
Changes in Plasma Pyruvate and TCA Cycle Metabolites upon Increased Hepatic Fatty Acid Oxidation and Ketogenesis in Male Wistar Rats

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results
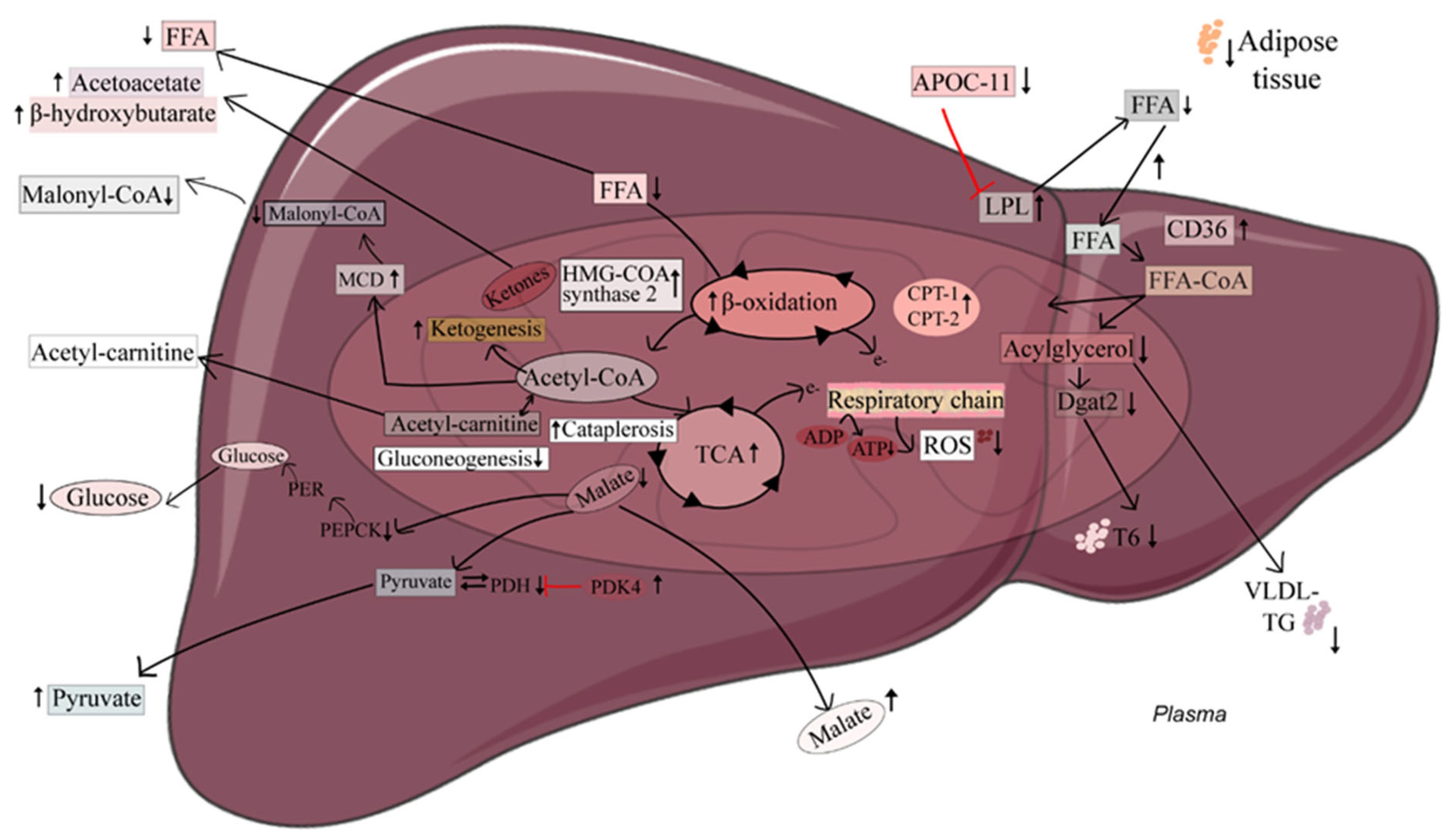
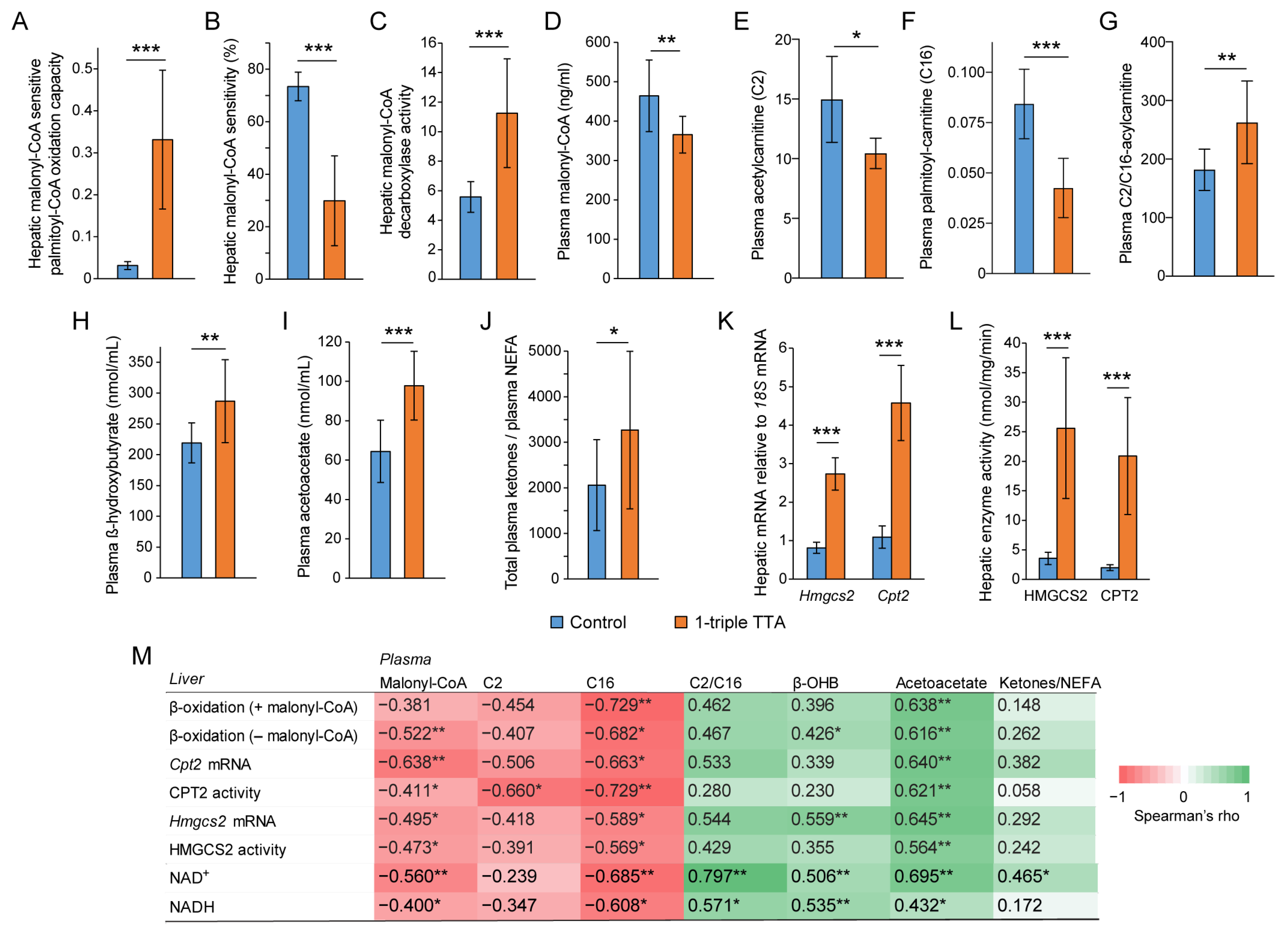
