
Compromised Muscle Properties in a Severe Hypophosphatasia Murine Model

 and
and
Abstract
1. Introduction
2. Results
2.1. Quantitative Assessment of Overall Phenotype in HPP Mice
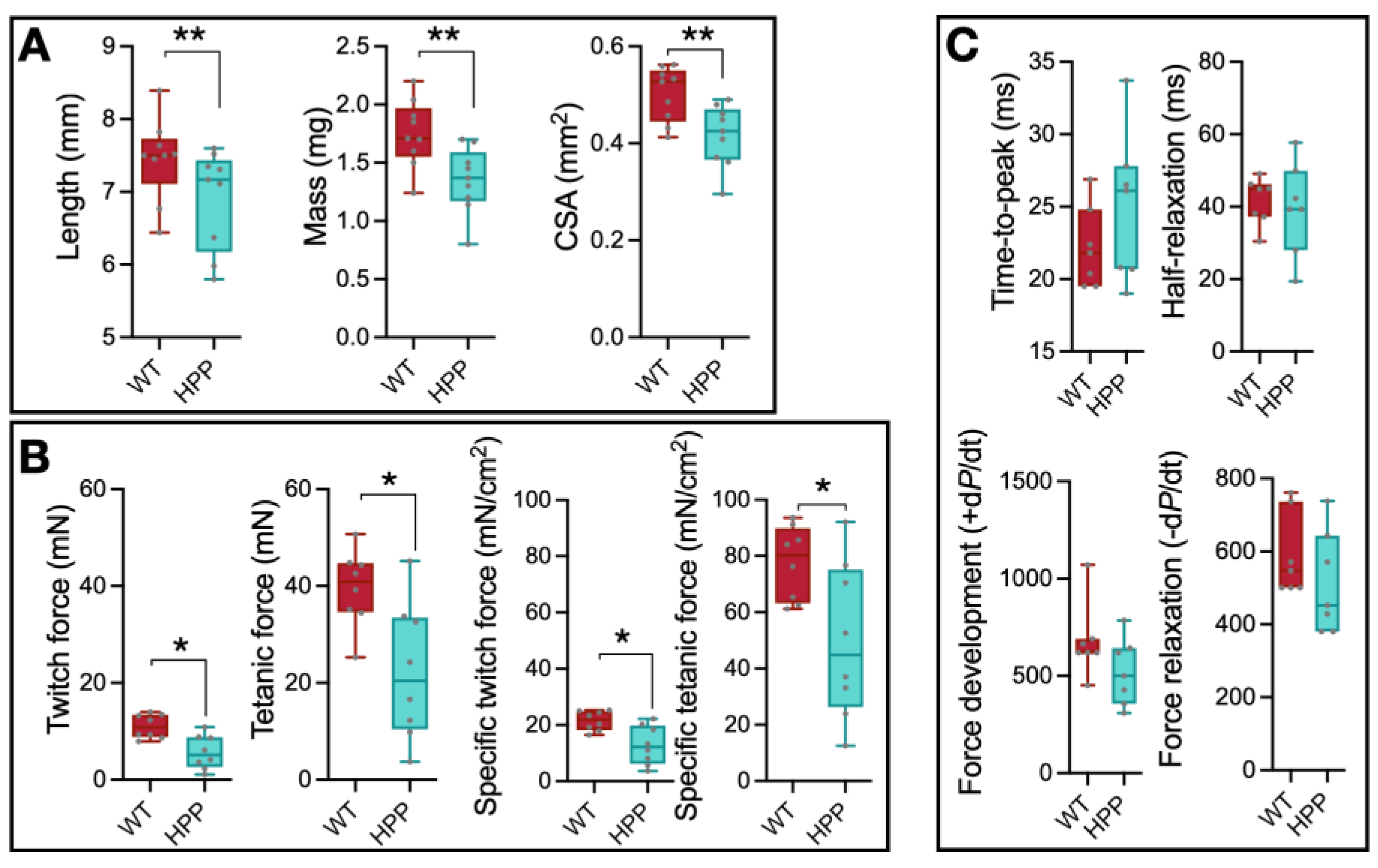
2.2. Muscle Size and Contractile Function
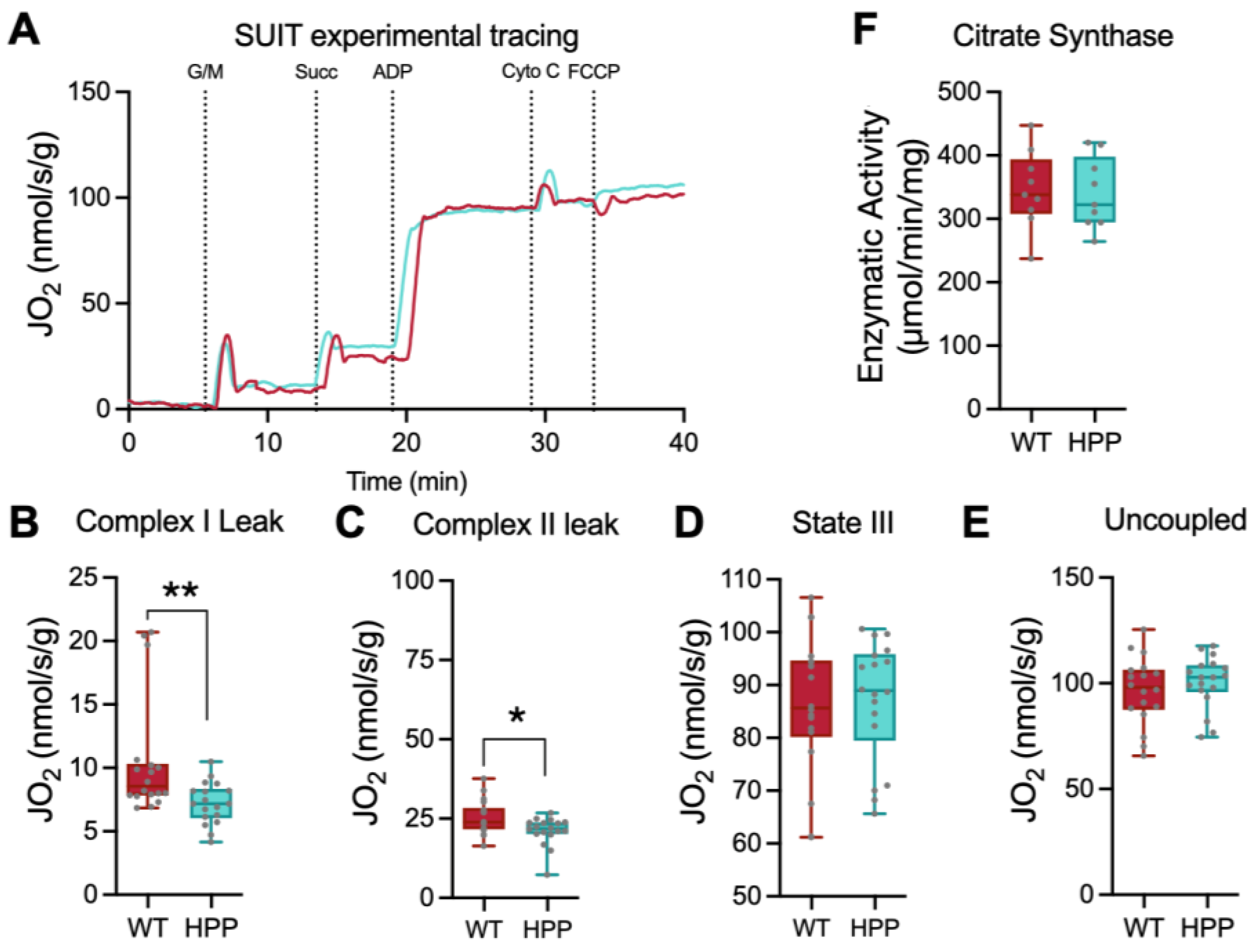
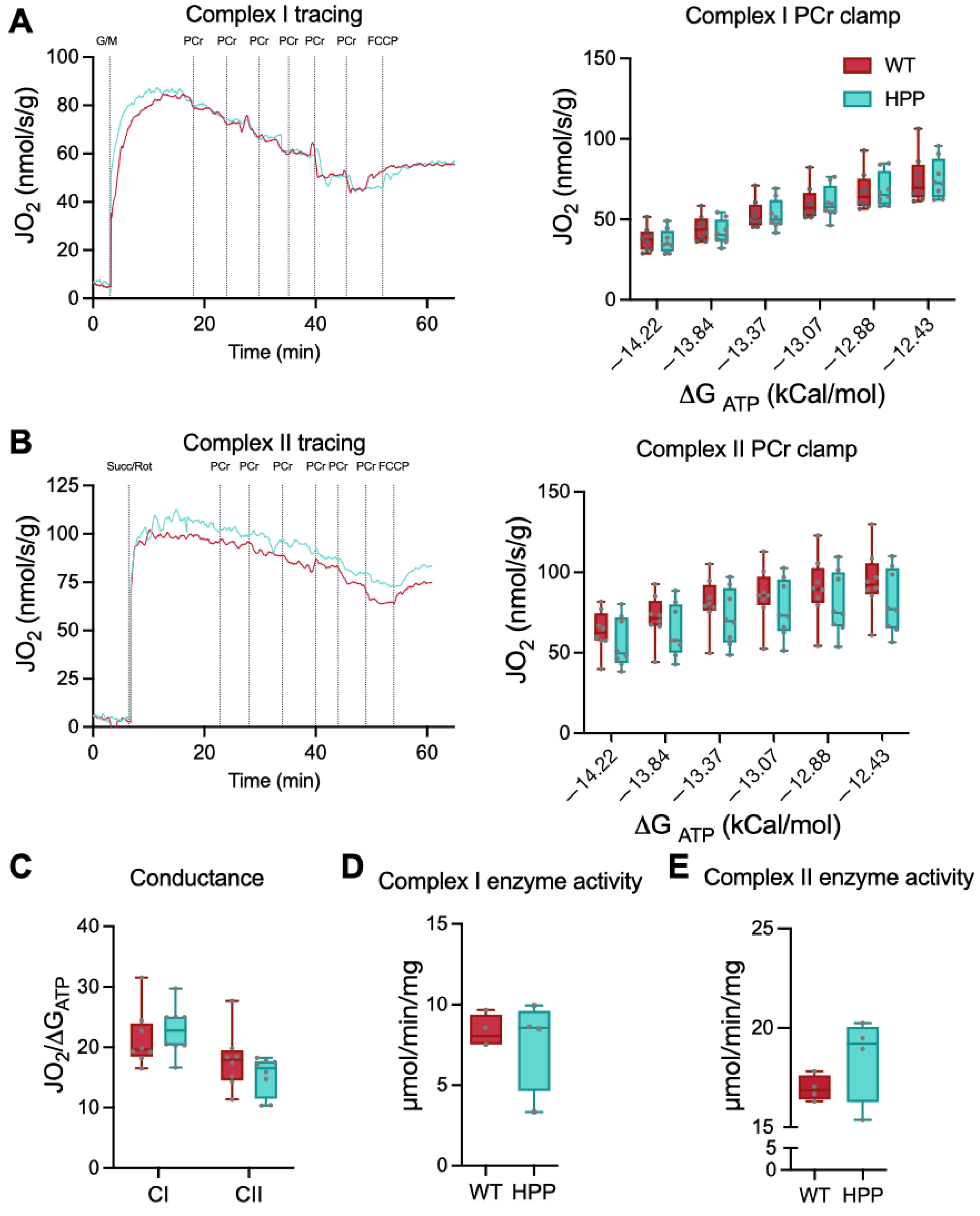
2.3. Mitochondrial Function
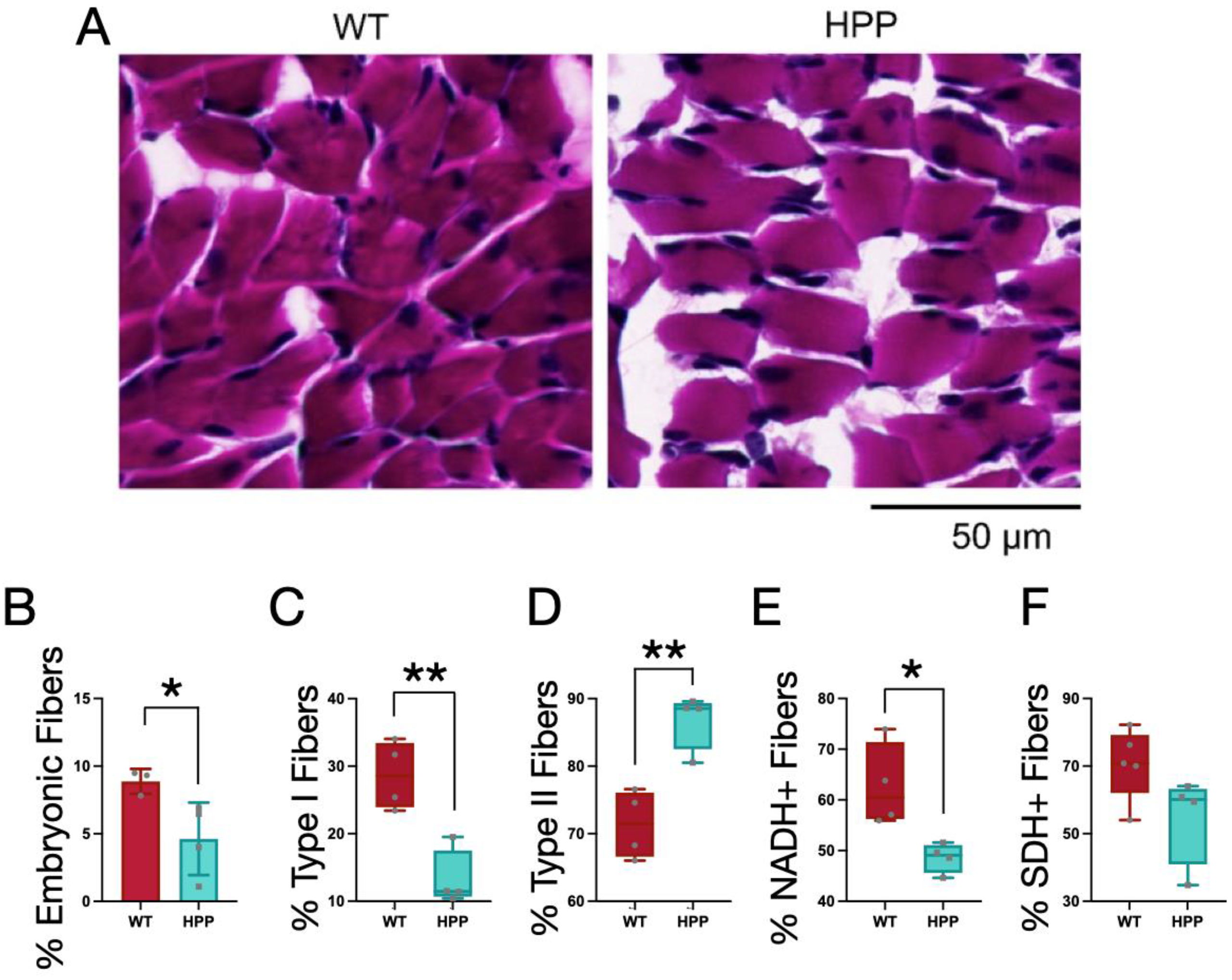
2.4. Muscle Histological Characteristics
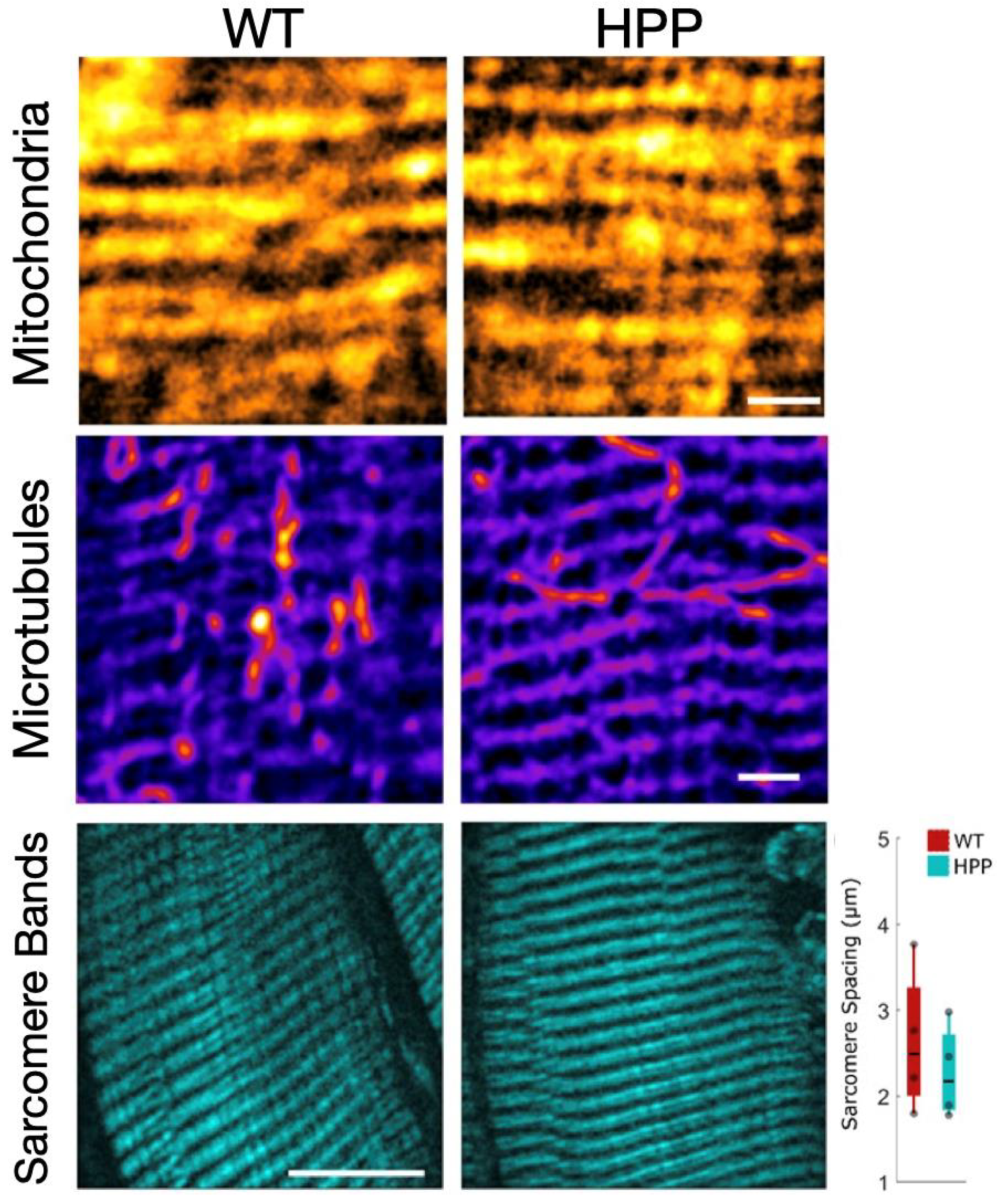
2.5. Muscle Morphological Characteristics
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Study Design
4.3. Experimental Methodology
4.3.1. Muscle Isolation and Preparation for Mitochondrial Experiments
4.3.2. In Vitro Assessment of Muscle Function
4.4. Histological Analyses of Skeletal Muscle Fibers
4.5. Microscopic Evaluation of Muscle Structure
4.6. Mitochondrial Oxygen Consumption
4.7. Mitochondrial Content (Citrate Synthase Activity)
4.8. Phosphocreatine Creatine Kinase Clamp Respiration
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bianchi, M.L. Hypophosphatasia: An overview of the disease and its treatment. Osteoporos. Int. 2015, 26, 2743–2757. [Google Scholar] [CrossRef]
- Millan, J.L.; Whyte, M.P. Alkaline Phosphatase and Hypophosphatasia. Calcif. Tissue Int. 2016, 98, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D. Hypophosphatasia. Am. J. Med. 1957, 22, 730–746. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E.; Yvard, A.; Taillandier, A.; Fauvert, D.; Simon-Bouy, B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann. Hum. Genet. 2011, 75, 439–445. [Google Scholar] [CrossRef] [PubMed]
- García-Fontana, C.; Villa-Suárez, J.M.; Andújar-Vera, F.; González-Salvatierra, S.; Martínez-Navajas, G.; Real, P.J.; Gómez Vida, J.M.; de Haro, T.; García-Fontana, B.; Muñoz-Torres, M. Epidemiological, Clinical and Genetic Study of Hypophosphatasia in A Spanish Population: Identification of Two Novel Mutations in The Alpl Gene. Sci. Rep. 2019, 9, 9569. [Google Scholar] [CrossRef]
