
Atherosclerosis and Its Related Laboratory Biomarkers


Abstract
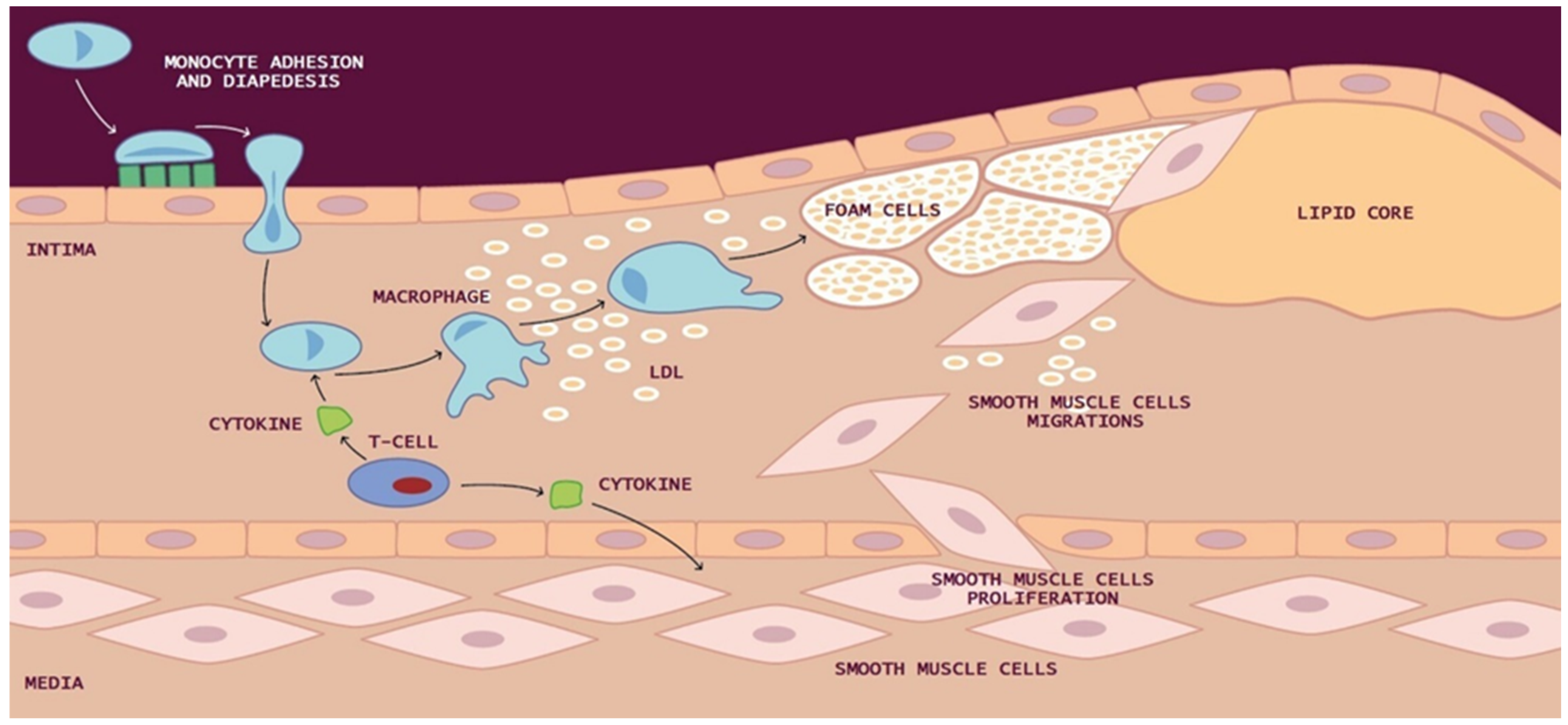
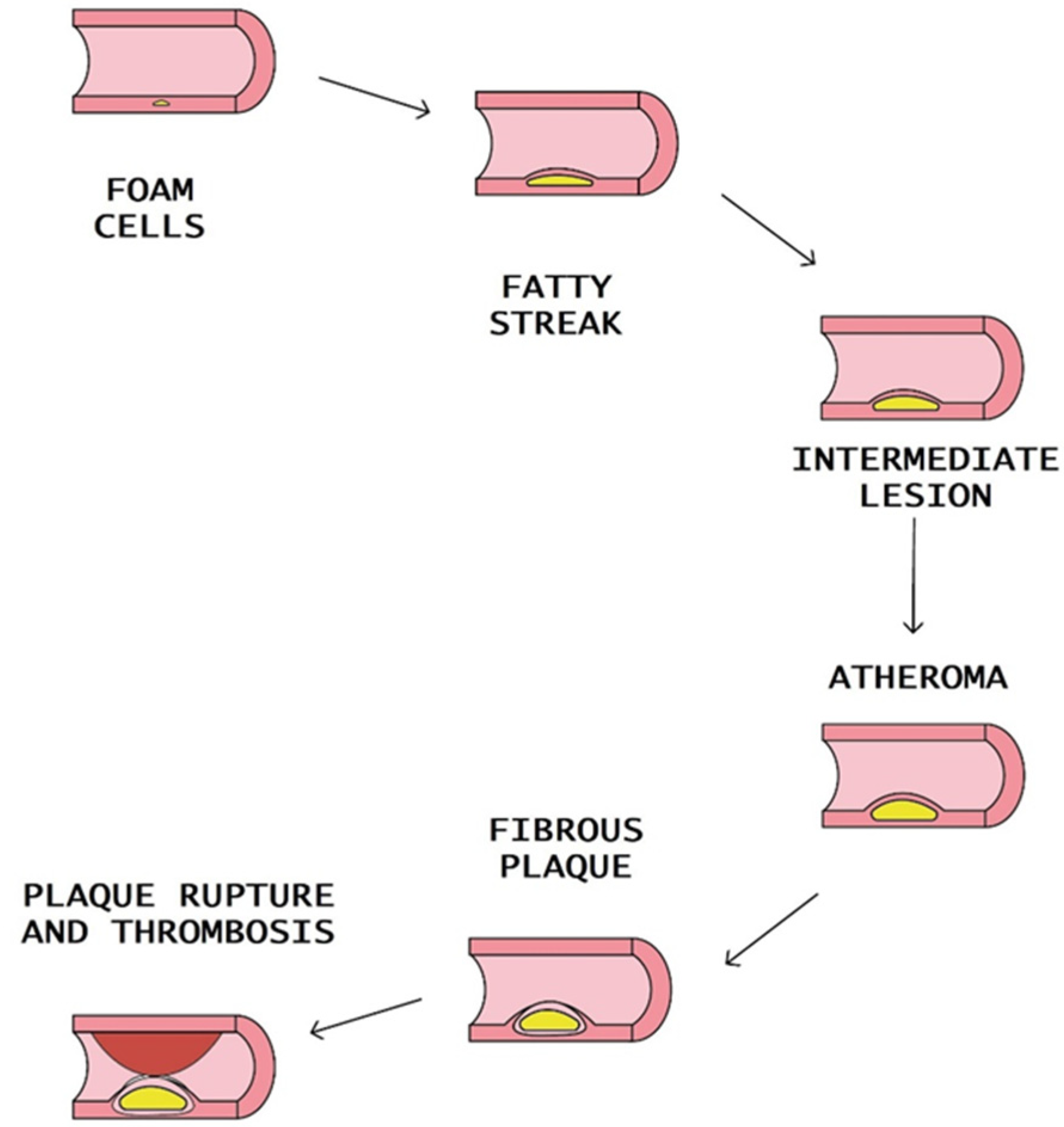
:1. Introduction
2. Biomarkers of Atherosclerosis
2.1. Lipoproteins
2.2. Microparticles and Exosomes
2.3. C-Reactive Protein
2.4. Inflammatory Markers
2.5. Cells
2.6. Micro-RNA (miRNA)
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAD | coronary artery disease |
| PAD | peripheral artery disease |
| CVD | cardiovascular disease |
| LDL | low-density lipoprotein |
| HDL | high-density lipoprotein |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| ox-LDL | oxidized LDL |
| ECs | endothelial cells |
| VCAM-1 | vascular cell adhesion molecule-1 |
| ICAM-1 | intercellular adhesion molecule-1 |
| IL-8 | interleukin-8 |
| MCP-1 | monocyte chemotactic protein-1 |
| M-CSF | macrophage colony-stimulating factor |
| SMCs | smooth muscle cells |
| LbLDL | large buoyant LDL |
| SdLDL | small dense LDL |
| CHD | coronary heart disease |
| TG | triglyceride |
| HDL-C | HDL cholesterol |
| NCEPIII | National Cholesterol Education Program |
| Lp(a) | lipoprotein(a) |
| ASCVD | atherosclerotic cardiovascular disease |
| AHA | American Heart Association |
| ACC | American College of Cardiology |
| hsCRP | high-sensitivity C-reactive protein |
| MACE | major adverse cardiovascular events |
| MPs | microparticles |
| EVs | extracellular vesicles |
| MVBs | multivesicular bodies |
| BAT | brown adipose tissue |
| miRNAs | micro-RNAs |
| lncRNAs | long non-coding RNAs |
| circRNAs | circulating RNAs |
| ACADVL | very long-chain acyl-CoA dehydrogenase |
| ABCA1 | ATP-binding cassette transporter A1 |
| IL-1 | interleukin-1 |
| IL-6 | interleukin-6 |
| TNF | tumor necrosis factor |
| IL-1β | interleukin-1β |
| NLRP3 | NLR family pyrin domain-containing 3 |
| NF-κB | nuclear factor kappa B |
| MHC | major histocompatibility complex |
| APCs | antigen-presenting cells |
| TCR | T cell receptor |
| scRNA-seq | single-cell RNA sequencing |
| CyTOF | mass cytometry |
| CITE-seq | cellular indexing of transcriptomes and epitopes by sequencing |
| NKT | natural killer T |
| iNKT | invariant NKT |
| TCRs | T cell receptors |
| IFNs | interferons |
| TLR | toll-like receptor |
| IL-4/IL-13 | interleukin-4/interleukin-13 |
| IL-12 | interleukin-12 |
| IL-23 | interleukin-23 |
| NOS | NOsynthase |
| ETS-1 | E26 transformation-specific sequence |
References
- Orekhov, A.N.; Ivanova, E.A. Introduction of the special issue “Atherosclerosis and Related Diseases”. Vessel Plus 2017, 1, 163–165. [Google Scholar] [CrossRef]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2019, 10, 1568. [Google Scholar] [CrossRef] [PubMed]
- Alipov, V.I.; Sukhorukov, V.N.; Karagodin, V.P.; Grechko, A.V.; Orekhov, A.N. Chemical composition of circulating native and desialylated low density lipoprotein: What is the difference? Vessel Plus 2017, 1, 107–115. [Google Scholar] [CrossRef]