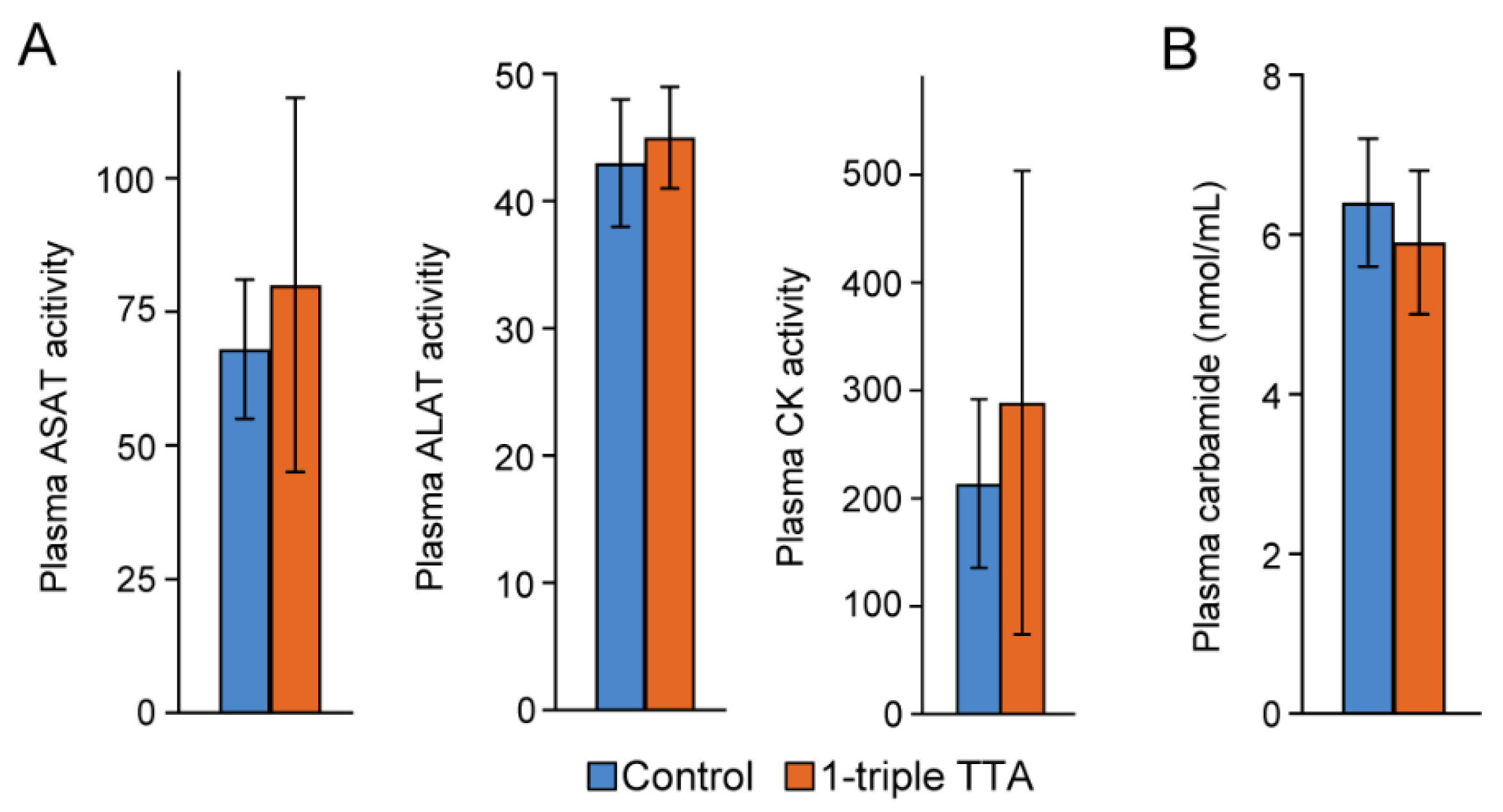
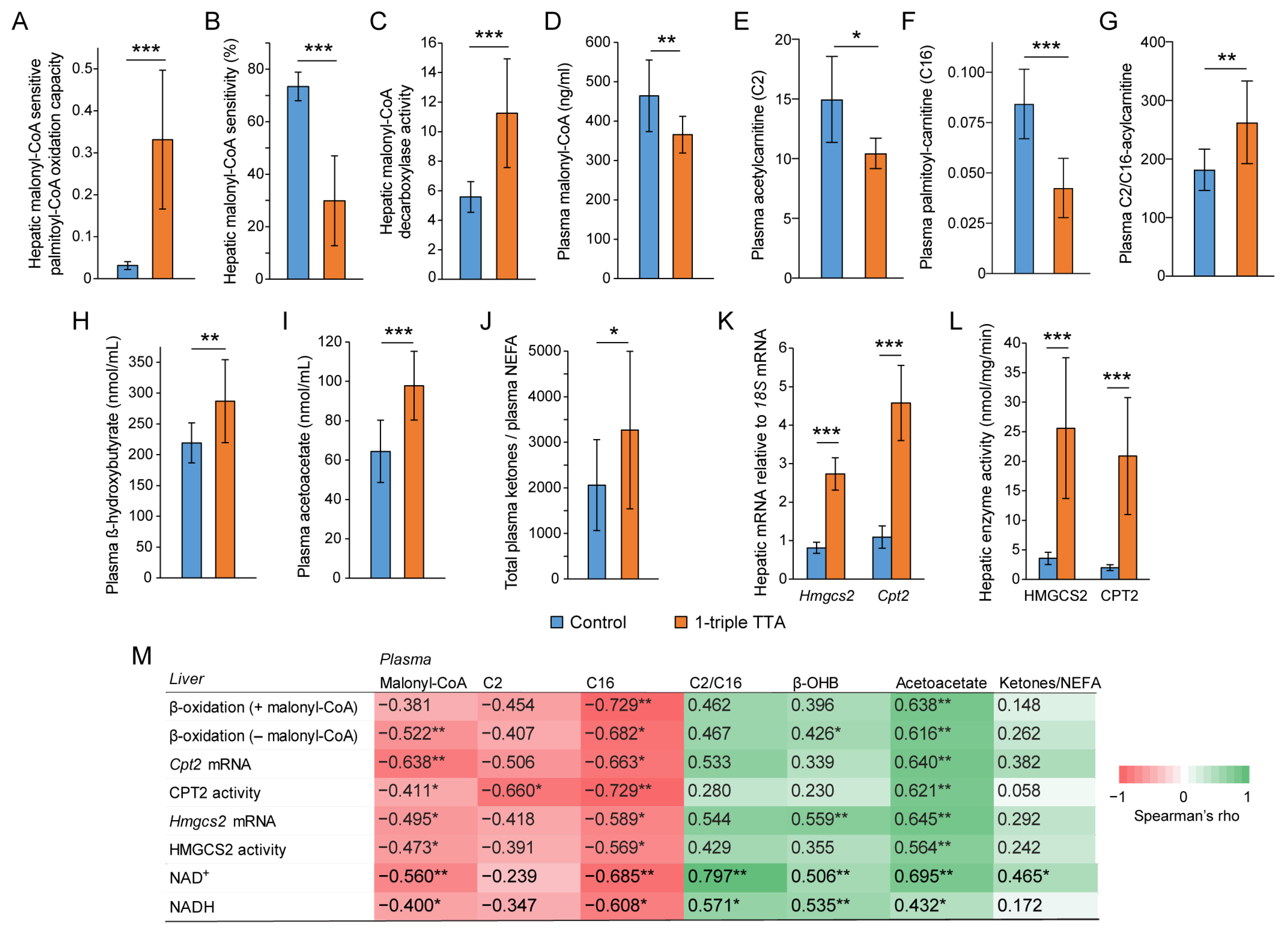
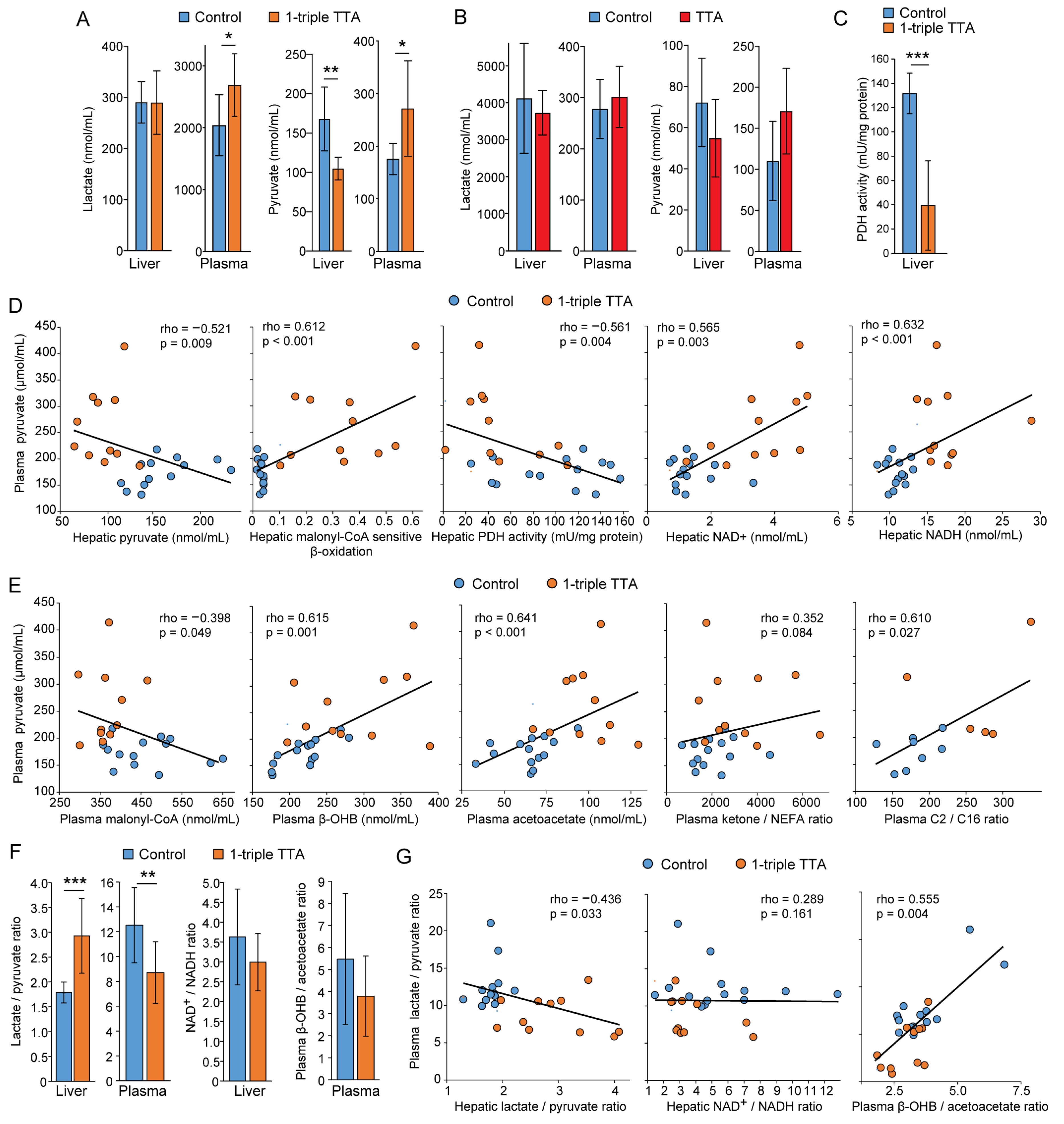