- González-Cejudo, T.; Villa-Suárez, J.M.; Ferrer-Millán, M.; Andújar-Vera, F.; Contreras-Bolívar, V.; Andreo-López, M.C.; Gómez-Vida, J.M.; Martínez-Heredia, L.; González-Salvatierra, S.; de Haro Muñoz, T.; et al. Mild hypophosphatasia may be twice as prevalent as previously estimated: An effective clinical algorithm to detect undiagnosed cases. Clin. Chem. Lab. Med. (CCLM) 2023. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia and the Role of Alkaline Phosphatase in Skeletal Mineralization. Endocr. Rev. 1994, 15, 439–461. [Google Scholar] [CrossRef]
- Shapiro, J.R.; Lewiecki, E.M. Hypophosphatasia in Adults: Clinical Assessment and Treatment Considerations. J. Bone Miner. Res. 2017, 32, 1977–1980. [Google Scholar] [CrossRef]
- Linglart, A.; Biosse-Duplan, M. Hypophosphatasia. Curr. Osteoporos. Rep. 2016, 14, 95–105. [Google Scholar] [CrossRef]
- Seshia, S.S.; Derbyshire, G.; Haworth, J.C.; Hoogstraten, J. Myopathy with hypophosphatasia. Arch. Dis. Child. 1990, 65, 130–131. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef]
- Whyte, M.P.; Madson, K.L.; Phillips, D.; Reeves, A.L.; McAlister, W.H.; Yakimoski, A.; Mack, K.E.; Hamilton, K.; Kagan, K.; Fujita, K.P.; et al. Asfotase alfa therapy for children with hypophosphatasia. JCI Insight 2016, 1, e85971. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Rockman-Greenberg, C.; Rauch, F.; Bhatti, M.T.; Moseley, S.; Denker, A.E.; Watsky, E.; Whyte, M.P. Five-year efficacy and safety of asfotase alfa therapy for adults and adolescents with hypophosphatasia. Bone 2019, 121, 149–162. [Google Scholar] [CrossRef]
- Magdaleno, A.L.; Singh, S.; Venkataraman, S.; Perilli, G.A.; Lee, Y.-Y. Adult-Onset Hypophosphatasia: Before and After Treatment with Asfotase ALFA. AACE Clin. Case Rep. 2019, 5, e344–e348. [Google Scholar] [CrossRef]
- Remde, H.; Cooper, M.S.; Quinkler, M. Successful Asfotase Alfa Treatment in an Adult Dialysis Patient With Childhood-Onset Hypophosphatasia. J. Endocr. Soc. 2017, 1, 1188–1193. [Google Scholar] [CrossRef][Green Version]
