Irinotecan in DMSO solution based on NMR measurements. J. Mol. Struct. 2012, 1013, 26–35. [Google Scholar] [CrossRef]
- di Nunzio, M.R.; Wang, Y.; Douhal, A. Spectroscopy and dynamics of topotecan anti-cancer drug comprised within cyclodextrins. J. Photochem. Photobiol. A Chem. 2013, 266, 12–21. [Google Scholar] [CrossRef]
- Hyz, K.; Kawȩcki, R.; Bednarek, E.; Bocian, W.; Sitkowski, J.; Kozerski, L. Topotecan dynamics, tautomerism and reactivity—1H/13C NMR and ESI MS study. Magn. Reson. Chem. 2010, 48, 575–584. [Google Scholar] [CrossRef]
- Dufrêne, Y.F.; Ando, T.; Garcia, R.; Alsteens, D.; Martinez-Martin, D.; Engel, A.; Gerber, C.; Müller, D.J. Imaging modes of atomic force microscopy for application in molecular and cell biology. Nat. Nanotechnol. 2017, 12, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Pinho, A.C.; Piedade, A.P. Zeta Potential, Contact Angles, and AFM Imaging of Protein Conformation Adsorbed on Hybrid Nanocomposite Surfaces. ACS Appl. Mater. Interfaces 2013, 5, 8187–8194. [Google Scholar] [CrossRef] [PubMed]
- Sigdel, K.P.; Wilt, L.A.; Marsh, B.P.; Roberts, A.G.; King, G.M. The conformation and dynamics of P-glycoprotein in a lipid bilayer investigated by atomic force microscopy. Biochem. Pharmacol. 2018, 156, 302–311. [Google Scholar] [CrossRef]
- Kondrashov, D.A.; Van Wynsberghe, A.W.; Bannen, R.M.; Cui, Q.; Phillips, G.N. Protein Structural Variation in Computational Models and Crystallographic Data. Structure 2007, 15, 169–177. [Google Scholar] [CrossRef]
- Li, M.; Dang, D.; Liu, L.; Xi, N.; Wang, Y. Imaging and Force Recognition of Single Molecular Behaviors Using Atomic Force Microscopy. Sensors 2017, 17, 200. [Google Scholar] [CrossRef]
- Al-Shawi, M.K.; Omote, H. The remarkable transport mechanism of P-glycoprotein; a multidrug transporter. J. Bioenerg. Biomembr. 2005, 37, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.K.; Kwon, H.; Atkins, W.M. Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid Nanodiscs and inhibition by the antibodies MRK16 and UIC2. J. Biol. Chem. 2011, 286, 39489–39496. [Google Scholar] [CrossRef]
- Verhalen, B.; Ernst, S.; Börsch, M.; Wilkens, S. Dynamic Ligand-induced Conformational Rearrangements in P-glycoprotein as Probed by Fluorescence Resonance Energy Transfer Spectroscopy. J. Biol. Chem. 2012, 287, 1112–1127. [Google Scholar] [CrossRef]
- Safa, A.R. Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr. Med. Chem. Anticancer Agents 2004, 4, 1–17. [Google Scholar] [CrossRef]
- Chufan, E.E.; Kapoor, K.; Sim, H.-M.; Singh, S.; Talele, T.T.; Durell, S.R.; Ambudkar, S.V. Multiple Transport-Active Binding Sites Are Available for a Single Substrate on Human P-Glycoprotein (ABCB1). PLoS ONE 2013, 8, e82463. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Ling, V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997, 250, 130–137. [Google Scholar] [CrossRef]
- Frank, G.A.; Shukla, S.; Rao, P.; Borgnia, M.J.; Bartesaghi, A.; Merk, A.; Mobin, A.; Esser, L.; Earl, L.A.; Gottesman, M.M.; et al. Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol. Pharmacol. 2016, 90, 35–41. [Google Scholar] [CrossRef]
- O’Mara, M.L.; Mark, A.E. Structural characterization of two metastable ATP-bound states of P-glycoprotein. PLoS ONE 2014, 9, e91916. [Google Scholar] [CrossRef]
- Futamata, R.; Ogasawara, F.; Ichikawa, T.; Kodan, A.; Kimura, Y.; Kioka, N.; Ueda, K. In vivo FRET analyses reveal a role of ATP hydrolysis–associated conformational changes in human P-glycoprotein. J. Biol. Chem. 2020, 295, 5002–5011. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Sigdel, K.P.; Schaefer, K.G.; Mensah, G.A.K.; King, G.M.; Roberts, A.G. The effects of anthracycline drugs on the conformational distribution of mouse P-glycoprotein explains their transport rate differences. Biochem. Pharmacol. 2020, 174, 113813. [Google Scholar] [CrossRef]
