

Animal Models for the Study of Gaucher Disease

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
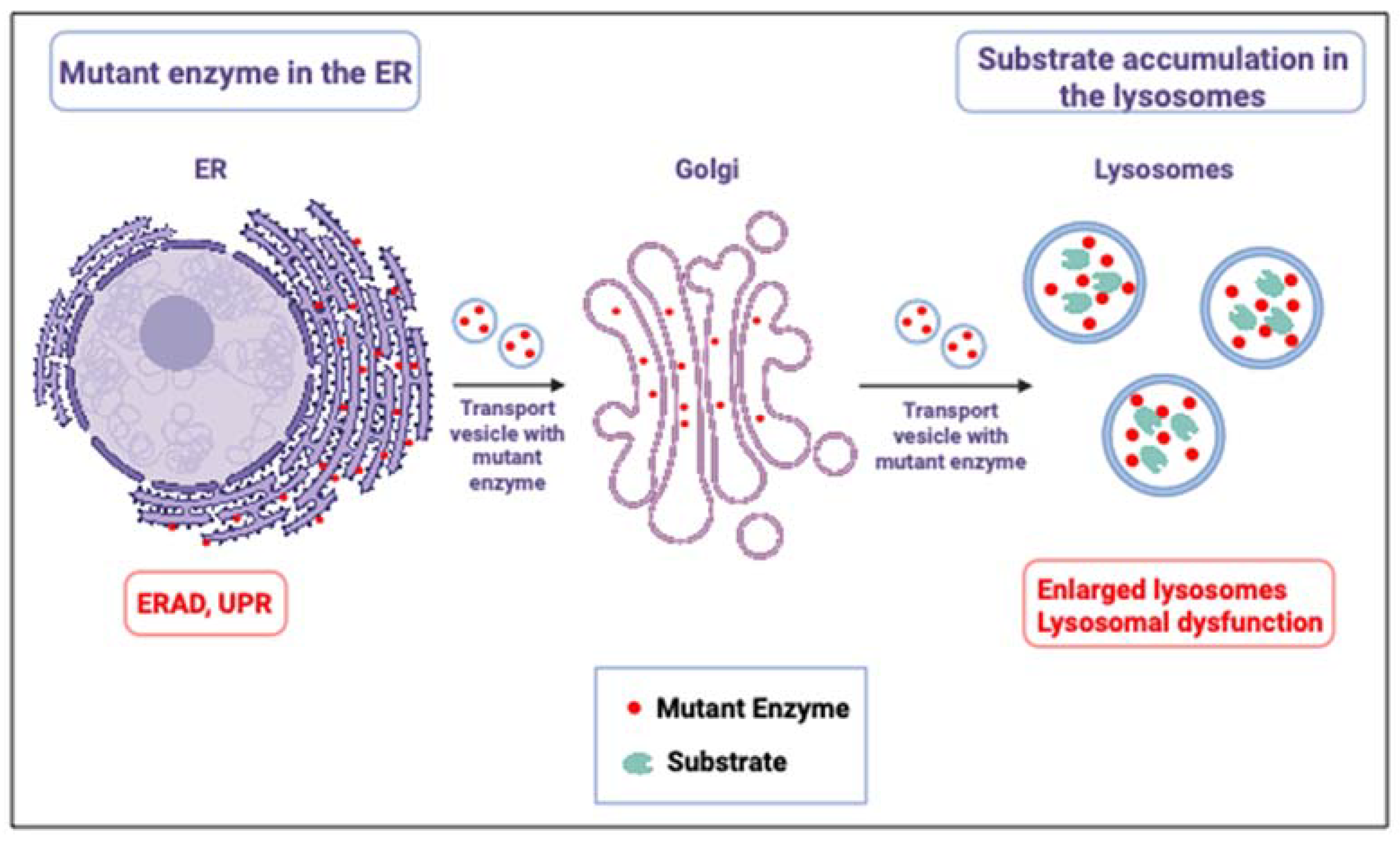
2. Mouse Models for Gaucher Disease
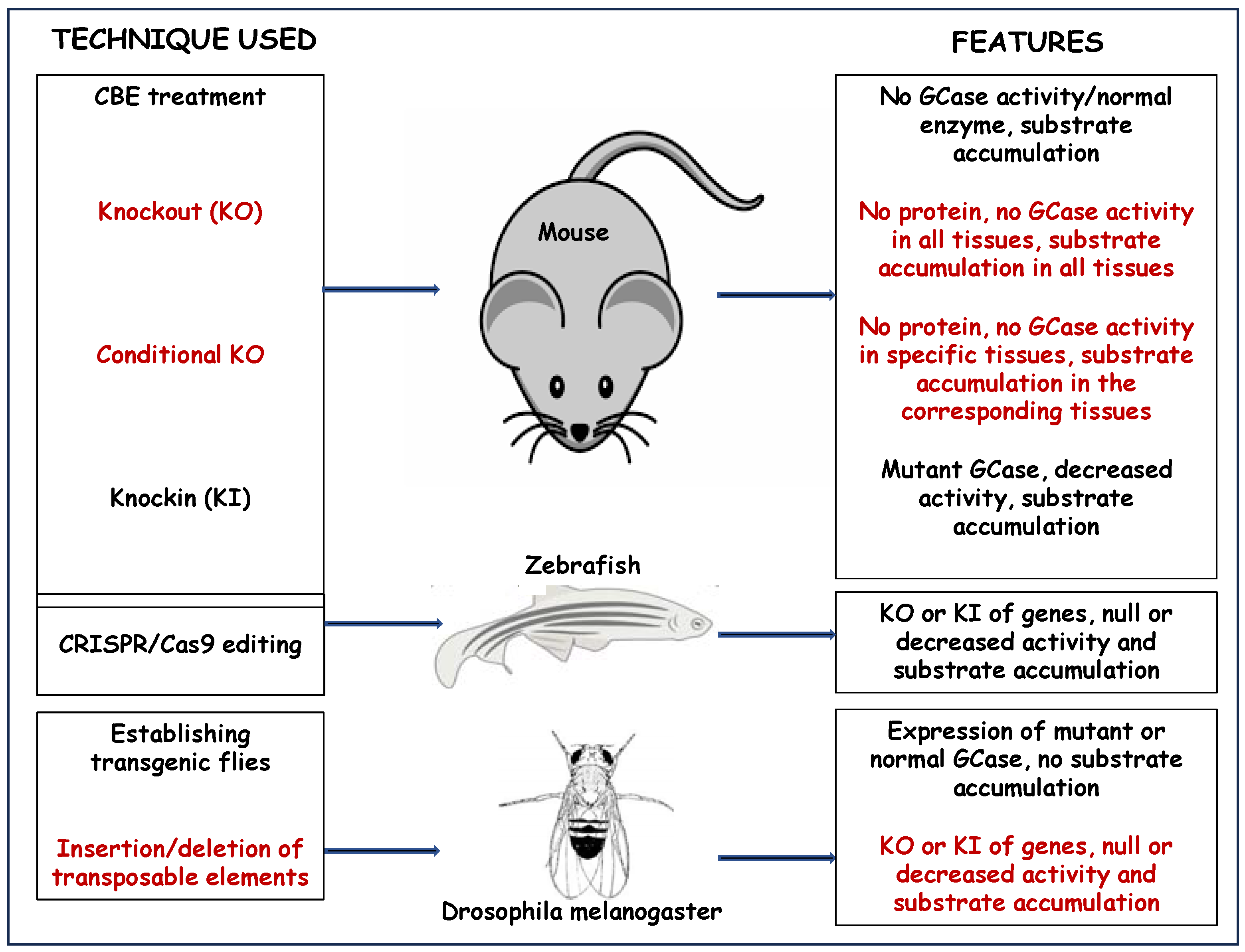
2.1. Chemically Induced Mice Models
2.2. Chimeric Mice Models
2.3. Knockout (KO) and Conditional Knockout GD Models
2.3.1. KO Mice Models
2.3.2. Conditional KO Mice Models
2.4. Knockin (KI) Mice Models
3. Fish Models
3.1. Pharmacological Zebrafish Models of Gaucher Disease
3.2. Genetic Models of Gaucher Disease
4. Medaka Fish Models of Gaucher Disease
5. Fly Models
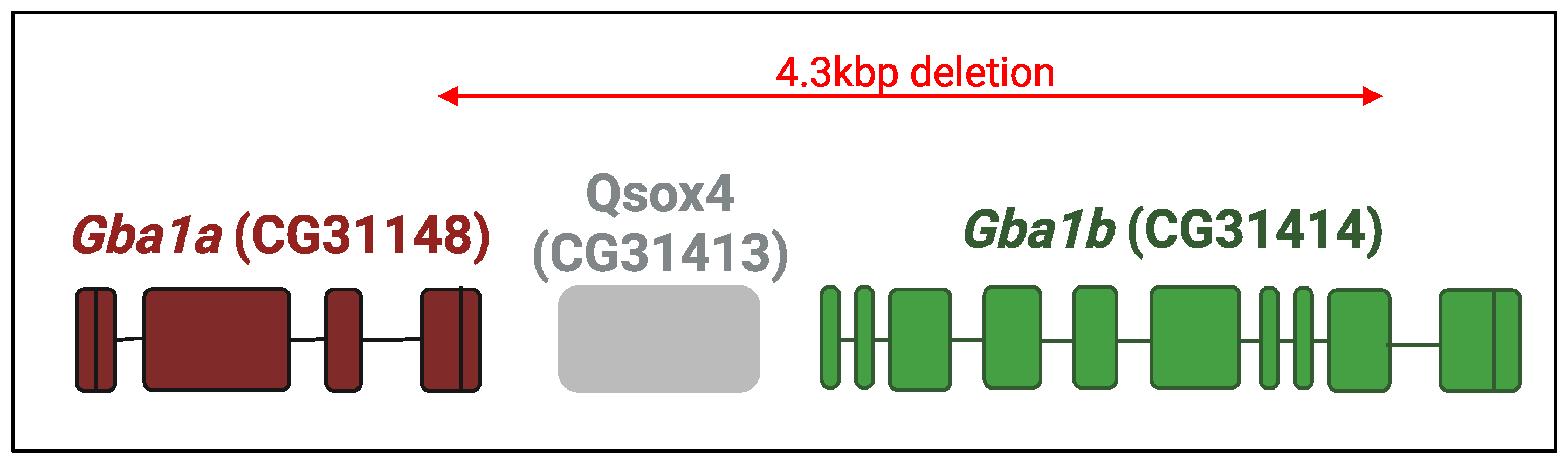
5.1. KO Drosophila Models
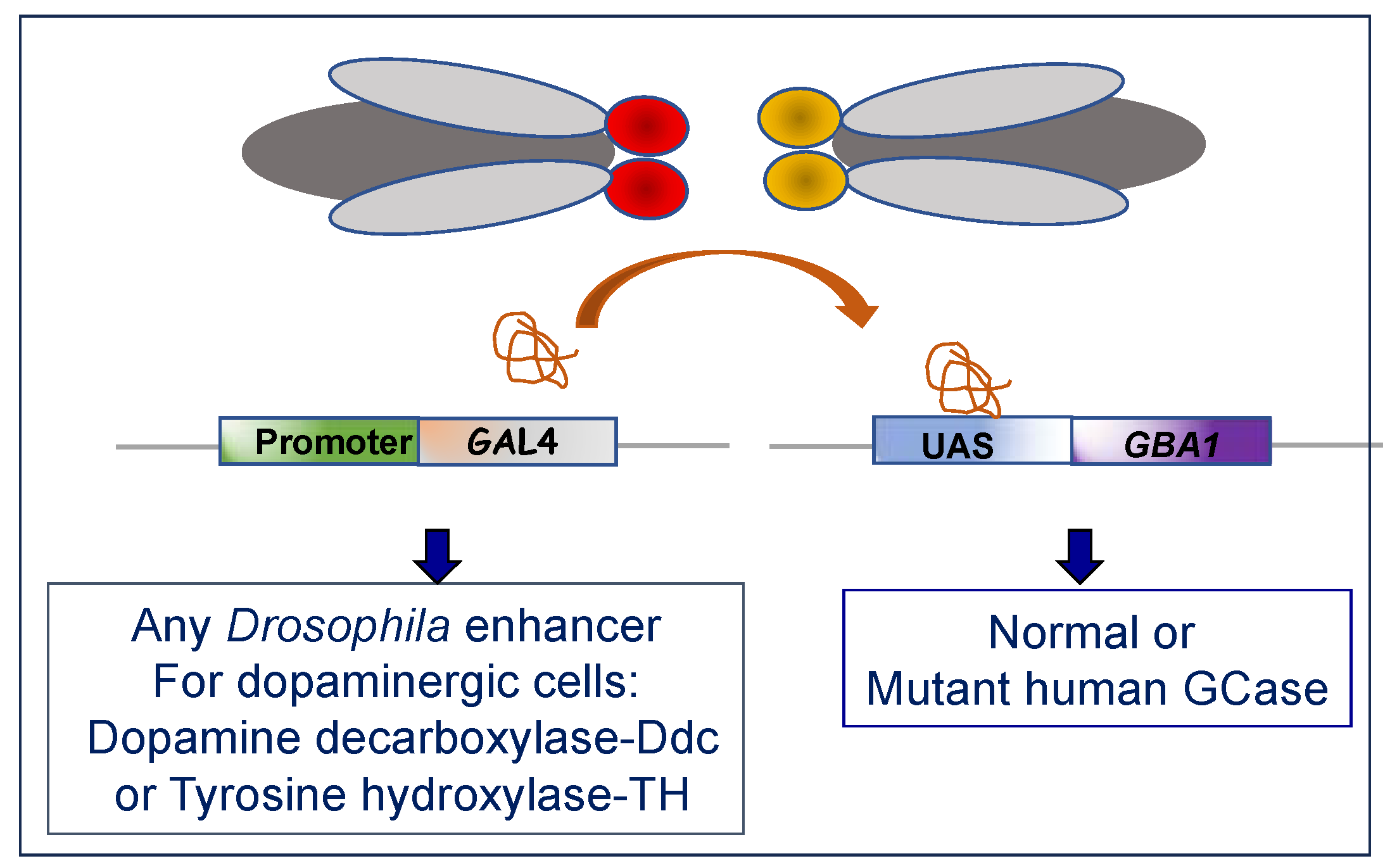
5.2. KI Drosophila Models
6. Other Animal Models of GD
6.1. Canine Model
6.2. Ovine Model
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neufeld, E.F. Lysosomal storage diseases. Annu. Rev. Biochem. 1991, 60, 257–280. [Google Scholar] [CrossRef]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef]
- Lieberman, R.L.; Wustman, B.A.; Huertas, P.; Powe, A.C., Jr.; Pine, C.W.; Khanna, R.; Schlossmacher, M.G.; Ringe, D.; Petsko, G.A. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 2007, 3, 101–107. [Google Scholar] [CrossRef]
- Brumshtein, B.; Aguilar-Moncayo, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Silman, I.; Shaaltiel, Y.; Aviezer, D.; Sussman, J.L.; Futerman, A.H. 6-Amino-6-deoxy-5,6-di-N-(N’-octyliminomethylidene)nojirimycin: Synthesis, biological evaluation, and crystal structure in complex with acid beta-glucosidase. Chembiochem 2009, 10, 1480–1485. [Google Scholar] [CrossRef]
- Kallemeijn, W.W.; Witte, M.D.; Voorn-Brouwer, T.M.; Walvoort, M.T.; Li, K.Y.; Codée, J.D.; van der Marel, G.A.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.M. A sensitive gel-based method combining distinct cyclophellitol-based probes for the identification of acid/base residues in human retaining β-glucosidases. J. Biol. Chem. 2014, 289, 35351–35362. [Google Scholar] [CrossRef]
- Tamaru, T.; Fujibayashi, S.; Brown, W.R.; Wenger, D.A. Immunocytochemical localization of sphingolipid activator protein-1, the sulfatide/GM1 ganglioside activator, to lysosomes in human liver and colon. Histochemistry 1986, 86, 195–200. [Google Scholar] [CrossRef]
- Vielhaber, G.; Hurwitz, R.; Sandhoff, K. Biosynthesis, processing, and targeting of sphingolipid activator protein (SAP) precursor in cultured human fibroblasts. Mannose 6-phosphate receptor-independent endocytosis of SAP precursor. J. Biol. Chem. 1996, 271, 32438–32446. [Google Scholar] [CrossRef]
- Leonova, T.; Qi, X.; Bencosme, A.; Ponce, E.; Sun, Y.; Grabowski, G.A. Proteolytic processing patterns of prosaposin in insect and mammalian cells. J. Biol. Chem. 1996, 271, 17312–17320. [Google Scholar] [CrossRef]
- Hineno, T.; Sano, A.; Kondoh, K.; Ueno, S.; Kakimoto, Y.; Yoshida, K. Secretion of sphingolipid hydrolase activator precursor, prosaposin. Biochem. Biophys. Res. Commun. 1991, 176, 668–674. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Carson, G.S.; Seo, H.C.; Hiraiwa, M.; Weiler, S.; Tomich, J.M.; Barranger, J.A.; Kahn, M.; Azuma, N.; Kishimoto, Y. Identification of the neurotrophic factor sequence of prosaposin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 681–685. [Google Scholar] [CrossRef]
- Tian, R.; Abarientos, A.; Hong, J.; Hashemi, S.H.; Yan, R.; Drager, N.; Leng, K.; Nalls, M.A.; Singleton, A.B.; Xu, K.; et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 2021, 24, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Hiraiwa, M.; O’Brien, J.S. Saposins: Structure, function, distribution, and molecular genetics. J. Lipid Res. 1992, 33, 1255–1267. [Google Scholar] [CrossRef]
- Matsuda, J.; Yoneshige, A.; Suzuki, K. The function of sphingolipids in the nervous system: Lessons learnt from mouse models of specific sphingolipid activator protein deficiencies. J. Neurochem. 2007, 103 (Suppl. S1), 32–38. [Google Scholar] [CrossRef]