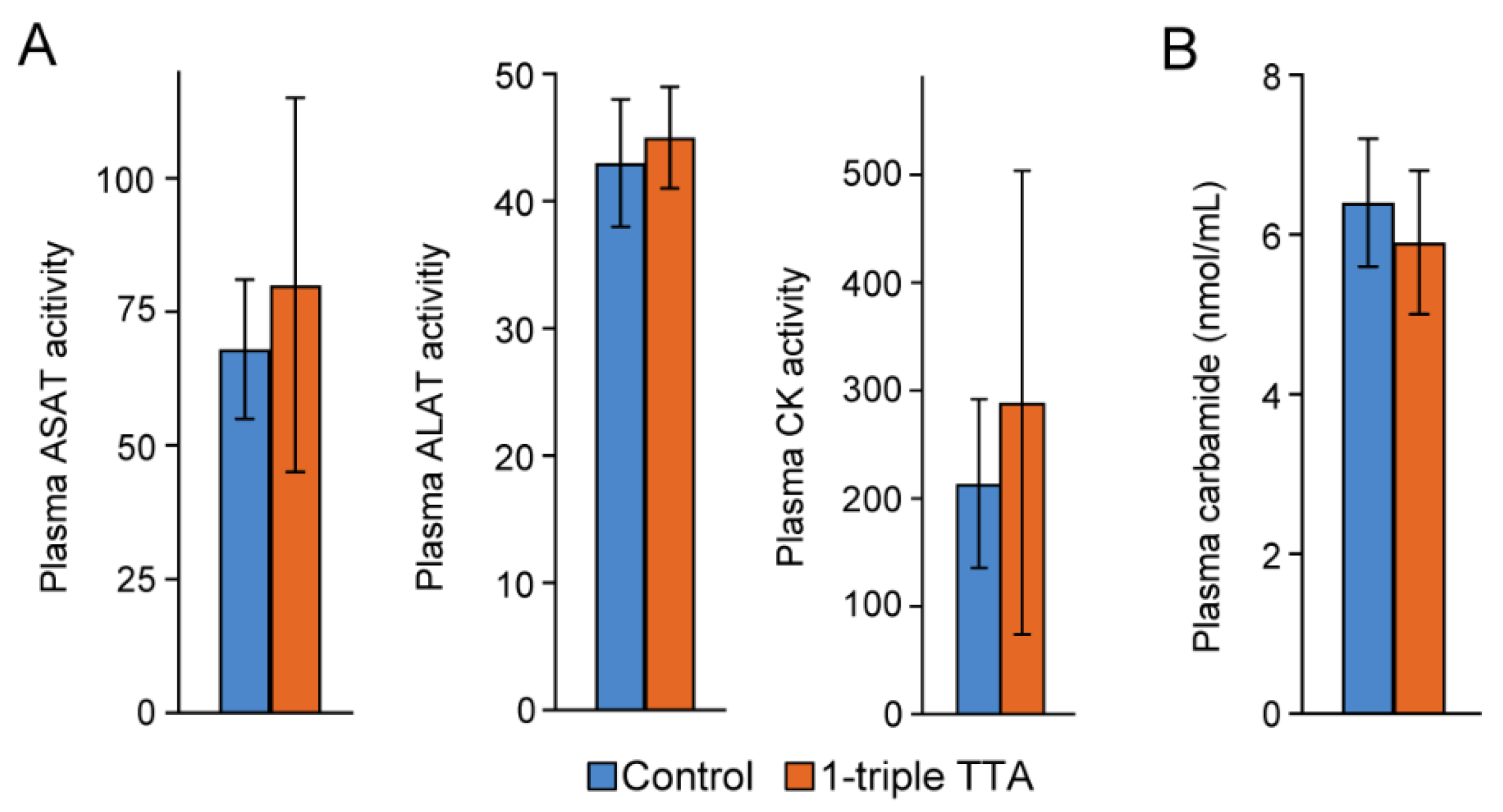
2.1. The 3-Thia Fatty-Acid-Induced Alterations