- Orekhov, A.N. Modified lipoproteins as biomarkers of atherosclerosis. Front. Biosci. 2018, 23, 1422–1444. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, S.B.; Eftedal, I.; Pedersen, M.; Jeppesen, P.B.; Nørregaard, R.; Matchkov, V.V. Endothelial dysfunction in small arteries and early signs of atherosclerosis in ApoE knockout rats. Sci. Rep. 2020, 10, 15296. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. Lipoprotein subfractions and cardiovascular disease risk. Curr. Opin. Infect. Dis. 2010, 21, 305–311. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2020, 11, 613780. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Qiao, Y.-N.; Zou, Y.-L.; Guo, S.-D. Low-density lipoprotein particles in atherosclerosis. Front. Physiol. 2022, 13, 931931. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1273042. [Google Scholar] [CrossRef] [PubMed]
- Brunzell, J.D.; Zambon, A.; Deeb, S.S. The effect of hepatic lipase on coronary artery disease in humans is influenced by the underlying lipoprotein phenotype. Biochim. Biophys. Acta 2012, 1821, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Epstein, F.H.; Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N. Desialylated low density lipoprotein--naturally occurring modified lipoprotein with atherogenic potency. Atherosclerosis 1991, 86, 153–161. [Google Scholar] [CrossRef] [PubMed]
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002, 106, 3143–3421. [Google Scholar] [CrossRef]
- Schmitz, G.; Orsó, E. Lipoprotein(a) hyperlipidemia as cardiovascular risk factor: Pathophysiological aspects. Clin. Res. Cardiol. Suppl. 2015, 10, 21–25. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M. Residual Cardiovascular Risk at Low LDL: Remnants, Lipoprotein(a), and Inflammation. Clin. Chem. 2021, 67, 143–153. [Google Scholar] [CrossRef]
- Tokgozoglu, L.; Kayikcioglu, M. Familial Hypercholesterolemia: Global Burden and Ap-proaches. Curr. Cardiol. Rep. 2021, 23, 151. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 74, e177–e232. [Google Scholar] [CrossRef]
- Della Corte, V.; Tuttolomondo, A.; Pecoraro, R.; Di Raimondo, D.; Vassallo, V.; Pinto, A. Inflammation, Endothelial Dysfunction and Arterial Stiffness as Therapeutic Targets in Cardiovascular Medicine. Curr. Pharm. Des. 2016, 22, 4658–4668. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Nissen, S.E.; Arsenault, B.J.; St John, J.; Riesmeyer, J.S.; Ruotolo, G.; McErlean, E.; Menon, V.; Cho, L.; Wolski, K.; et al. Effect of C-Reactive Protein on Lipoprotein(a)-Associated Cardiovascular Risk in Optimally Treated Patients With High-Risk Vascular Disease: A Prespecified Secondary Analysis of the ACCELERATE Trial. JAMA Cardiol. 2020, 5, 1136–1143. [Google Scholar] [CrossRef]
- Heo, J.; Kang, H. Exosome-Based Treatment for Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 1002. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signaling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, N.; Yan, T.; Shi, Y.-N.; Chen, J.; Zhang, C.-J.; Xie, X.-J.; Liao, D.-F.; Qin, L. The crosstalk: Exosomes and lipid metabolism. Cell Commun. Signal. 2020, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Z.; Qi, M.; Zhao, P.; Duan, Y.; Yang, G.; Yuan, L. Corresponding address, Lijun Yuan. Brown adipose tissue-derived exosomes mitigate the metabolic syndrome in high fat diet mice. Theranostics 2020, 10, 8197–8210. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Z.; Liu, Y.; Yuan, L. Exosomes in atherosclerosis: Performers, bystanders, biomarkers, and therapeutic targets. Theranostics 2021, 11, 3996–4010. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Proebsting, S.; Hoelscher, M.; Przybilla, D.; Baumann, K.; Schmitz, T.; Dolf, A.; Endl, E.; Franklin, B.S.; et al. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J. Am. Heart Assoc. 2014, 3, e001249. [Google Scholar] [CrossRef]