- Pan, L.; Aller, S.G. Allosteric Role of Substrate Occupancy toward the Alignment of P-glycoprotein Nucleotide Binding Domains. Sci. Rep. 2018, 8, 14643. [Google Scholar] [CrossRef]
- Garrigues, A.; Escargueil, A.E.; Orlowski, S. The multidrug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. Proc. Natl. Acad. Sci. USA 2002, 99, 10347–10352. [Google Scholar] [CrossRef]
- Tran, N.N.B.; Bui, A.T.A.; Jaramillo-Martinez, V.; Weber, J.; Zhang, Q.; Urbatsch, I.L. Lipid environment determines the drug-stimulated ATPase activity of P-glycoprotein. Front. Mol. Biosci. 2023, 10, 1141081. [Google Scholar] [CrossRef] [PubMed]
- Al-Shawi, M.K.; Polar, M.K.; Omote, H.; Figler, R.A. Transition State Analysis of the Coupling of Drug Transport to ATP Hydrolysis by P-glycoprotein. J. Biol. Chem. 2003, 278, 52629–52640. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: Its role in modulating protein function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; Kapoor, K.; Tajkhorshid, E. Probing cholesterol binding and translocation in P-glycoprotein. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183090. [Google Scholar] [CrossRef]
- Ahmed Juvale, I.I.; Abdul Hamid, A.A.; Abd Halim, K.B.; Che Has, A.T. P-glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777. [Google Scholar] [CrossRef]
- Bársony, O.; Szalóki, G.; Türk, D.; Tarapcsák, S.; Gutay-Tóth, Z.; Bacsó, Z.; Holb, I.J.; Székvölgyi, L.; Szabó, G.; Csanády, L.; et al. A single active catalytic site is sufficient to promote transport in P-glycoprotein. Sci. Rep. 2016, 6, 24810. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.M. Camptothecin: Roles of the D and E rings in binding to the topoisomerase I-DNA covalent binary complex. Curr. Med. Chem. Anticancer Agents 2005, 5, 353–362. [Google Scholar] [CrossRef]
- Lerner-Marmarosh, N.; Gimi, K.; Urbatsch, I.L.; Gros, P.; Senior, A.E. Large scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, walker A, and walker B mutant proteins. J. Biol. Chem. 1999, 274, 34711–34718. [Google Scholar] [CrossRef] [PubMed]
- Urbatsch, I.L.; Beaudet, L.; Carrier, I.; Gros, P. Mutations in either nucleotide-binding site of P-glycoprotein (Mdr3) prevent vanadate trapping of nucleotide at both sites. Biochemistry 1998, 37, 4592–4602. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.; Wobbe, C.R. Preparation of protein extracts from yeast. Curr. Protoc. Mol. Biol. 1993, 23, 13.13.1–13.13.9. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Swartz, D.J.; Protasevich, I.I.; Brouillette, C.G.; Harrell, P.M.; Hildebrandt, E.; Gasser, B.; Mattanovich, D.; Ward, A.; Chang, G.; et al. A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLoS ONE 2011, 6, e22577. [Google Scholar] [CrossRef]
- Chifflet, S.; Torriglia, A.; Chiesa, R.; Tolosa, S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: Application to lens ATPases. Anal. Biochem. 1988, 168, 1–4. [Google Scholar] [CrossRef]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; Wiley-Interscience: Hoboken, NJ, USA, 1993. [Google Scholar]
- Sharom, F.J.; Russell, P.L.; Qu, Q.; Lu, P. Fluorescence techniques for studying membrane transport proteins: The P-glycoprotein multidrug transporter. Methods Mol. Biol. 2003, 227, 109–128. [Google Scholar] [CrossRef] [PubMed]
- Cioni, P.; Strambini, G.B. Acrylamide Quenching of Protein Phosphorescence as a Monitor of Structural Fluctuations in the Globular Fold. J. Am. Chem. Soc. 1998, 120, 11749–11757. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. Functional characterization of Escherichia coli MsbA: Interaction with nucleotides and substrates. J. Biol. Chem. 2008, 283, 12840–12850. [Google Scholar] [CrossRef] [PubMed]
