
REV7 Monomer Is Unable to Participate in Double Strand Break Repair and Translesion Synthesis but Suppresses Mitotic Errors

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
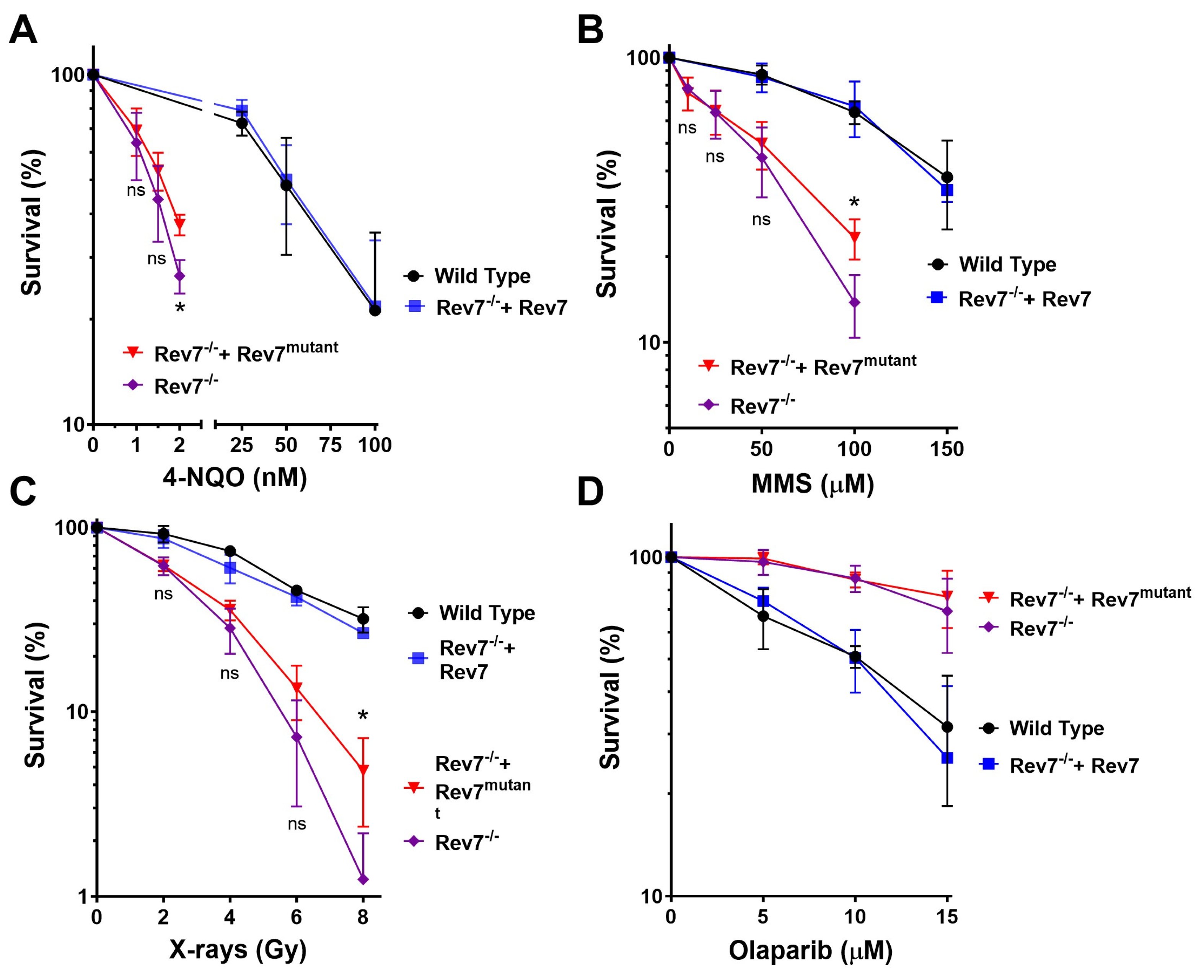
2.1. Rev7 Dimerization Is Required for Resistance to DNA-Damaging Agents
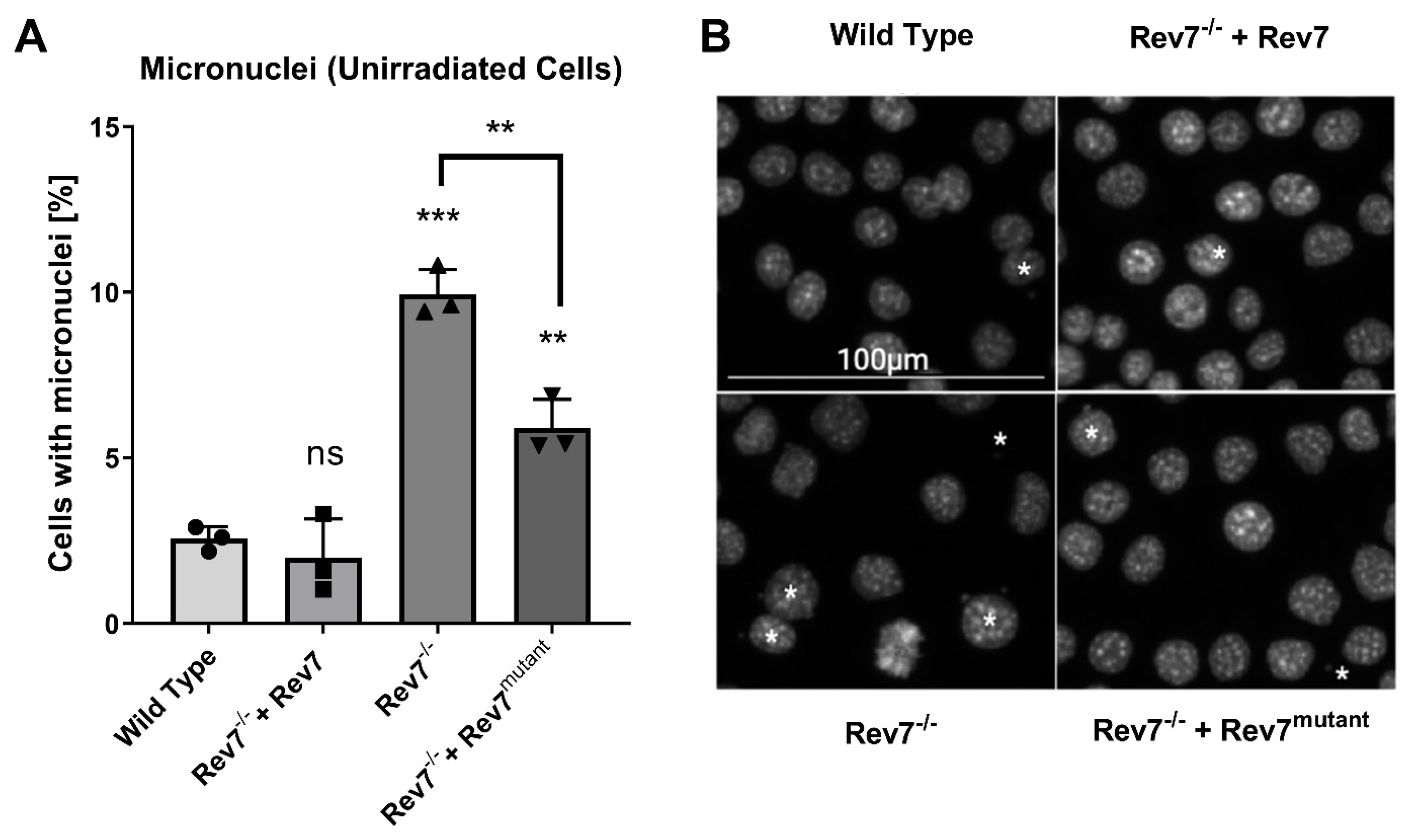
2.2. The Rev7 Homodimer Suppresses Genomic Instability
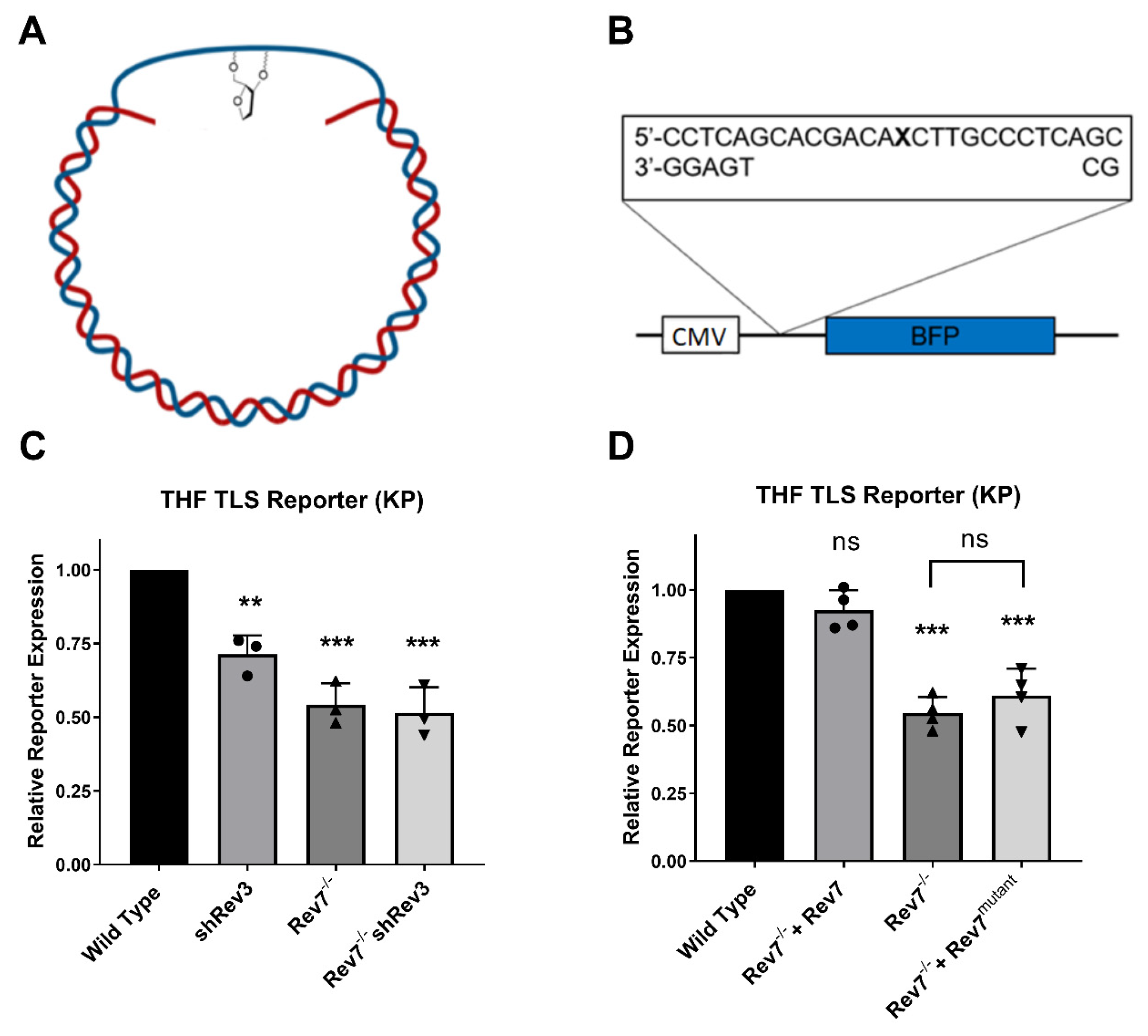
2.3. Rev7 Dimerization Is Required for Pol Zeta Function in Gap-Filling TLS
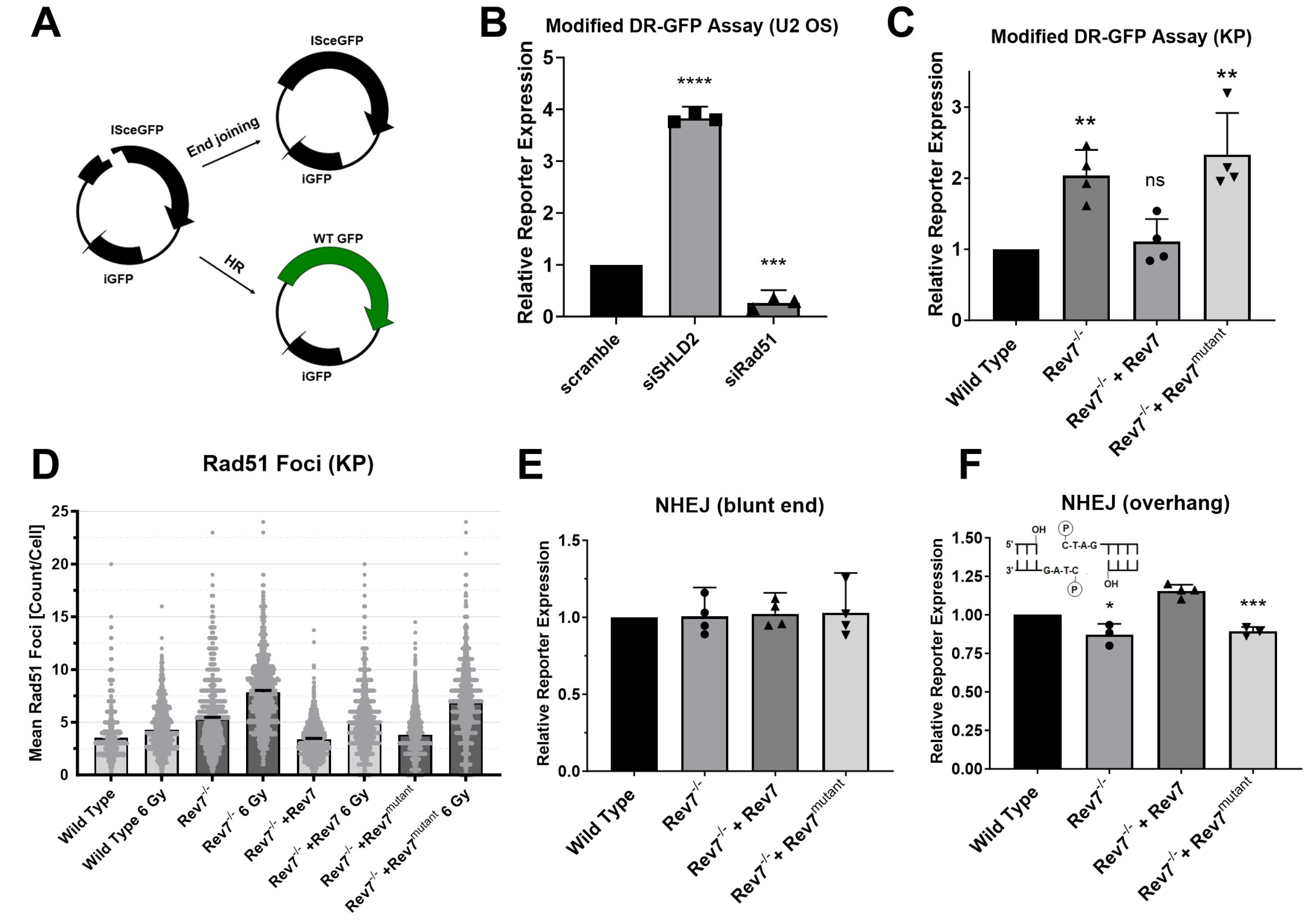
2.4. Rev7 Dimerization Is Required for Suppression of End Resection by Shieldin
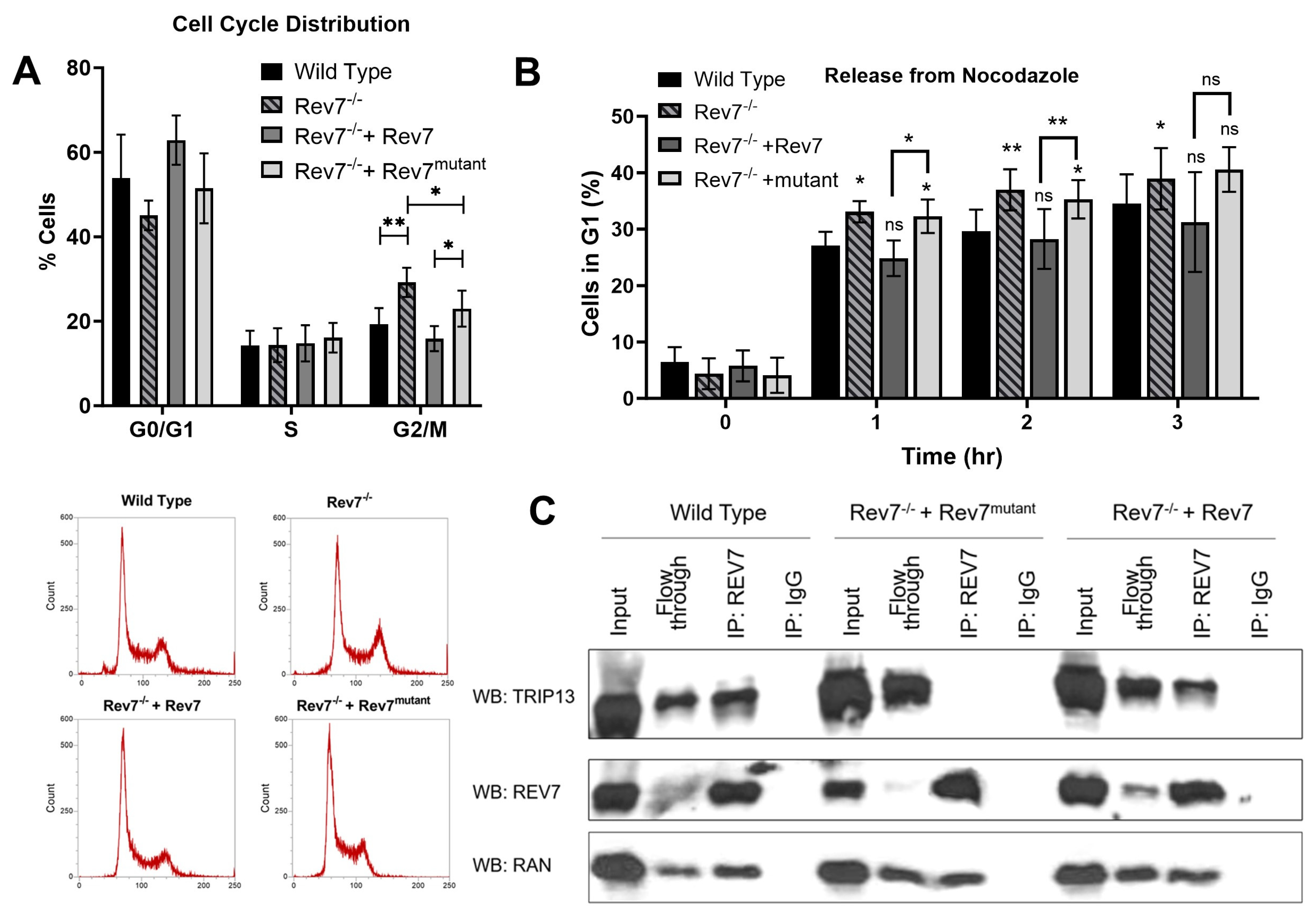
2.5. Rev7 Homodimerization Is Partially Dispensable in Cell Cycle Regulation
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Culture
4.3. Chemicals
4.4. Enzymes
4.5. Oligonucleotides
4.6. Western Blotting
4.7. Antibodies
4.8. Clonogenic Survival Assays
4.9. Micronucleus Assay
4.10. Rad51 and γH2AX Focus Assays
4.11. Cell Cycle Profiling
4.12. Co-Immunoprecipitation
4.13. Reporter Plasmid Cocktails
4.14. Fluorescent Host-Cell Reactivation Assays
4.15. Incorporation of THF into the Modified Pmax BFP Plasmid
4.16. Generation of Gapped Reporter Plasmids for TLS
4.17. DSB Rejoining Reporters (NHEJ)
4.18. Modified DR-GFP Reporter Assay
4.19. CellTiterGlo Viability Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cybulla, E.; Vindigni, A. Leveraging the replication stress response to optimize cancer therapy. Nat. Rev. Cancer 2023, 23, 6–24. [Google Scholar] [CrossRef]