- Raju, S.; Fish, J.E.; Howe, K.L. MicroRNAs as sentinels and protagonists of carotid artery thromboembolism. Clin. Sci. 2020, 134, 169–192. [Google Scholar] [CrossRef]
- Jiang, H.; Toscano, J.F.; Song, S.S.; Schlick, K.H.; Dumitrascu, O.M.; Pan, J.; Lyden, P.D.; Saver, J.L.; Gonzalez, N.R. Differential expression of circulating exosomal microRNAs in refractory intracranial atherosclerosis associated with antiangiogenesis. Sci. Rep. 2019, 9, 19429. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.; Hosen, M.R.; Zietzer, A.; Flender, A.; Levermann, P.; Schmitz, T.; Frühwald, D.; Goody, P.; Nickenig, G.; et al. Atherosclerotic Conditions Promote the Packaging of Functional MicroRNA-92a-3p into Endothelial Microvesicles. Circ. Res. 2019, 124, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Zhang, S.; Yan, S.; Wang, Z.; Wang, C.; Zhang, X. MiR-30e and miR-92a are related to atherosclerosis by targeting ABCA1. Mol. Med. Rep. 2019, 19, 3298–3304. [Google Scholar] [CrossRef] [PubMed]
- Hajibabaie, F.; Kouhpayeh, S.; Mirian, M.; Rahimmanesh, I.; Boshtam, M.; Sadeghian, L.; Gheibi, A.; Khanahmad, H.; Shariati, L. MicroRNAs as the actors in the atherosclerosis scenario. J. Physiol. Biochem. 2020, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, T.A.; Duong, P.; Bouchareychas, L.; Chen, M.; Chung, A.; Schaller, M.S.; Oskowitz, A.; Raffai, R.L.; Conte, M.S. Circulating exosomes from patients with peripheral artery disease influence vascular cell migration and contain distinct microRNA cargo. JVS Vasc. Sci. 2020, 1, 28–41. [Google Scholar] [CrossRef]
- Gandham, S.; Su, X.; Wood, J.; Nocera, A.L.; Alli, S.C.; Milane, L.; Zimmerman, A.; Amiji, M.; Ivanov, A.R. Technologies and Standardization in Research on Extracellular Vesicles. Trends Biotechnol. 2020, 38, 1066–1098. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; on behalf of the CANTOS Trial Group. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomized controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Zhang, W.; Speiser, J.L.; Ye, F.; Tsai, M.Y.; Cainzos-Achirica, M.; Nasir, K.; Herrington, D.M.; Shapiro, M.D. High-sensitivity C-Reactive Protein Modifies the Cardiovascular Risk of Lipoprotein(a): Multi-Ethnic Study of Atherosclerosis. J. Am. Coll. Cardiol. 2021, 78, 1083–1094. [Google Scholar] [CrossRef]
- Ngwa, D.N.; Pathak, A.; Agrawal, A. IL-6 regulates induction of C-reactive protein gene expression by activating STAT3 isoforms. Mol. Immunol. 2022, 146, 50–56. [Google Scholar] [CrossRef]