- Koyama, H.; Yasuda, S.; Kakoi, S.; Ohata, Y.; Shimizu, Y.; Hasegawa, C.; Hayakawa, A.; Akiyama, T.; Yagi, T.; Aotani, D.; et al. Effect of Asfotase Alfa on Muscle Weakness in a Japanese Adult Patient of Hypophosphatasia with Low ALP Levels. Intern. Med. 2020, 59, 811–815. [Google Scholar] [CrossRef]
- Nishizawa, H.; Sato, Y.; Ishikawa, M.; Arakawa, Y.; Iijima, M.; Akiyama, T.; Takano, K.; Watanabe, A.; Kosho, T. Marked motor function improvement in a 32-year-old woman with childhood-onset hypophosphatasia by asfotase alfa therapy: Evaluation based on standardized testing batteries used in Duchenne muscular dystrophy clinical trials. Mol. Genet. Metab. Rep. 2020, 25, 100643. [Google Scholar] [CrossRef]
- Kitoh, H.; Izawa, M.; Kaneko, H.; Kitamura, A.; Matsuyama, S.; Kato, K.; Ogi, T. Two children with hypophosphatasia with a heterozygous c.1559delT variant in the ALPL gene, the most common variant in Japanese populations. Bone Rep. 2022, 17, 101626. [Google Scholar] [CrossRef]
- Williams, D.K.; Pinzón, C.; Huggins, S.; Pryor, J.H.; Falck, A.; Herman, F.; Oldeschulte, J.; Chavez, M.B.; Foster, B.L.; White, S.H.; et al. Genetic engineering a large animal model of human hypophosphatasia in sheep. Sci. Rep. 2018, 8, 16945. [Google Scholar] [CrossRef]
- Zhang, Z.; Nam, H.K.; Crouch, S.; Hatch, N.E. Tissue Nonspecific Alkaline Phosphatase Function in Bone and Muscle Progenitor Cells: Control of Mitochondrial Respiration and ATP Production. Int. J. Mol. Sci. 2021, 22, 1140. [Google Scholar] [CrossRef]
- Narisawa, S.; Fröhlander, N.; Millán, J.L. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn. 1997, 208, 432–446. [Google Scholar] [CrossRef]