- Rafi, M.A.; Amini, S.; Zhang, X.L.; Wenger, D.A. Correction of sulfatide metabolism after transfer of prosaposin cDNA to cultured cells from a patient with SAP-1 deficiency. Am. J. Hum. Genet. 1992, 50, 1252–1258. [Google Scholar]
- Kuchar, L.; Ledvinova, J.; Hrebicek, M.; Myskova, H.; Dvorakova, L.; Berna, L.; Chrastina, P.; Asfaw, B.; Elleder, M.; Petermoller, M.; et al. Prosaposin deficiency and saposin B deficiency (activator-deficient metachromatic leukodystrophy): Report on two patients detected by analysis of urinary sphingolipids and carrying novel PSAP gene mutations. Am. J. Med. Genet. A 2009, 149A, 613–621. [Google Scholar] [CrossRef]
- Matzner, U.; Breiden, B.; Schwarzmann, G.; Yaghootfam, A.; Fluharty, A.L.; Hasilik, A.; Sandhoff, K.; Gieselmann, V. Saposin B-dependent reconstitution of arylsulfatase A activity in vitro and in cell culture models of metachromatic leukodystrophy. J. Biol. Chem. 2009, 284, 9372–9381. [Google Scholar] [CrossRef]
- Ho, M.W.; O’Brien, J.S. Gaucher’s disease: Deficiency of ‘acid’-glucosidase and reconstitution of enzyme activity in vitro. Proc. Natl. Acad. Sci. USA 1971, 68, 2810–2813. [Google Scholar] [CrossRef]
- Matsuda, J.; Kido, M.; Tadano-Aritomi, K.; Ishizuka, I.; Tominaga, K.; Toida, K.; Takeda, E.; Suzuki, K.; Kuroda, Y. Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Hum. Mol. Genet. 2004, 13, 2709–2723. [Google Scholar] [CrossRef]
- Schnabel, D.; Schröder, M.; Sandhoff, K. Mutation in the sphingolipid activator protein 2 in a patient with a variant of Gaucher disease. FEBS Lett. 1991, 284, 57–59. [Google Scholar] [CrossRef]
- Vaccaro, A.M.; Motta, M.; Tatti, M.; Scarpa, S.; Masuelli, L.; Bhat, M.; Vanier, M.T.; Tylki-Szymanska, A.; Salvioli, R. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet. 2010, 19, 2987–2997. [Google Scholar] [CrossRef]
- Tamargo, R.J.; Velayati, A.; Goldin, E.; Sidransky, E. The role of saposin C in Gaucher disease. Mol. Genet. Metab. 2012, 106, 257–263. [Google Scholar] [CrossRef]
- Tylki-Szymańska, A.; Czartoryska, B.; Vanier, M.T.; Poorthuis, B.J.; Groener, J.A.; Ługowska, A.; Millat, G.; Vaccaro, A.M.; Jurkiewicz, E. Non-neuronopathic Gaucher disease due to saposin C deficiency. Clin. Genet. 2007, 72, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Schwake, M.; Schroder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Balreira, A.; Gaspar, P.; Caiola, D.; Chaves, J.; Beirao, I.; Lima, J.L.; Azevedo, J.E.; Miranda, M.C. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum. Mol. Genet. 2008, 17, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Malini, E.; Zampieri, S.; Deganuto, M.; Romanello, M.; Sechi, A.; Bembi, B.; Dardis, A. Role of LIMP-2 in the intracellular trafficking of beta-glucosidase in different human cellular models. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3839–3852. [Google Scholar] [CrossRef]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef]
- Maor, G.; Filocamo, M.; Horowitz, M. ITCH regulates degradation of mutant glucocerebrosidase: Implications to Gaucher disease. Hum. Mol. Genet. 2013, 22, 1316–1327. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Back, S.H.; Song, B.; Han, J.; Hassler, J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in beta-cells. Diabetes Obes. Metab. 2010, 12 (Suppl. S2), 99–107. [Google Scholar] [CrossRef]
- Brady, R.O.; Kanfer, J.N.; Shapiro, D. Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher’s Disease. Biochem. Biophys. Res. Commun. 1965, 18, 221–225. [Google Scholar] [CrossRef]
- Raghavan, S.S.; Mumford, R.A.; Kanfer, J.N. Isolation and characterization of glucosylsphingosine from Gaucher’s spleen. J. Lipid Res. 1974, 15, 484–490. [Google Scholar] [CrossRef]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr.; Kaplan, K.W. Genetics of Sphingolipidosis. In Cerebral Sphingolipidosis; Aronson, S.M., Volk, B.W., Eds.; Academic Press: New York, NY, USA, 1962; pp. 395–411. [Google Scholar]