- Braig, D.; Nero, T.L.; Koch, H.-G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A.; et al. Transitional changes in the CRP structure lead to the exposure of pro-inflammatory binding sites. Nat. Commun. 2017, 8, 14188. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Shih, J.; Matias, M.; Hennekens, C.H. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998, 98, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rossouw, J.E.; Siscovick, D.S.; Mouton, C.P.; Rifai, N.; Wallace, R.B.; Jackson, R.D.; Pettinger, M.B.; Ridke, P.M. Inflammatory biomarkers, hormone replacement therapy, and incident coronary heart disease: Prospective analysis from the Women’s Health Initiative observational study. JAMA 2002, 288, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Pai, J.K.; Pischon, T.; Ma, J.; Manson, J.E.; Hankinson, S.E.; Joshipura, K.; Curhan, G.C.; Rifai, N.; Cannuscio, C.C.; Stampfer, M.J.; et al. Inflammatory markers and the risk of coronary heart disease in men and women. N. Engl. J. Med. 2004, 351, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream to Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Danesh, J.; Wheeler, J.G.; Hirschfield, G.M.; Eda, S.; Eiriksdottir, G.; Rumley, A.; Lowe, G.D.; Pepys, M.B.; Gudnason, V. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N. Engl. J. Med. 2004, 350, 1387–1397. [Google Scholar] [CrossRef]
- Miller, M.; Zhan, M.; Havas, S. High attributable risk of elevated C-reactive protein level to conventional coronary heart disease risk factors: The Third National Health and Nutrition Examination Survey. Arch. Intern. Med. 2005, 165, 2063–2068. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Cannon, R.O., III; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myers, G.L.; et al. Markers of inflammation and cardiovascular disease: Application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003, 107, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Morrow, D.A.; Rose, L.M.; Rifai, N.; Cannon, C.P.; Braunwald, E. Relative efficacy of atorvastatin 80 mg and pravastatin 40 mg in achieving the dual goals of low-density lipoprotein cholesterol <70 mg/dL and C-reactive protein <2 mg/L: An analysis of the PROVE-IT TIMI-22 trial. J. Am. Coll. Cardiol. 2005, 45, 1644–1648. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Lehti, S.; Nguyen, S.D.; Belevich, I.; Vihinen, H.; Heikkilä, H.M.; Soliymani, R.; Käkelä, R.; Saksi, J.; Jauhiainen, M.; Grabowski, G.A.; et al. Extracellular Lipids Accumulate in Human Carotid Arteries as Distinct Three-Dimensional Structures and Have Pro-inflammatory Properties. Am. J. Pathol. 2018, 188, 525–538. [Google Scholar] [CrossRef]
- Lorey, M.B.; Öörni, K.; Kovanen, P.T. Modified Lipoproteins Induce Arterial Wall Inflammation during Atherogenesis. Front. Cardiovasc. Med. 2022, 9, 841545. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Gautier, E.L.; Ganda, A.; Molusky, M.M.; Wang, W.; Fotakis, P.; Wang, N.; Randolph, G.J.; D’Agati, V.D.; Yvan-Charvet, L.; et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab. 2017, 25, 1294–1304.e6. [Google Scholar] [CrossRef]