- Anand, J.; Chiou, L.; Sciandra, C.; Zhang, X.; Hong, J.; Wu, D.; Zhou, P.; Vaziri, C. Roles of trans-lesion synthesis (TLS) DNA polymerases in tumorigenesis and cancer therapy. NAR Cancer 2023, 5, zcad005. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.M.; Dash, R.C.; Hadden, M.K. Translesion synthesis inhibitors as a new class of cancer chemotherapeutics. Expert Opin. Investig. Drugs 2021, 30, 13–24. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Stallons, L.J.; McGregor, W.G. Translesion synthesis polymerases in the prevention and promotion of carcinogenesis. J. Nucleic Acids 2010, 2010, 643857. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, C.; Rogozin, I.B.; Gu, Q.; Wu, D.; Day, T.A. Unravelling roles of error-prone DNA polymerases in shaping cancer genomes. Oncogene 2021, 40, 6549–6565. [Google Scholar] [CrossRef]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef]
- Lee, Y.S.; Gregory, M.T.; Yang, W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. USA 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Hara, K.; Shimizu, T.; Sato, M.; Hashimoto, H. Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J. Biol. Chem. 2012, 287, 33847–33852. [Google Scholar] [CrossRef]
- Pustovalova, Y.; Bezsonova, I.; Korzhnev, D.M. The C-terminal domain of human Rev1 contains independent binding sites for DNA polymerase η and Rev7 subunit of polymerase ζ. FEBS Lett. 2012, 586, 3051–3056. [Google Scholar] [CrossRef]
- Ross, A.L.; Simpson, L.J.; Sale, J.E. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005, 33, 1280–1289. [Google Scholar] [CrossRef]
- Malik, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Cryo-EM structure of translesion DNA synthesis polymerase ζ with a base pair mismatch. Nat. Commun. 2022, 13, 1050. [Google Scholar] [CrossRef]