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef]
- Stone, D.L.; Carey, W.F.; Christodoulou, J.; Sillence, D.; Nelson, P.; Callahan, M.; Tayebi, N.; Sidransky, E. Type 2 Gaucher disease: The collodion baby phenotype revisited. Arch. Dis. Child. Fetal Neonatal Ed. 2000, 82, F163–F166. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Barton, N.W.; Grabowski, G.A. The role of neurogenetics in Gaucher disease. Arch. Neurol. 1993, 50, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M. The unfolded protein response. Mol. Biotechnol. 2006, 34, 279–290. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Legler, G. Studies on the action mechanism of glycoside splitting anzymes, I. Presentation and properties of specific inhibitors. Hoppe Seylers Z. Physiol. Chem. 1966, 345, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Kanfer, J.N.; Legler, G.; Sullivan, J.; Raghavan, S.S.; Mumford, R.A. The Gaucher mouse. Biochem. Biophys. Res. Commun. 1975, 67, 85–90. [Google Scholar] [CrossRef]
- Stephens, M.C.; Bernatsky, A.; Burachinsky, V.; Legler, G.; Kanfer, J.N. The Gaucher mouse: Differential action of conduritol B epoxide and reversibility of its effects. J. Neurochem. 1978, 30, 1023–1027. [Google Scholar] [CrossRef]
- Marshall, J.; McEachern, K.A.; Kyros, J.A.; Nietupski, J.B.; Budzinski, T.; Ziegler, R.J.; Yew, N.S.; Sullivan, J.; Scaria, A.; van Rooijen, N.; et al. Demonstration of feasibility of in vivo gene therapy for Gaucher disease using a chemically induced mouse model. Mol. Ther. 2002, 6, 179–189. [Google Scholar] [CrossRef]
- Vitner, E.B.; Salomon, R.; Farfel-Becker, T.; Meshcheriakova, A.; Ali, M.; Klein, A.D.; Platt, F.M.; Cox, T.M.; Futerman, A.H. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 2014, 20, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Zigdon, H.; Meshcheriakova, A.; Klein, A.D.; Yaacobi, C.; Eilam, R.; Kenwood, B.M.; Rahim, A.A.; Massaro, G.; Merrill, A.H., Jr.; et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J. Pathol. 2016, 239, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.D.; Ferreira, N.S.; Ben-Dor, S.; Duan, J.; Hardy, J.; Cox, T.M.; Merrill, A.H., Jr.; Futerman, A.H. Identification of Modifier Genes in a Mouse Model of Gaucher Disease. Cell Rep. 2016, 16, 2546–2553. [Google Scholar] [CrossRef]
- Aerts, J.M.; van Breemen, M.J.; Bussink, A.P.; Ghauharali, K.; Sprenger, R.; Boot, R.G.; Groener, J.E.; Hollak, C.E.; Maas, M.; Smit, S.; et al. Biomarkers for lysosomal storage disorders: Identification and application as exemplified by chitotriosidase in Gaucher disease. Acta Paediatr. 2008, 97, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kramer, G.; Wegdam, W.; Donker-Koopman, W.; Ottenhoff, R.; Gaspar, P.; Verhoek, M.; Nelson, J.; Gabriel, T.; Kallemeijn, W.; Boot, R.G.; et al. Elevation of glycoprotein nonmetastatic melanoma protein B in type 1 Gaucher disease patients and mouse models. FEBS Open Bio 2016, 6, 902–913. [Google Scholar] [CrossRef]
- Dahl, M.; Doyle, A.; Olsson, K.; Mansson, J.E.; Marques, A.R.A.; Mirzaian, M.; Aerts, J.M.; Ehinger, M.; Rothe, M.; Modlich, U.; et al. Lentiviral gene therapy using cellular promoters cures type 1 Gaucher disease in mice. Mol. Ther. 2015, 23, 835–844. [Google Scholar] [CrossRef]
- Zigdon, H.; Savidor, A.; Levin, Y.; Meshcheriakova, A.; Schiffmann, R.; Futerman, A.H. Identification of a biomarker in cerebrospinal fluid for neuronopathic forms of Gaucher disease. PLoS ONE 2015, 10, e0120194. [Google Scholar] [CrossRef]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.; Dekel, H.; Leshem, N.; Enquist, I.B.; Karlsson, S.; Futerman, A.H. No evidence for activation of the unfolded protein response in neuronopathic models of Gaucher disease. Hum. Mol. Genet. 2009, 18, 1482–1488. [Google Scholar] [CrossRef]
- Beutler, E.; West, C.; Torbett, B.E.; Deguchi, H. A chimeric mouse model of Gaucher disease. Mol. Med. 2002, 8, 247–250. [Google Scholar] [CrossRef]