- Fedde, K.N.; Blair, L.; Silverstein, J.; Coburn, S.P.; Ryan, L.M.; Weinstein, R.S.; Waymire, K.; Narisawa, S.; Millán, J.L.; Macgregor, G.R.; et al. Alkaline Phosphatase Knock-Out Mice Recapitulate the Metabolic and Skeletal Defects of Infantile Hypophosphatasia. J. Bone Miner. Res. 1999, 14, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Ullman-Cullere, M.H.; Foltz, C.J. Body condition scoring: A rapid and accurate method for assessing health status in mice. Lab. Anim. Sci. 1999, 49, 319–323. [Google Scholar]
- Brooks, S.V.; Faulkner, J.A. Contractile properties of skeletal muscles from young, adult and aged mice. J. Physiol. 1988, 404, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Hayes, D.A.; Lowe, D.A.; Williams, J.H.; Armstrong, R.B. Eccentric contraction-induced injury in normal and hindlimb-suspended mouse soleus and EDL muscles. J. Appl. Physiol. 1994, 77, 1421–1430. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Davidson, M.T.; Narowski, T.M.; Lin, C.T.; Koves, T.R.; Muoio, D.M. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 2018, 24, 3593–3606.e3510. [Google Scholar] [CrossRef]
- McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Nelson, M.A.M.; Kew, K.A.; Wooten, A.R.; Fisher-Wellman, K.H. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci. Rep. 2020, 10, 17599. [Google Scholar] [CrossRef]
- McFaline-Figueroa, J.; Hunda, E.T.; Heo, J.; Winders, E.A.; Greising, S.M.; Call, J.A. The bioenergetic “CK Clamp” technique detects substrate-specific changes in mitochondrial respiration and membrane potential during early VML injury pathology. Front. Physiol. 2023, 14, 571. [Google Scholar] [CrossRef] [PubMed]
- Sébastien, M.; Giannesini, B.; Aubin, P.; Brocard, J.; Chivet, M.; Pietrangelo, L.; Boncompagni, S.; Bosc, C.; Brocard, J.; Rendu, J.; et al. Deletion of the microtubule-associated protein 6 (MAP6) results in skeletal muscle dysfunction. Skelet. Muscle 2018, 8, 30. [Google Scholar] [CrossRef]
- Khairallah, R.J.; Shi, G.; Sbrana, F.; Prosser, B.L.; Borroto, C.; Mazaitis, M.J.; Hoffman, E.P.; Mahurkar, A.; Sachs, F.; Sun, Y.; et al. Microtubules Underlie Dysfunction in Duchenne Muscular Dystrophy. Sci. Signal. 2012, 5, ra56. [Google Scholar] [CrossRef]