- Akagi, J.; Masutani, C.; Kataoka, Y.; Kan, T.; Ohashi, E.; Mori, T.; Ohmori, H.; Hanaoka, F. Interaction with DNA polymerase eta is required for nuclear accumulation of REV1 and suppression of spontaneous mutations in human cells. DNA Repair 2009, 8, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Washington, M.T.; Haracska, L.; Prakash, S.; Prakash, L. Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 2000, 406, 1015–1019. [Google Scholar] [CrossRef]
- Yoon, J.H.; Bhatia, G.; Prakash, S.; Prakash, L. Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases kappa and zeta in human cells. Proc. Natl. Acad. Sci. USA 2010, 107, 14116–14121. [Google Scholar] [CrossRef] [PubMed]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; McArthur, M.J.; Park, J.; Basu, D.; Wakamiya, M.; Prakash, L.; Prakash, S. Error-Prone Replication through UV Lesions by DNA Polymerase θ Protects against Skin Cancers. Cell 2019, 176, 1295–1309.e1215. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e923. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of PRIMPOL-dependent ssDNA gaps. Mol. Cell 2021, 81, 4008–4025.e4007. [Google Scholar] [CrossRef] [PubMed]
- Paniagua, I.; Tayeh, Z.; Falcone, M.; Hernández Pérez, S.; Cerutti, A.; Jacobs, J.J.L. MAD2L2 promotes replication fork protection and recovery in a shieldin-independent and REV3L-dependent manner. Nat. Commun. 2022, 13, 5167. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, R.O.; Willis, N.A.; Elia, A.E.H.; Clairmont, C.; Li, S.; Wu, X.; D’Andrea, A.D.; Scully, R.; Elledge, S.J. The Protexin complex counters resection on stalled forks to promote homologous recombination and crosslink repair. Mol. Cell 2021, 81, 4440–4456.e4447. [Google Scholar] [CrossRef]