- Tybulewicz, V.L.; Tremblay, M.L.; LaMarca, M.E.; Willemsen, R.; Stubblefield, B.K.; Winfield, S.; Zablocka, B.; Sidransky, E.; Martin, B.M.; Huang, S.P.; et al. Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 1992, 357, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Holleran, W.M.; Takagi, Y.; Imokawa, G.; Jackson, S.; Lee, J.M.; Elias, P.M. beta-Glucocerebrosidase activity in murine epidermis: Characterization and localization in relation to differentiation. J. Lipid Res. 1992, 33, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Hong, Y.B.; Kim, E.Y.; Jung, S.C. Upregulation of proinflammatory cytokines in the fetal brain of the Gaucher mouse. J. Korean Med. Sci. 2006, 21, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Hong, Y.B.; Go, S.H.; Lee, B.; Jung, S.C. Downregulation of neurotrophic factors in the brain of a mouse model of Gaucher disease; implications for neuronal loss in Gaucher disease. Exp. Mol. Med. 2006, 38, 348–356. [Google Scholar] [CrossRef]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; McKay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Enquist, I.B.; Nilsson, E.; Ooka, A.; Mansson, J.E.; Olsson, K.; Ehinger, M.; Brady, R.O.; Richter, J.; Karlsson, S. Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13819–13824. [Google Scholar] [CrossRef]
- Pavlova, E.V.; Wang, S.Z.; Archer, J.; Dekker, N.; Aerts, J.M.; Karlsson, S.; Cox, T.M. B cell lymphoma and myeloma in murine Gaucher’s disease. J. Pathol. 2013, 231, 88–97. [Google Scholar] [CrossRef]
- Pavlova, E.V.; Archer, J.; Wang, S.; Dekker, N.; Aerts, J.M.; Karlsson, S.; Cox, T.M. Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J. Pathol. 2015, 235, 113–124. [Google Scholar] [CrossRef]
- Sinclair, G.B.; Jevon, G.; Colobong, K.E.; Randall, D.R.; Choy, F.Y.; Clarke, L.A. Generation of a conditional knockout of murine glucocerebrosidase: Utility for the study of Gaucher disease. Mol. Genet. Metab. 2007, 90, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Salazar, M.A.; Bercury, S.D.; Ziegler, R.J.; Marshall, J.; Hodges, B.L.; Chuang, W.L.; Pacheco, J.; Li, L.; Cheng, S.H.; Scheule, R.K. Intracerebroventricular delivery of glucocerebrosidase reduces substrates and increases lifespan in a mouse model of neuronopathic Gaucher disease. Exp. Neurol. 2010, 225, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Salazar, M.A.; Deriso, M.; Bercury, S.D.; Li, L.; Lydon, J.T.; Weber, W.; Pande, N.; Cromwell, M.A.; Copeland, D.; Leonard, J.; et al. Systemic delivery of a glucosylceramide synthase inhibitor reduces CNS substrates and increases lifespan in a mouse model of type 2 Gaucher disease. PLoS ONE 2012, 7, e43310. [Google Scholar] [CrossRef] [PubMed]
- Enquist, I.B.; Lo Bianco, C.; Ooka, A.; Nilsson, E.; Mansson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Vitner, E.B.; Pressey, S.N.; Eilam, R.; Cooper, J.D.; Futerman, A.H. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum. Mol. Genet. 2011, 20, 1375–1386. [Google Scholar] [CrossRef]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 2012, 135, 1724–1735. [Google Scholar] [CrossRef]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef]
- van Weely, S.; Brandsma, M.; Strijland, A.; Tager, J.M.; Aerts, J.M. Demonstration of the existence of a second, non-lysosomal glucocerebrosidase that is not deficient in Gaucher disease. Biochim. Biophys. Acta 1993, 1181, 55–62. [Google Scholar] [CrossRef]
- Yildiz, Y.; Matern, H.; Thompson, B.; Allegood, J.C.; Warren, R.L.; Ramirez, D.M.; Hammer, R.E.; Hamra, F.K.; Matern, S.; Russell, D.W. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Investig. 2006, 116, 2985–2994. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; Donker-Koopman, W.; Strijland, A.; van Marle, J.; Overkleeft, H.S.; Wennekes, T.; Aerts, J.M. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J. Biol. Chem. 2007, 282, 1305–1312. [Google Scholar] [CrossRef]