- Vahabi, S.; Nazemi Salman, B.; Javanmard, A. Atomic Force Microscopy Application in Biological Research: A Review Study. Iran. J. Med. Sci. 2013, 38, 76–83. [Google Scholar]
- Marsh, B.P.; Chada, N.; Gari, R.R.S.; Sigdel, K.P.; King, G.M. The hessian blob algorithm: Precise particle detection in atomic force microscopy imagery. Sci. Rep. 2018, 8, 978. [Google Scholar] [CrossRef]
- Chu, C.Y.; Henderson, D.J.; Parmeter, C.F. On discrete Epanechnikov kernel functions. Comput. Stat. Data Anal. 2017, 116, 79–105. [Google Scholar] [CrossRef]
- Schaefer, K.G.; Roberts, A.G.; King, G.M. Advantages and potential limitations of applying AFM kymograph analysis to pharmaceutically relevant membrane proteins in lipid bilayers. Sci. Rep. 2023, 13, 11427. [Google Scholar] [CrossRef]
- Viegas, A.; Manso, J.; Nobrega, F.L.; Cabrita, E.J. Saturation-Transfer Difference (STD) NMR: A Simple and Fast Method for Ligand Screening and Characterization of Protein Binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Roberts, A.G.; Sjogren, S.E.; Fomina, N.; Vu, K.T.; Almutairi, A.; Halpert, J.R. NMR-derived models of amidopyrine and its metabolites in complexes with rabbit cytochrome P450 2B4 reveal a structural mechanism of sequential N-dealkylation. Biochemistry 2011, 50, 2123–2134. [Google Scholar] [CrossRef]
- Claasen, B.; Axmann, M.; Meinecke, R.; Meyer, B. Direct observation of ligand binding to membrane proteins in living cells by a saturation transfer double difference (STDD) NMR spectroscopy method shows a significantly higher affinity of integrin αIIbβ3 in native platelets than in liposomes. J. Am. Chem. Soc. 2005, 127, 916–919. [Google Scholar] [CrossRef]
- Haselhorst, T.; Munster-Kuhnel, A.K.; Oschlies, M.; Tiralongo, J.; Gerardy-Schahn, R.; von Itzstein, M. Direct detection of ligand binding to Sepharose-immobilised protein using saturation transfer double difference (STDD) NMR spectroscopy. Biochem. Biophys. Res. Commun. 2007, 359, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Venkitakrishnan, R.P.; Benard, O.; Max, M.; Markley, J.L.; Assadi-Porter, F.M. Use of NMR saturation transfer difference spectroscopy to study ligand binding to membrane proteins. Membr. Protein Struct. Dyn. Methods Protoc. 2012, 914, 47–63. [Google Scholar]
- Piotto, M.; Saudek, V.; Sklenář, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Mao, X.A.; Ye, C.H.; Huang, H.; Nicholson, J.K.; Lindon, J.C. Improved WATERGATE pulse sequences for solvent suppression in NMR spectroscopy. J. Magn. Reson. 1998, 132, 125–129. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mensah, G.A.K.; Schaefer, K.G.; Bartlett, M.G.; Roberts, A.G.; King, G.M. Drug-Induced Conformational Dynamics of P-Glycoprotein Underlies the Transport of Camptothecin Analogs. Int. J. Mol. Sci. 2023, 24, 16058. https://doi.org/10.3390/ijms242216058
Mensah GAK, Schaefer KG, Bartlett MG, Roberts AG, King GM. Drug-Induced Conformational Dynamics of P-Glycoprotein Underlies the Transport of Camptothecin Analogs. International Journal of Molecular Sciences. 2023; 24(22):16058. https://doi.org/10.3390/ijms242216058
Chicago/Turabian StyleMensah, Gershon A. K., Katherine G. Schaefer, Michael G. Bartlett, Arthur G. Roberts, and Gavin M. King. 2023. "Drug-Induced Conformational Dynamics of P-Glycoprotein Underlies the Transport of Camptothecin Analogs" International Journal of Molecular Sciences 24, no. 22: 16058. https://doi.org/10.3390/ijms242216058
APA StyleMensah, G. A. K., Schaefer, K. G., Bartlett, M. G., Roberts, A. G., & King, G. M. (2023). Drug-Induced Conformational Dynamics of P-Glycoprotein Underlies the Transport of Camptothecin Analogs. International Journal of Molecular Sciences, 24(22), 16058. https://doi.org/10.3390/ijms242216058