in Fatty Acid Oxidation
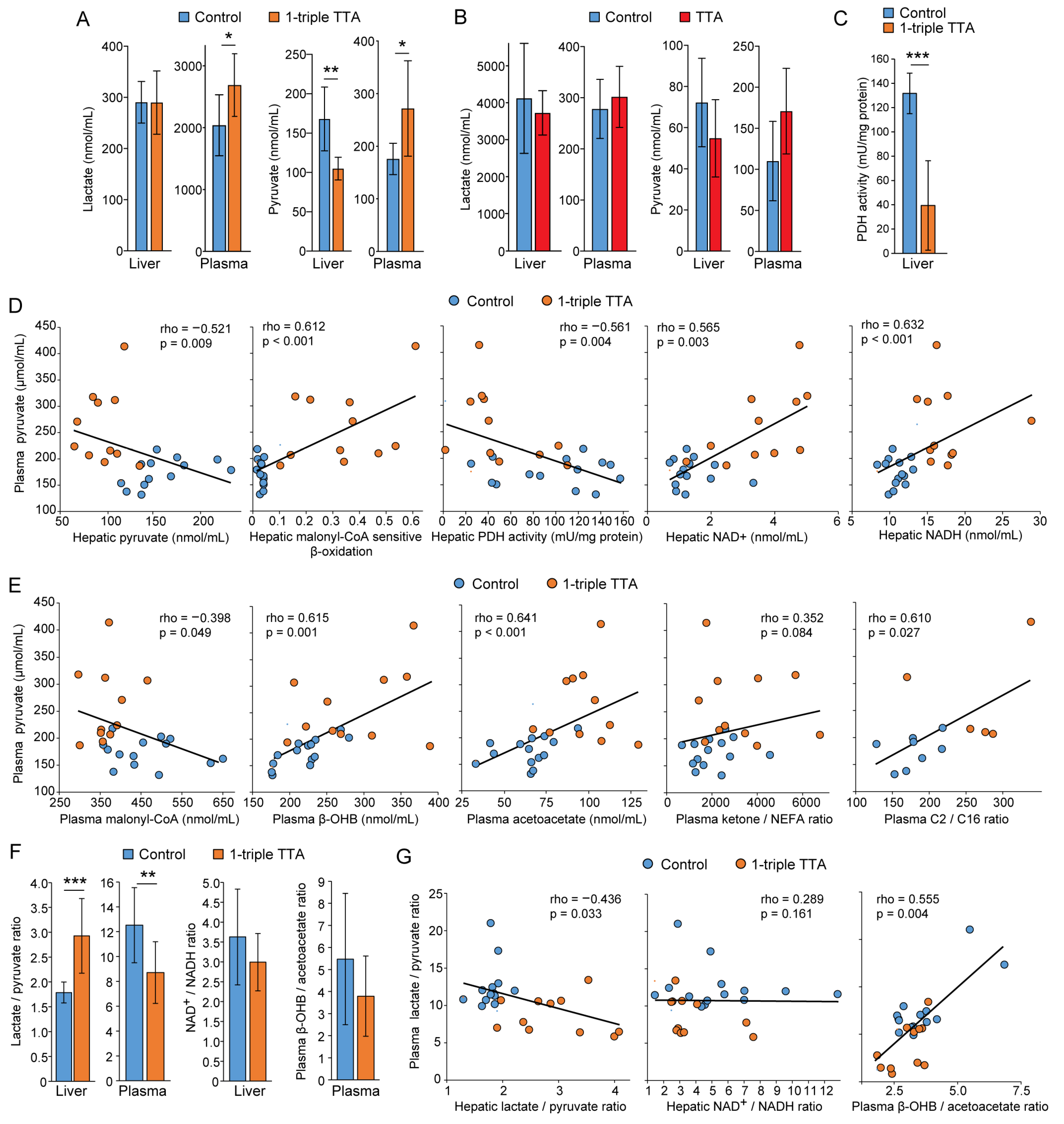
2.2. Relationship between Hepatic