- Marques, A.R.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Ghauharali-van der Vlugt, K.; Meijer, R.; Giraldo, P.; Alfonso, P.; et al. Glucosylated cholesterol in mammalian cells and tissues: Formation and degradation by multiple cellular beta-glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J.; et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef] [PubMed]
- Pewzner-Jung, Y.; Joseph, T.; Blumenreich, S.; Vardi, A.; Ferreira, N.S.; Cho, S.M.; Eilam, R.; Tsoory, M.; Biton, I.E.; Brumfeld, V.; et al. Brain pathology and cerebellar purkinje cell loss in a mouse model of chronic neuronopathic Gaucher disease. Prog. Neurobiol. 2021, 197, 101939. [Google Scholar] [CrossRef] [PubMed]
- Boddupalli, C.S.; Nair, S.; Belinsky, G.; Gans, J.; Teeple, E.; Nguyen, T.H.; Mehta, S.; Guo, L.; Kramer, M.L.; Ruan, J.; et al. Neuroinflammation in neuronopathic Gaucher disease: Role of microglia and NK cells, biomarkers, and response to substrate reduction therapy. eLife 2022, 11, e79830. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Suzuki, K.; Reed, J.D.; Grinberg, A.; Westphal, H.; Hoffmann, A.; Doring, T.; Sandhoff, K.; Proia, R.L. Mice with type 2 and 3 Gaucher disease point mutations generated by a single insertion mutagenesis procedure. Proc. Natl. Acad. Sci. USA 1998, 95, 2503–2508. [Google Scholar] [CrossRef]
- Tsuji, S.; Choudary, P.V.; Martin, B.M.; Stubblefield, B.K.; Mayor, J.A.; Barranger, J.A.; Ginns, E.I. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N. Engl. J. Med. 1987, 316, 570–575. [Google Scholar] [CrossRef]
- Eyal, N.; Wilder, S.; Horowitz, M. Prevalent and rare mutations among Gaucher patients. Gene 1990, 96, 277–283. [Google Scholar] [CrossRef]
- Mizukami, H.; Mi, Y.; Wada, R.; Kono, M.; Yamashita, T.; Liu, Y.; Werth, N.; Sandhoff, R.; Sandhoff, K.; Proia, R.L. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Investig. 2002, 109, 1215–1221. [Google Scholar] [CrossRef]
- Xu, Y.H.; Quinn, B.; Witte, D.; Grabowski, G.A. Viable mouse models of acid beta-glucosidase deficiency: The defect in Gaucher disease. Am. J. Pathol. 2003, 163, 2093–2101. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Xu, Y.H.; Sun, Y.; Barnes, S.; Grabowski, G.A. Comparative therapeutic effects of velaglucerase alfa and imiglucerase in a Gaucher disease mouse model. PLoS ONE 2010, 5, e10750. [Google Scholar] [CrossRef]
- Xu, Y.H.; Jia, L.; Quinn, B.; Zamzow, M.; Stringer, K.; Aronow, B.; Sun, Y.; Zhang, W.; Setchell, K.D.; Grabowski, G.A. Global gene expression profile progression in Gaucher disease mouse models. BMC Genom. 2011, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- McEachern, K.A.; Fung, J.; Komarnitsky, S.; Siegel, C.S.; Chuang, W.L.; Hutto, E.; Shayman, J.A.; Grabowski, G.A.; Aerts, J.M.; Cheng, S.H.; et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol. Genet. Metab. 2007, 91, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; McEachern, K.A.; Chuang, W.L.; Hutto, E.; Siegel, C.S.; Shayman, J.A.; Grabowski, G.A.; Scheule, R.K.; Copeland, D.P.; Cheng, S.H. Improved management of lysosomal glucosylceramide levels in a mouse model of type 1 Gaucher disease using enzyme and substrate reduction therapy. J. Inherit. Metab. Dis. 2010, 33, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef]
- Marshall, J.; Sun, Y.; Bangari, D.S.; Budman, E.; Park, H.; Nietupski, J.B.; Allaire, A.; Cromwell, M.A.; Wang, B.; Grabowski, G.A.; et al. CNS-accessible Inhibitor of Glucosylceramide Synthase for Substrate Reduction Therapy of Neuronopathic Gaucher Disease. Mol. Ther. 2016, 24, 1019–1029. [Google Scholar] [CrossRef]
- Chang, H.H.; Asano, N.; Ishii, S.; Ichikawa, Y.; Fan, J.Q. Hydrophilic iminosugar active-site-specific chaperones increase residual glucocerebrosidase activity in fibroblasts from Gaucher patients. FEBS J. 2006, 273, 4082–4092. [Google Scholar] [CrossRef]