- Ferrucci, L.; Baroni, M.; Ranchelli, A.; Lauretani, F.; Maggio, M.; Mecocci, P.; Ruggiero, C. Interaction between bone and muscle in older persons with mobility limitations. Curr. Pharm. Des. 2014, 20, 3178–3197. [Google Scholar] [CrossRef] [PubMed]
- Amanzadeh, J.; Reilly, R.F., Jr. Hypophosphatemia: An evidence-based approach to its clinical consequences and management. Nat. Clin. Pract. Nephrol. 2006, 2, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Gentry, B.A.; Ferreira, J.A.; McCambridge, A.J.; Brown, M.; Phillips, C.L. Skeletal muscle weakness in osteogenesis imperfecta mice. Matrix Biol. 2010, 29, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Veilleux, L.N.; Rauch, F. Muscle-Bone Interactions in Pediatric Bone Diseases. Curr. Osteoporos. Rep. 2017, 15, 425–432. [Google Scholar] [CrossRef]
- Seeman, E.; Hopper, J.L.; Young, N.R.; Formica, C.; Goss, P.; Tsalamandris, C. Do genetic factors explain associations between muscle strength, lean mass, and bone density? A twin study. Am. J. Physiol. 1996, 270, E320–E327. [Google Scholar] [CrossRef]
- Marcell, T.J. Sarcopenia: Causes, consequences, and preventions. J. Gerontology. Ser. A Biol. Sci. Med. Sci. 2003, 58, M911–M916. [Google Scholar] [CrossRef]
- Fricke, O.; Schoenau, E. The ‘Functional Muscle-Bone Unit’: Probing the relevance of mechanical signals for bone development in children and adolescents. Growth Horm. IGF Res. 2007, 17, 1–9. [Google Scholar] [CrossRef]
- Tsouni, P.; Kuntzer, T.; Ochsner, F. Neurological manifestations in musculoskeletal disorders. Rev. Medicale Suisse 2014, 10, 966–969. [Google Scholar]
- Sirola, J.; Kröger, H. Similarities in acquired factors related to postmenopausal osteoporosis and sarcopenia. J. Osteoporos. 2011, 2011, 536735. [Google Scholar] [CrossRef][Green Version]
- Taketani, T.; Oyama, C.; Mihara, A.; Tanabe, Y.; Abe, M.; Hirade, T.; Yamamoto, S.; Bo, R.; Kanai, R.; Tadenuma, T.; et al. Ex Vivo Expanded Allogeneic Mesenchymal Stem Cells With Bone Marrow Transplantation Improved Osteogenesis in Infants With Severe Hypophosphatasia. Cell Transplant. 2015, 24, 1931–1943. [Google Scholar] [CrossRef]