Fatty Acid Oxidation and Circulating Pyruvate, Lactate, and Ketones
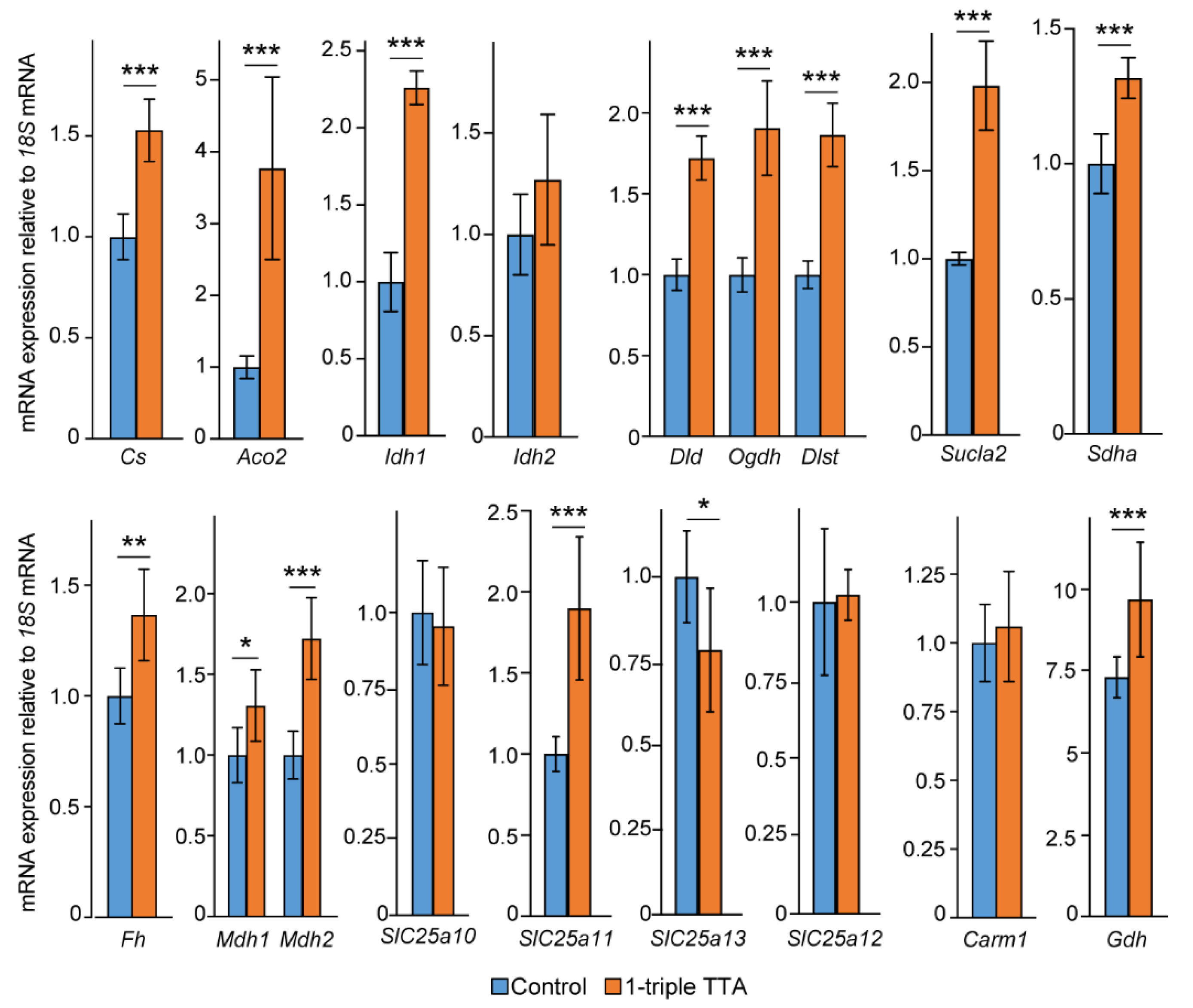