- Steet, R.; Chung, S.; Lee, W.S.; Pine, C.W.; Do, H.; Kornfeld, S. Selective action of the iminosugar isofagomine, a pharmacological chaperone for mutant forms of acid-beta-glucosidase. Biochem. Pharmacol. 2007, 73, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Kornhaber, G.J.; Tropak, M.B.; Maegawa, G.H.; Tuske, S.J.; Coales, S.J.; Mahuran, D.J.; Hamuro, Y. Isofagomine induced stabilization of glucocerebrosidase. Chembiochem 2008, 9, 2643–2649. [Google Scholar] [CrossRef]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. FEBS J. 2010, 277, 1618–1638. [Google Scholar] [CrossRef]
- Sun, Y.; Ran, H.; Liou, B.; Quinn, B.; Zamzow, M.; Zhang, W.; Bielawski, J.; Kitatani, K.; Setchell, K.D.; Hannun, Y.A.; et al. Isofagomine in vivo effects in a neuronopathic Gaucher disease mouse. PLoS ONE 2011, 6, e19037. [Google Scholar] [CrossRef]
- Meyers, J.R. Zebrafish: Development of a Vertebrate Model Organism. Curr. Protoc. Essent. Lab. Tech. 2018, 16, e19. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Gerhard, G.S.; Kauffman, E.J.; Wang, X.; Stewart, R.; Moore, J.L.; Kasales, C.J.; Demidenko, E.; Cheng, K.C. Life spans and senescent phenotypes in two strains of Zebrafish (Danio rerio). Exp. Gerontol. 2002, 37, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.L.; Spitsbergen, J.M.; Wolterbeek, A.P.; Woutersen, R.A. Normal anatomy and histology of the adult zebrafish. Toxicol. Pathol. 2011, 39, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Udvadia, A.J.; Linney, E. Windows into development: Historic, current, and future perspectives on transgenic zebrafish. Dev. Biol. 2003, 256, 1–17. [Google Scholar] [CrossRef]
- Felker, A.; Mosimann, C. Contemporary zebrafish transgenesis with Tol2 and application for Cre/lox recombination experiments. Methods Cell Biol. 2016, 135, 219–244. [Google Scholar] [CrossRef]
- Artola, M.; Kuo, C.L.; Lelieveld, L.T.; Rowland, R.J.; van der Marel, G.A.; Codee, J.D.C.; Boot, R.G.; Davies, G.J.; Aerts, J.; Overkleeft, H.S. Functionalized Cyclophellitols Are Selective Glucocerebrosidase Inhibitors and Induce a Bona Fide Neuropathic Gaucher Model in Zebrafish. J. Am. Chem. Soc. 2019, 141, 4214–4218. [Google Scholar] [CrossRef]
- Witte, M.D.; Kallemeijn, W.W.; Aten, J.; Li, K.Y.; Strijland, A.; Donker-Koopman, W.E.; van den Nieuwendijk, A.M.; Bleijlevens, B.; Kramer, G.; Florea, B.I.; et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat. Chem. Biol. 2010, 6, 907–913. [Google Scholar] [CrossRef]
- Kuo, C.L.; van Meel, E.; Kytidou, K.; Kallemeijn, W.W.; Witte, M.; Overkleeft, H.S.; Artola, M.E.; Aerts, J.M. Activity-Based Probes for Glycosidases: Profiling and Other Applications. Methods Enzym. 2018, 598, 217–235. [Google Scholar] [CrossRef]
- Eisen, J.S.; Smith, J.C. Controlling morpholino experiments: Don’t stop making antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Marques, A.R.A.; Gaspar, P.; Oussoren, S.V.; Kytidou, K.; Codee, J.D.C.; van der Marel, G.; Overkleeft, H.S.; et al. Simultaneous quantitation of sphingoid bases by UPLC-ESI-MS/MS with identical (13)C-encoded internal standards. Clin. Chim. Acta 2017, 466, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, L.T.; Mirzaian, M.; Kuo, C.L.; Artola, M.; Ferraz, M.J.; Peter, R.E.A.; Akiyama, H.; Greimel, P.; van den Berg, R.; Overkleeft, H.S.; et al. Role of beta-glucosidase 2 in aberrant glycosphingolipid metabolism: Model of glucocerebrosidase deficiency in zebrafish. J. Lipid Res. 2019, 60, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Zancan, I.; Bellesso, S.; Costa, R.; Salvalaio, M.; Stroppiano, M.; Hammond, C.; Argenton, F.; Filocamo, M.; Moro, E. Glucocerebrosidase deficiency in zebrafish affects primary bone ossification through increased oxidative stress and reduced Wnt/beta-catenin signaling. Hum. Mol. Genet. 2015, 24, 1280–1294. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef]
- Lelieveld, L.T.; Gerhardt, S.; Maas, S.; Zwiers, K.C.; de Wit, C.; Beijk, E.H.; Ferraz, M.J.; Artola, M.; Meijer, A.H.; Tudorache, C.; et al. Consequences of excessive glucosylsphingosine in glucocerebrosidase-deficient zebrafish. J. Lipid Res. 2022, 63, 100199. [Google Scholar] [CrossRef]