- Rajamäki, K.; Mäyränpää, M.I.; Risco, A.; Tuimala, J.; Nurmi, K.; Cuenda, A.; Eklund, K.K.; Öörni, K.; Kovanen, P.T. p38δ MAPK: A Novel Regulator of NLRP3 Inflammasome Activation With Increased Expression in Coronary Atherogenesis. Arter. Thromb. Vasc. Biol. 2016, 36, 1937–1946. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.N.; Carroll, R.G.; Galván-Peña, S.; Mills, E.L.; Olden, R.; Triantafilou, M.; Wolf, A.I.; Bryant, C.E.; Triantafilou, K.; Masters, S.L. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol. Med. 2017, 23, 165–180. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Varghese, G.P.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Demkes, E.J.; Fiolet, A.T.L.; Dekker, M.; Bosch, L.; van Hout, G.P.J.; Timmers, L.; de Kleijn, D.P.V. Immunomodulation of the NLRP3 Inflammasome in Atherosclerosis, Coronary Artery Disease, and Acute Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 23–34. [Google Scholar] [CrossRef]
- Abe, J.-I.; Berk, B.C. Atheroprone flow activation of the sterol regulatory element binding protein 2 and nod-like receptor protein 3 inflammasome mediates focal atherosclerosis. Circulation 2013, 128, 579–582. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Wheeler, M.E.; Cotran, R.S.; Gimbrone, M.A. Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am. J. Pathol. 1985, 121, 394–403. [Google Scholar] [PubMed]
- Libby, P.; Warner, S.J.; Friedman, G.B. Interleukin 1: A mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J. Clin. Investig. 1988, 81, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A. Directing transition from innate to acquired immunity: Defining a role for IL-6. J. Immunol. 2005, 175, 3463–3468. [Google Scholar] [CrossRef]
- Aker, S.; Bantis, C.; Reis, P.; Kuhr, N.; Schwandt, C.; Grabensee, B.; Heering, P.; Ivens, K. Influence of interleukin-6 G-174C gene polymorphism on coronary artery disease, cardiovascular complications and mortality in dialysis patients. Nephrol. Dial. Transplant. 2009, 24, 2847–2851. [Google Scholar] [CrossRef]
- Held, C.; White, H.D.; Stewart, R.A.H.; Budaj, A.; Cannon, C.P.; Hochman, J.S.; Koenig, W.; Siegbahn, A.; Steg, P.G.; Soffer, J.; et al. Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences From the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. J. Am. Heart Assoc. 2017, 6, e005077. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Mazzarino, M.C.; Spandidos, D.A.; Malaponte, G. Proinflammatory circulating molecules in peripheral arterial disease. Int. J. Mol. Med. 2007, 20, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, S.S.; Marino, E.; Scuto, S.; Di Raimondo, D. Pathophysiology of Peripheral Arterial Disease (PAD): A Review on Oxidative Disorders. Int. J. Mol. Sci. 2020, 21, 4393. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, S.S.; Li Volsi, G.; Fiore, V.; Mangiafico, M.; Barbagallo, I.; Parenti, R.; Rizzo, M.; Li Volti, G. Plasma heme oxygenase-1 is decreased in peripheral artery disease patients. Mol. Med. Rep. 2016, 14, 3459–3463. [Google Scholar] [CrossRef] [PubMed]
- Wachsmann-Maga, A.; Kaszuba, M.; Maga, M.; Włodarczyk, A.; Krężel, J.; Kaczmarczyk, P.; Bogucka, K.; Maga, P. Leukotrienes in the atherosclerotic cardiovascular diseases—A systematic review. Acta Angiol. 2022, 28, 147–153. [Google Scholar] [CrossRef]
- Maga, P.; Sanak, M.; Rewerska, B.; Maga, M.; Jawien, J.; Wachsmann, A.; Rewerski, P.; Szczeklik, W.; Celejewska-Wójcik, N. Urinary cysteinyl leukotrienes in one-year follow-up of percutaneous transluminal angioplasty for peripheral arterial occlusive disease. Atherosclerosis 2016, 249, 174–180. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef]