- Listovsky, T.; Sale, J.E. Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J. Cell Biol. 2013, 203, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Wu, Z.; Maher, V.M.; McCormick, J.J.; Xiao, W. Rev7/Mad2B plays a critical role in the assembly of a functional mitotic spindle. Cell Cycle 2015, 14, 3929–3938. [Google Scholar] [CrossRef]
- Wang, X.; Pernicone, N.; Pertz, L.; Hua, D.; Zhang, T.; Listovsky, T.; Xie, W. REV7 has a dynamic adaptor region to accommodate small GTPase RAN/Shigella IpaB ligands, and its activity is regulated by the RanGTP/GDP switch. J. Biol. Chem. 2019, 294, 15733–15742. [Google Scholar] [CrossRef]
- Malik, R.; Kopylov, M.; Gomez-Llorente, Y.; Jain, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Structure and mechanism of B-family DNA polymerase ζ specialized for translesion DNA synthesis. Nat. Struct. Mol. Biol. 2020, 27, 913–924. [Google Scholar] [CrossRef]
- Tomida, J.; Takata, K.; Lange, S.S.; Schibler, A.C.; Yousefzadeh, M.J.; Bhetawal, S.; Dent, S.Y.; Wood, R.D. REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase ζ. Nucleic Acids Res. 2015, 43, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Shimizu, T.; Unzai, S.; Akashi, S.; Sato, M.; Hashimoto, H. Purification, crystallization and initial X-ray diffraction study of human REV7 in complex with a REV3 fragment. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 1302–1305. [Google Scholar] [CrossRef]