- Fan, J.; Hale, V.L.; Lelieveld, L.T.; Whitworth, L.J.; Busch-Nentwich, E.M.; Troll, M.; Edelstein, P.H.; Cox, T.M.; Roca, F.J.; Aerts, J.; et al. Gaucher disease protects against tuberculosis. Proc. Natl. Acad. Sci. USA 2023, 120, e2217673120. [Google Scholar] [CrossRef]
- Schartl, M. Beyond the zebrafish: Diverse fish species for modeling human disease. Dis. Models Mech. 2014, 7, 181–192. [Google Scholar] [CrossRef]
- Witten, P.E.; Harris, M.P.; Huysseune, A.; Winkler, C. Small teleost fish provide new insights into human skeletal diseases. Methods Cell Biol. 2017, 138, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Bajoghli, B.; Dick, A.M.; Claasen, A.; Doll, L.; Aghaallaei, N. Zebrafish and Medaka: Two Teleost Models of T-Cell and Thymic Development. Int. J. Mol. Sci. 2019, 20, 4179. [Google Scholar] [CrossRef] [PubMed]
- Lleras-Forero, L.; Winkler, C.; Schulte-Merker, S. Zebrafish and medaka as models for biomedical research of bone diseases. Dev. Biol. 2020, 457, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Koike, M.; Ansai, S.; Kinoshita, M.; Ishikawa-Fujiwara, T.; Matsui, H.; Naruse, K.; Sakamoto, N.; Uchiyama, Y.; Todo, T.; et al. Viable neuronopathic Gaucher disease model in Medaka (Oryzias latipes) displays axonal accumulation of alpha-synuclein. PLoS Genet. 2015, 11, e1005065. [Google Scholar] [CrossRef]
- Nakanishi, E.; Uemura, N.; Akiyama, H.; Kinoshita, M.; Masanori, S.; Taruno, Y.; Yamakado, H.; Matsuzawa, S.I.; Takeda, S.; Hirabayashi, Y.; et al. Impact of Gba2 on neuronopathic Gaucher’s disease and alpha-synuclein accumulation in medaka (Oryzias latipes). Mol. Brain 2021, 14, 80. [Google Scholar] [CrossRef]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Models Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef]
- Wagner, C.; Isermann, K.; Fehrenbach, H.; Roeder, T. Molecular architecture of the fruit fly’s airway epithelial immune system. BMC Genom. 2008, 9, 446. [Google Scholar] [CrossRef]
- Charng, W.L.; Yamamoto, S.; Bellen, H.J. Shared mechanisms between Drosophila peripheral nervous system development and human neurodegenerative diseases. Curr. Opin. Neurobiol. 2014, 27, 158–164. [Google Scholar] [CrossRef]
- Apidianakis, Y.; Rahme, L.G. Drosophila melanogaster as a model for human intestinal infection and pathology. Dis. Models Mech. 2011, 4, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Bellen, H.J.; Levis, R.W.; Liao, G.; He, Y.; Carlson, J.W.; Tsang, G.; Evans-Holm, M.; Hiesinger, P.R.; Schulze, K.L.; Rubin, G.M.; et al. The BDGP gene disruption project: Single transposon insertions associated with 40% of Drosophila genes. Genetics 2004, 167, 761–781. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef]
- Duffy, J.B. GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genesis 2002, 34, 1–15. [Google Scholar] [CrossRef]
- Hales, K.G.; Korey, C.A.; Larracuente, A.M.; Roberts, D.M. Genetics on the Fly: A Primer on the Drosophila Model System. Genetics 2015, 201, 815–842. [Google Scholar] [CrossRef]
- Kaufman, T.C. A Short History and Description of Drosophila melanogaster Classical Genetics: Chromosome Aberrations, Forward Genetic Screens, and the Nature of Mutations. Genetics 2017, 206, 665–689. [Google Scholar] [CrossRef]
- Cabasso, O.; Paul, S.; Dorot, O.; Maor, G.; Krivoruk, O.; Pasmanik-Chor, M.; Mirzaian, M.; Ferraz, M.; Aerts, J.; Horowitz, M. Drosophila melanogaster Mutated in its GBA1b Ortholog Recapitulates Neuronopathic Gaucher Disease. J. Clin. Med. 2019, 8, 1420. [Google Scholar] [CrossRef]
- Cabasso, O.; Paul, S.; Maor, G.; Pasmanik-Chor, M.; Kallemeijn, W.; Aerts, J.; Horowitz, M. The Uncovered Function of the Drosophila GBA1a-Encoded Protein. Cells 2021, 10, 630. [Google Scholar] [CrossRef]