- He, S.; Kahles, F.; Rattik, S.; Nairz, M.; McAlpine, C.S.; Anzai, A.; Selgrade, D.; Fenn, A.M.; Chan, C.T.; Mindur, J.E.; et al. Gut intraepithelial T cells calibrate metabolism and accelerate cardiovascular disease. Nature 2019, 566, 115–119. [Google Scholar] [CrossRef]
- Kimura, T.; Kobiyama, K.; Winkels, H.; Tse, K.; Miller, J.; Vassallo, M.; Wolf, D.; Ryden, C.; Orecchioni, M.; Dileepan, T.; et al. Regulatory CD4+ T Cells Recognize Major Histocompatibility Complex Class II Molecule-Restricted Peptide Epitopes of Apolipoprotein B. Circulation 2018, 138, 1130–1143. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Koltsova, E.K.; Garcia, Z.; Chodaczek, G.; Landau, M.; McArdle, S.; Scott, S.R.; von Vietinghoff, S.; Galkina, E.; Miller, Y.I.; Acton, S.T.; et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J. Clin. Investig. 2012, 122, 3114–3126. [Google Scholar] [CrossRef]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.; Cochain, C.; Vafadarnejad, E.; Saliba, A.-E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef]
- Hansson, G.; Jonasson, L.; Lojsthed, B.; Stemme, S.; Kocher, O.; Gabbiani, G. Localization of T lymphocytes and macrophages in fibrous and complicated human atherosclerotic plaques. Atherosclerosis 1988, 72, 135–141. [Google Scholar] [CrossRef]
- Jonasson, L.; Holm, J.; Skalli, O.; Bondjers, G.; Hansson, G.K. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arterioscler. Dallas Tex 1986, 6, 131–138. [Google Scholar] [CrossRef]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.-E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef]
- Cole, J.E.; Park, I.; Ahern, D.J.; Kassiteridi, C.; Abeam, D.D.; Goddard, M.E.; Green, P.; Maffia, P.; Monaco, C. Immune cell census in murine atherosclerosis: Cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc. Res. 2018, 114, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.-A.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Major, A.S.; Wilson, M.T.; McCaleb, J.L.; Su, Y.R.; Stanic, A.K.; Joyce, S.; Van Kaer, L.; Fazio, S.; Linton, M.F. Quantitative and qualitative differences in proatherogenic NKT cells in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2351–2357. [Google Scholar] [CrossRef]
- van Puijvelde, G.H.M.; van Wanrooij, E.J.A.; Hauer, A.D.; de Vos, P.; van Berkel, T.J.C.; Kuiper, J. Effect of natural killer T cell activation on the initiation of atherosclerosis. Thromb. Haemost. 2009, 102, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; To, K.; Kanellakis, P.; Hosseini, H.; Deswaerte, V.; Tipping, P.; Smyth, M.J.; Toh, B.-H.; Bobik, A.; Kyaw, T.; et al. CD4+ natural killer T cells potently augment aortic root atherosclerosis by perforin- and granzyme B-dependent cytotoxicity. Circ. Res. 2015, 116, 245–254. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.-R.; Yang, S.-M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Bories, G.F.P.; Leitinger, N. Macrophage metabolism in atherosclerosis. FEBS Lett. 2017, 591, 3042–3060. [Google Scholar] [CrossRef]