- Liu, J.; Nam, H.K.; Campbell, C.; Gasque, K.C.d.S.; Millán, J.L.; Hatch, N.E. Tissue-nonspecific Alkaline Phosphatase Deficiency Causes Abnormal Craniofacial Bone Development in the Alpl(−/−) Mouse Model of Infantile Hypophosphatasia. Bone 2014, 67, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Brooke, M.; Kaiser, K. Three human myosin ATPase systems and their importance in muscle pathology. Neurology 1970, 20, 404–405. [Google Scholar]
- Peter, J.B.; Barnard, R.J.; Edgerton, V.R.; Gillespie, C.A.; Stempel, K.E. Metabolic profiles of three fiber types of skeletal muscle in guinea pigs and rabbits. Biochemistry 1972, 11, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Agbulut, O.; Noirez, P.; Beaumont, F.; Butler-Browne, G. Myosin heavy chain isoforms in postnatal muscle development of mice. Biol. Cell 2003, 95, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.L.; Leinwand, L.A. Postnatal Myosin Heavy Chain Isoform Expression in Normal Mice and Mice Null for IIb or IId Myosin Heavy Chains. Dev. Biol. 2001, 229, 383–395. [Google Scholar] [CrossRef]
- Schiaffino, S.; Rossi, A.C.; Smerdu, V.; Leinwand, L.A.; Reggiani, C. Developmental myosins: Expression patterns and functional significance. Skelet. Muscle 2015, 5, 22. [Google Scholar] [CrossRef]
- Staron, R.S. Human skeletal muscle fiber types: Delineation, development, and distribution. Can. J. Appl. Physiol. 1997, 22, 307–327. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, L.; Xuan, K.; Hu, C.; Liu, S.; Liao, L.; Li, B.; Jin, F.; Shi, S.; Jin, Y. Alpl prevents bone ageing sensitivity by specifically regulating senescence and differentiation in mesenchymal stem cells. Bone Res. 2018, 6, 27. [Google Scholar] [CrossRef]
- Cairns, S.P.; Chin, E.R.; Renaud, J.-M. Stimulation pulse characteristics and electrode configuration determine site of excitation in isolated mammalian skeletal muscle: Implications for fatigue. J. Appl. Physiol. 2007, 103, 359–368. [Google Scholar] [CrossRef]