- Rizzo, A.A.; Vassel, F.M.; Chatterjee, N.; D’Souza, S.; Li, Y.; Hao, B.; Hemann, M.T.; Walker, G.C.; Korzhnev, D.M. Rev7 dimerization is important for assembly and function of the Rev1/Polζ translesion synthesis complex. Proc. Natl. Acad. Sci. USA 2018, 115, E8191–E8200. [Google Scholar] [CrossRef]
- DuPage, M.; Dooley, A.L.; Jacks, T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 2009, 4, 1064–1072. [Google Scholar] [CrossRef]
- Vassel, F.M.; Bian, K.; Walker, G.C.; Hemann, M.T. Rev7 loss alters cisplatin response and increases drug efficacy in chemotherapy-resistant lung cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 28922–28924. [Google Scholar] [CrossRef]
- Wittschieben, J.P.; Reshmi, S.C.; Gollin, S.M.; Wood, R.D. Loss of DNA polymerase zeta causes chromosomal instability in mammalian cells. Cancer Res. 2006, 66, 134–142. [Google Scholar] [CrossRef]
- Wiltrout, M.E.; Walker, G.C. The DNA polymerase activity of Saccharomyces cerevisiae Rev1 is biologically significant. Genetics 2011, 187, 21–35. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Jdey, W.; Thierry, S.; Popova, T.; Stern, M.H.; Dutreix, M. Micronuclei Frequency in Tumors Is a Predictive Biomarker for Genetic Instability and Sensitivity to the DNA Repair Inhibitor AsiDNA. Cancer Res. 2017, 77, 4207–4216. [Google Scholar] [CrossRef] [PubMed]
- Iarmarcovai, G.; Ceppi, M.; Botta, A.; Orsière, T.; Bonassi, S. Micronuclei frequency in peripheral blood lymphocytes of cancer patients: A meta-analysis. Mutat. Res. 2008, 659, 274–283. [Google Scholar] [CrossRef]
- Tomer, G.; Livneh, Z. Analysis of unassisted translesion replication by the DNA polymerase III holoenzyme. Biochemistry 1999, 38, 5948–5958. [Google Scholar] [CrossRef] [PubMed]