- Dasari, S.K.; Schejter, E.; Bialik, S.; Shkedy, A.; Levin-Salomon, V.; Levin-Zaidman, S.; Kimchi, A. Death by over-eating: The Gaucher disease associated gene GBA1, identified in a screen for mediators of autophagic cell death, is necessary for developmental cell death in Drosophila midgut. Cell Cycle 2017, 16, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J.; Gronke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.Y.; Trinh, K.; Thomas, R.E.; Yu, S.; Germanos, A.A.; Whitley, B.N.; Sardi, S.P.; Montine, T.J.; Pallanck, L.J. Glucocerebrosidase Deficiency in Drosophila Results in alpha-Synuclein-Independent Protein Aggregation and Neurodegeneration. PLoS Genet. 2016, 12, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Atilano, M.L.; Hull, A.; Romila, C.-A.; Adams, M.L.; Wildfire, J.; Ureña, E.; Dyson, M.; Ivan-Castillo-Quan, J.; Partridge, L.; Kinghorn, K.J. Gba1 deletion causes immune hyperactivation and microbial dysbiosis through autophagic defects. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kawasaki, H.; Suzuki, T.; Ito, K.; Takahara, T.; Goto-Inoue, N.; Setou, M.; Sakata, K.; Ishida, N. Minos-insertion mutant of the Drosophila GBA gene homologue showed abnormal phenotypes of climbing ability, sleep and life span with accumulation of hydroxy-glucocerebroside. Gene 2017, 614, 49–55. [Google Scholar] [CrossRef]
- Cao, Y.; Chtarbanova, S.; Petersen, A.J.; Ganetzky, B. Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc. Natl. Acad. Sci. USA 2013, 110, E1752–E1760. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Hartley, W.J.; Blakemore, W.F. Neurovisceral glucocerebroside storage (Gaucher’s disease) in a dog. Vet. Pathol. 1973, 10, 191–201. [Google Scholar] [CrossRef]
- Van De Water, N.S.; Jolly, R.D.; Farrow, B.R. Canine Gaucher disease--the enzymic defect. Aust. J. Exp. Biol. Med. Sci. 1979, 57, 551–554. [Google Scholar] [CrossRef]
- Karageorgos, L.; Lancaster, M.J.; Nimmo, J.S.; Hopwood, J.J. Gaucher disease in sheep. J. Inherit. Metab. Dis. 2011, 34, 209–215. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Suter, R.; Gong, H.; Fang, Q.; Zhou, P.; Hickford, J.G.H. A nucleotide substitution in exon 8 of the glucosylceramidase beta gene is associated with Gaucher disease in sheep. Anim. Genet. 2017, 48, 733–734. [Google Scholar] [CrossRef] [PubMed]
- Karageorgos, L.; Hein, L.; Rozaklis, T.; Adams, M.; Duplock, S.; Snel, M.; Hemsley, K.; Kuchel, T.; Smith, N.; Hopwood, J.J. Glycosphingolipid analysis in a naturally occurring ovine model of acute neuronopathic Gaucher disease. Neurobiol. Dis. 2016, 91, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.K.; Rozaklis, T.; Adams, M.K.; Hopwood, J.J.; Karageorgos, L. Lipid composition of microdomains is altered in neuronopathic Gaucher disease sheep brain and spleen. Mol. Genet. Metab. 2017, 121, 259–270. [Google Scholar] [CrossRef]
- Winner, L.K.; Beard, H.; Karageorgos, L.; Smith, N.J.; Hopwood, J.J.; Hemsley, K.M. The ovine Type II Gaucher disease model recapitulates aspects of human brain disease. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166658. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabasso, O.; Kuppuramalingam, A.; Lelieveld, L.; Van der Lienden, M.; Boot, R.; Aerts, J.M.; Horowitz, M. Animal Models for the Study of Gaucher Disease. Int. J. Mol. Sci. 2023, 24, 16035. https://doi.org/10.3390/ijms242216035
Cabasso O, Kuppuramalingam A, Lelieveld L, Van der Lienden M, Boot R, Aerts JM, Horowitz M. Animal Models for the Study of Gaucher Disease. International Journal of Molecular Sciences. 2023; 24(22):16035. https://doi.org/10.3390/ijms242216035
Chicago/Turabian StyleCabasso, Or, Aparna Kuppuramalingam, Lindsey Lelieveld, Martijn Van der Lienden, Rolf Boot, Johannes M. Aerts, and Mia Horowitz. 2023. "Animal Models for the Study of Gaucher Disease" International Journal of Molecular Sciences 24, no. 22: 16035. https://doi.org/10.3390/ijms242216035
APA StyleCabasso, O., Kuppuramalingam, A., Lelieveld, L., Van der Lienden, M., Boot, R., Aerts, J. M., & Horowitz, M. (2023). Animal Models for the Study of Gaucher Disease. International Journal of Molecular Sciences, 24(22), 16035. https://doi.org/10.3390/ijms242216035