- Ohkubo, S.; Kimura, J.; Matsuoka, I. Ecto-alkaline phosphatase in NG108-15 cells: A key enzyme mediating P1 antagonist-sensitive ATP response. Br. J. Pharmacol. 2000, 131, 1667–1672. [Google Scholar] [CrossRef]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef]
- Millán, J.L.; Narisawa, S.; Lemire, I.; Loisel, T.P.; Boileau, G.; Leonard, P.; Gramatikova, S.; Terkeltaub, R.; Camacho, N.P.; McKee, M.D.; et al. Enzyme Replacement Therapy for Murine Hypophosphatasia. J. Bone Miner. Res. 2008, 23, 777–787. [Google Scholar] [CrossRef]
- Call, J.A.; Lowe, D.A. Eccentric Contraction-Induced Muscle Injury: Reproducible, Quantitative, Physiological Models to Impair Skeletal Muscle’s Capacity to Generate Force. Methods Mol. Biol. 2016, 1460, 3–18. [Google Scholar] [CrossRef]
- Allen, D.L.; Harrison, B.C.; Maass, A.; Bell, M.L.; Byrnes, W.C.; Leinwand, L.A. Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. J. Appl. Physiol. 2001, 90, 1900–1908. [Google Scholar] [CrossRef]
- Punsoni, M.; Mangray, S.; Lombardo, K.A.; Heath, N.; Stopa, E.G.; Yakirevich, E. Succinate Dehydrogenase B (SDHB) Immunohistochemistry for the Evaluation of Muscle Biopsies. Appl. Immunohistochem. Mol. Morphol. AIMM 2017, 25, 645–650. [Google Scholar] [CrossRef]
- Tehrani, K.F.; Pendleton, E.G.; Southern, W.M.; Call, J.A.; Mortensen, L.J. Two-photon deep-tissue spatially resolved mitochondrial imaging using membrane potential fluorescence fluctuations. Biomed. Opt. Express 2018, 9, 254–259. [Google Scholar] [CrossRef]
- Nichenko, A.S.; Southern, W.M.; Tehrani, K.F.; Qualls, A.E.; Flemington, A.B.; Mercer, G.H.; Yin, A.; Mortensen, L.J.; Yin, H.; Call, J.A. Mitochondrial-specific autophagy linked to mitochondrial dysfunction following traumatic freeze injury in mice. Am. J. Physiol. Cell Physiol. 2020, 318, C242–C252. [Google Scholar] [CrossRef]