- Avkin, S.; Goldsmith, M.; Velasco-Miguel, S.; Geacintov, N.; Friedberg, E.C.; Livneh, Z. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: The role of DNA polymerase kappa. J. Biol. Chem. 2004, 279, 53298–53305. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Z.D.; Margulies, C.M.; Chaim, I.A.; McRee, S.K.; Mazzucato, P.; Ahmad, A.; Abo, R.P.; Butty, V.L.; Forget, A.L.; Samson, L.D. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E1823–E1832. [Google Scholar] [CrossRef] [PubMed]
- de Krijger, I.; Föhr, B.; Pérez, S.H.; Vincendeau, E.; Serrat, J.; Thouin, A.M.; Susvirkar, V.; Lescale, C.; Paniagua, I.; Hoekman, L.; et al. MAD2L2 dimerization and TRIP13 control shieldin activity in DNA repair. Nat. Commun. 2021, 12, 5421. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, J.; Lee, C.J.; D’Souza, S.; Minesinger, B.; Kim, H.; D’Andrea, A.D.; Walker, G.C.; Zhou, P. Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) ζ, and Pol κ. J. Biol. Chem. 2012, 287, 33836–33846. [Google Scholar] [CrossRef]
- Gan, G.N.; Wittschieben, J.P.; Wittschieben, B.; Wood, R.D. DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 2008, 18, 174–183. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e886. [Google Scholar] [CrossRef]
- Findlay, S.; Heath, J.; Luo, V.M.; Malina, A.; Morin, T.; Coulombe, Y.; Djerir, B.; Li, Z.; Samiei, A.; Simo-Cheyou, E.; et al. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 2018, 37, e100158. [Google Scholar] [CrossRef]