- Riksen, N.P.; Stienstra, R. Metabolism of innate immune cells: Impact on atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Ghanbari, M.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. Implications of microRNAs in the Pathogenesis of Atherosclerosis and Prospects for Therapy. Curr. Drug Targets 2021, 22, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005, 37, 766–770. [Google Scholar] [CrossRef]
- De Rosa, S.; Curcio, A.; Indolfi, C. Emerging role of microRNAs in cardiovascular diseases. Circ. J. 2014, 78, 567–575. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Santovito, D.; Mezzetti, A.; Cipollone, F. MicroRNAs and atherosclerosis: New actors for an old movie. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 937–943. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Shirafkan, N.; Mansoori, B.; Mohammadi, A.; Shomali, N.; Ghasbi, M.; Baradaran, B. MicroRNAs as novel biomarkers for colorectal cancer: New outlooks. Biomed. Pharmacother. 2018, 97, 1319–1330. [Google Scholar] [CrossRef]
- Sharma, A.R.; Sharma, G.; Bhattacharya, M.; Lee, S.-S.; Chakraborty, C. Circulating miRNA in Atherosclerosis: A Clinical Biomarker and Early Diagnostic Tool. Curr. Mol. Med. 2022, 22, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Pak, K.; Goh, T.S.; Jeong, D.C.; Han, M.-E.; Kim, J.; Oh, S.-O.; Kim, C.D.; Kim, Y.H. Prognostic Value of MicroRNAs in Coronary Artery Diseases: A Meta-Analysis. Yonsei Med. J. 2018, 59, 495–500. [Google Scholar] [CrossRef]
- Lu, Y.; Thavarajah, T.; Gu, W.; Cai, J.; Xu, Q. Impact of miRNA in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, E159–E170. [Google Scholar] [CrossRef]
- Singh, S.; de Ronde, M.W.J.; Kok, M.G.M.; Beijk, M.A.; De Winter, R.J.; van der Wal, A.C.; Sondermeijer, B.M.; Meijers, J.C.M.; Creemers, E.E.; Pinto-Sietsma, S.-J. MiR-223-3p and miR-122-5p as circulating biomarkers for plaque instability. Open Heart 2020, 7, e001223. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, Y.; Fang, T.; Wang, X.; Chen, L.; Zheng, C.; Kang, Y.; Jiang, L.; You, X.; Gai, S.; et al. Circulating MicroRNA-423-3p Improves the Prediction of Coronary Artery Disease in a General Population–Six-Year Follow-up Results From the China-Cardiovascular Disease Study. Circ. J. 2020, 84, 1155–1162. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, H.; Zhu, M.; Qian, Y.; Lin, S.; Li, X. Circulating microRNAs as biomarkers for severe coronary artery disease. Medicine 2020, 99, e19971. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Vanella, L.; Abraham, N.G.; Scuto, S.; Marino, E.; Rocic, P. Pathophysiology of chronic peripheral ischemia: New perspectives. Ther. Adv. Chronic Dis. 2020, 11, 2040622319894466. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Anzaldi, M.; Fiore, V. Inflammation in peripheral arterial disease (PAD). Curr. Pharm. Des. 2012, 18, 4350–4357. [Google Scholar] [CrossRef] [PubMed]
- Tabaei, S.; Tabaee, S.S. Implications for MicroRNA involvement in the prognosis and treatment of atherosclerosis. Mol. Cell. Biochem. 2021, 476, 1327–1336. [Google Scholar] [CrossRef]