- Nichenko, A.S.; Southern, W.M.; Atuan, M.; Luan, J.; Peissig, K.B.; Foltz, S.J.; Beedle, A.M.; Warren, G.L.; Call, J.A. Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. Am. J. Physiol. Cell Physiol. 2016, 311, C190–C200. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Postnatal Day | HPP Weight | HPP Weight as Percent of WT Littermate | HPP Body Condition Score | Abnormal Movement/HPP Total |
|---|---|---|---|---|
| P10 | 4.67 ± 0.82 g | 84.8 ± 18.2% | 2.63 ± 0.34 | 3/31 |
| P12 | 5.16 ± 0.88 g | 84.3 ± 16.3% | 2.61 ± 0.34 | 6/31 |
| P14 | 5.58 ± 0.99 g | 87.6 ± 27.6% | 2.64 ± 0.33 | 11/31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pendleton, E.G.; Nichenko, A.S.; Mcfaline-Figueroa, J.; Raymond-Pope, C.J.; Schifino, A.G.; Pigg, T.M.; Barrow, R.P.; Greising, S.M.; Call, J.A.; Mortensen, L.J. Compromised Muscle Properties in a Severe Hypophosphatasia Murine Model. Int. J. Mol. Sci. 2023, 24, 15905. https://doi.org/10.3390/ijms242115905
Pendleton EG, Nichenko AS, Mcfaline-Figueroa J, Raymond-Pope CJ, Schifino AG, Pigg TM, Barrow RP, Greising SM, Call JA, Mortensen LJ. Compromised Muscle Properties in a Severe Hypophosphatasia Murine Model. International Journal of Molecular Sciences. 2023; 24(21):15905. https://doi.org/10.3390/ijms242115905
Chicago/Turabian StylePendleton, Emily G., Anna S. Nichenko, Jennifer Mcfaline-Figueroa, Christiana J. Raymond-Pope, Albino G. Schifino, Taylor M. Pigg, Ruth P. Barrow, Sarah M. Greising, Jarrod A. Call, and Luke J. Mortensen. 2023. "Compromised Muscle Properties in a Severe Hypophosphatasia Murine Model" International Journal of Molecular Sciences 24, no. 21: 15905. https://doi.org/10.3390/ijms242115905
APA StylePendleton, E. G., Nichenko, A. S., Mcfaline-Figueroa, J., Raymond-Pope, C. J., Schifino, A. G., Pigg, T. M., Barrow, R. P., Greising, S. M., Call, J. A., & Mortensen, L. J. (2023). Compromised Muscle Properties in a Severe Hypophosphatasia Murine Model. International Journal of Molecular Sciences, 24(21), 15905. https://doi.org/10.3390/ijms242115905