- Gisler, S.; Gonçalves, J.P.; Akhtar, W.; de Jong, J.; Pindyurin, A.V.; Wessels, L.F.A.; van Lohuizen, M. Multiplexed Cas9 targeting reveals genomic location effects and gRNA-based staggered breaks influencing mutation efficiency. Nat. Commun. 2019, 10, 1598. [Google Scholar] [CrossRef]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, J.; Kool, H.; van Schendel, R.; Tijsterman, M. Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J. 2017, 36, 3634–3649. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Barwacz, S.A.; Bhowmick, R.; Lundgaard, K.; Gonçalves Dinis, M.M.; Clausen, M.; Kanemaki, M.T.; Liu, Y. Mitotic DNA synthesis in response to replication stress requires the sequential action of DNA polymerases zeta and delta in human cells. Nat. Commun. 2023, 14, 706. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Feng, J.; Zuo, P.; Yang, J.; Lu, Y.; Yin, Y. Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat. Commun. 2020, 11, 1972. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Tomimatsu, N.; Burma, S. Immunofluorescence-based methods to monitor DNA end resection. Stress Responses Methods Protoc. 2015, 1292, 67–75. [Google Scholar] [CrossRef]
- Cruz-García, A.; López-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Dickins, R.A.; Hemann, M.T.; Zilfou, J.T.; Simpson, D.R.; Ibarra, I.; Hannon, G.J.; Lowe, S.W. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat. Genet. 2005, 37, 1289–1295. [Google Scholar] [CrossRef]
- Piett, C.G.; Pecen, T.J.; Laverty, D.J.; Nagel, Z.D. Large-scale preparation of fluorescence multiplex host cell reactivation (FM-HCR) reporters. Nat. Protoc. 2021, 16, 4265–4298. [Google Scholar] [CrossRef]