- Toor, S.M.; Aldous, E.K.; Parray, A.; Akhtar, N.; Al-Sarraj, Y.; Abdelalim, E.M.; Arredouani, A.; El-Agnaf, O.; Thornalley, P.J.; Pananchikkal, S.V.; et al. Circulating MicroRNA Profiling Identifies Distinct MicroRNA Signatures in Acute Ischemic Stroke and Transient Ischemic Attack Patients. Int. J. Mol. Sci. 2022, 24, 108. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Biomarkers | Function in Atherosclerosis |
|---|---|
| Ox-LDL | initiation and progression of atherosclerosis by inducing dysfunction in endothelial cells (ECs) |
| sdLDL | powerful source of cholesterol for developing atherosclerotic plaque |
| Lp(a) | pro-inflammatory action on the arterial wall due to oxidized phospholipids, and atherogenic and pro-thrombotic activity due to the homology with plasminogen |
| CRP | upregulate cell adhesion molecules, enhance the release of MCP-1, facilitate the uptake of LDL into macrophages |
| M1-like macrophages | induce the recruitment and activation of other macrophages, T cells, B cells, and dendritic cells, promoting inflammation and advancing atherosclerotic plaque development |
| M2-like macrophages | secrete anti-inflammatory and pro-fibrotic mediators and help limit inflammation, thus inhibiting the progression of atherosclerosis |
| miRNAs | Function in Atherosclerosis and Their Potential as a Biomarker |
|---|---|
| Mir-122-5p, Mir-27b-3p, Mir-101-3p | related to ischemic events in intracranial atherosclerotic disease |
| Mir-30e, Mir-92, Mir-92a-3p miR-17-92 | released by endothelial cells involved in atherogenesis |
| Mir-133a, Mir-155, Mir-21, Mir-210, Mir-126, and Mir-499 | atherosclerotic lesion development |
| MiR-126 | modulating in negative vascular inflammation |
| MiR-155 and MiR-221/222 | down-regulate the expression of NOsynthase (NOS) |
| MiR-21 | promoter of VSMC proliferation |
| MiR-26a | induces VSMC proliferation and migration |
| MiR-145 and MiR-133a | inhibit proliferation and prevent the phenotypic switch |
| MiR-30c, MiR-148a, and MiR-21 | play atheroprotective role |
| MiR-223 | driving initiation, progression, and plaque rupture steps |
| MiR-155, MiR-222, MiR-424, and MiR-503 | promote monocyte differentiation |
| MiR-233-3 p, MiR-423-3p, MiR-32-3p, MiR-3149, and MiR-26a-5p | reliable as a biomarker in acute coronary syndrome |
| MiR-100, MiR-133a/b, and MiR-127 | could identify plaques with clinical evidence of vulnerability |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Corte, V.; Todaro, F.; Cataldi, M.; Tuttolomondo, A. Atherosclerosis and Its Related Laboratory Biomarkers. Int. J. Mol. Sci. 2023, 24, 15546. https://doi.org/10.3390/ijms242115546
Della Corte V, Todaro F, Cataldi M, Tuttolomondo A. Atherosclerosis and Its Related Laboratory Biomarkers. International Journal of Molecular Sciences. 2023; 24(21):15546. https://doi.org/10.3390/ijms242115546
Chicago/Turabian StyleDella Corte, Vittoriano, Federica Todaro, Marco Cataldi, and Antonino Tuttolomondo. 2023. "Atherosclerosis and Its Related Laboratory Biomarkers" International Journal of Molecular Sciences 24, no. 21: 15546. https://doi.org/10.3390/ijms242115546
APA StyleDella Corte, V., Todaro, F., Cataldi, M., & Tuttolomondo, A. (2023). Atherosclerosis and Its Related Laboratory Biomarkers. International Journal of Molecular Sciences, 24(21), 15546. https://doi.org/10.3390/ijms242115546