- Chaim, I.A.; Nagel, Z.D.; Jordan, J.J.; Mazzucato, P.; Ngo, L.P.; Samson, L.D. In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proc. Natl. Acad. Sci. USA 2017, 114, E10379–E10388. [Google Scholar] [CrossRef]
- Tomida, J.; Takata, K.I.; Bhetawal, S.; Person, M.D.; Chao, H.P.; Tang, D.G.; Wood, R.D. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 2018, 37, e99543. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassel, F.M.; Laverty, D.J.; Bian, K.; Piett, C.G.; Hemann, M.T.; Walker, G.C.; Nagel, Z.D. REV7 Monomer Is Unable to Participate in Double Strand Break Repair and Translesion Synthesis but Suppresses Mitotic Errors. Int. J. Mol. Sci. 2023, 24, 15799. https://doi.org/10.3390/ijms242115799
Vassel FM, Laverty DJ, Bian K, Piett CG, Hemann MT, Walker GC, Nagel ZD. REV7 Monomer Is Unable to Participate in Double Strand Break Repair and Translesion Synthesis but Suppresses Mitotic Errors. International Journal of Molecular Sciences. 2023; 24(21):15799. https://doi.org/10.3390/ijms242115799
Chicago/Turabian StyleVassel, Faye M., Daniel J. Laverty, Ke Bian, Cortt G. Piett, Michael T. Hemann, Graham C. Walker, and Zachary D. Nagel. 2023. "REV7 Monomer Is Unable to Participate in Double Strand Break Repair and Translesion Synthesis but Suppresses Mitotic Errors" International Journal of Molecular Sciences 24, no. 21: 15799. https://doi.org/10.3390/ijms242115799
APA StyleVassel, F. M., Laverty, D. J., Bian, K., Piett, C. G., Hemann, M. T., Walker, G. C., & Nagel, Z. D. (2023). REV7 Monomer Is Unable to Participate in Double Strand Break Repair and Translesion Synthesis but Suppresses Mitotic Errors. International Journal of Molecular Sciences, 24(21), 15799. https://doi.org/10.3390/ijms242115799