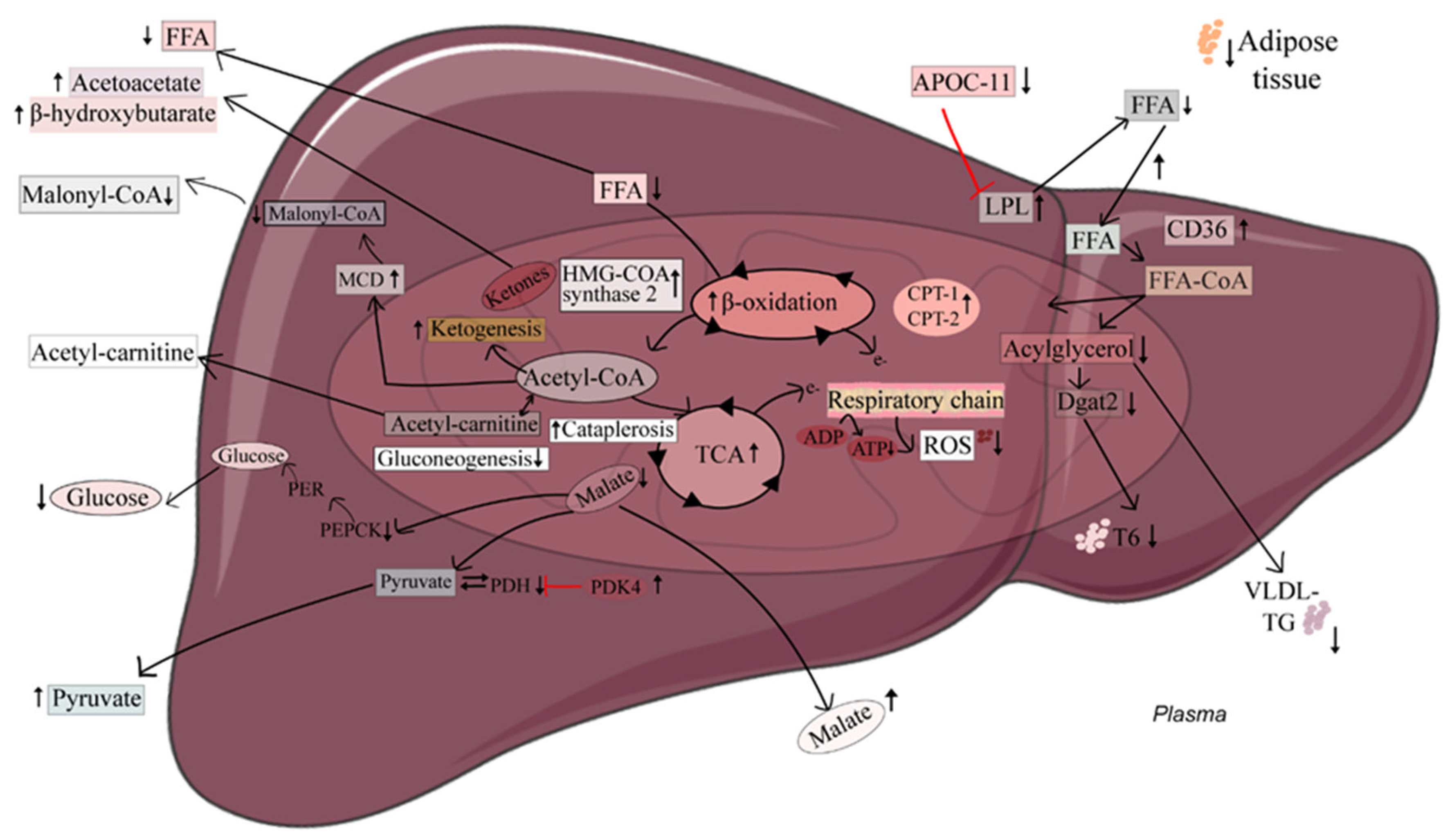
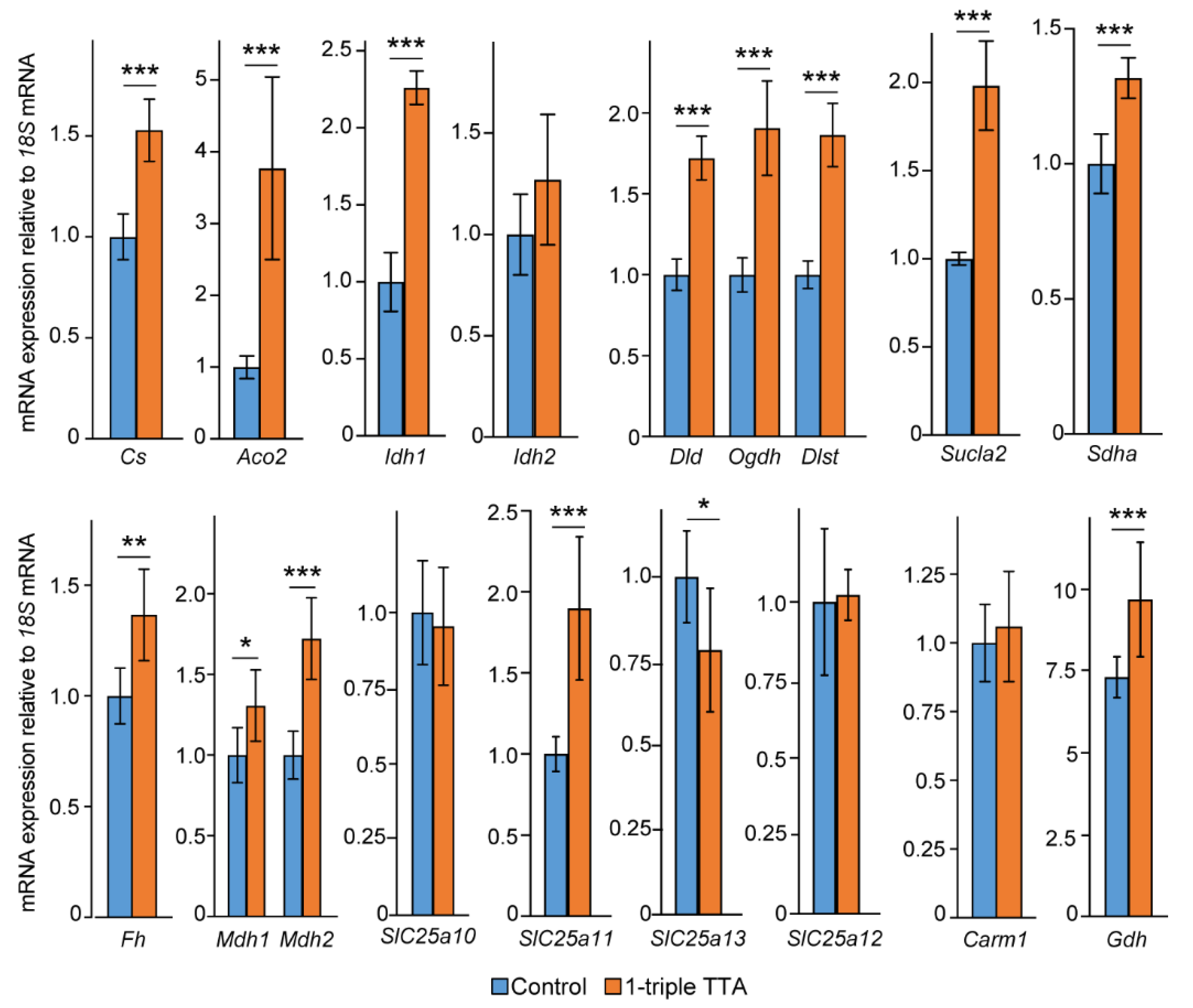
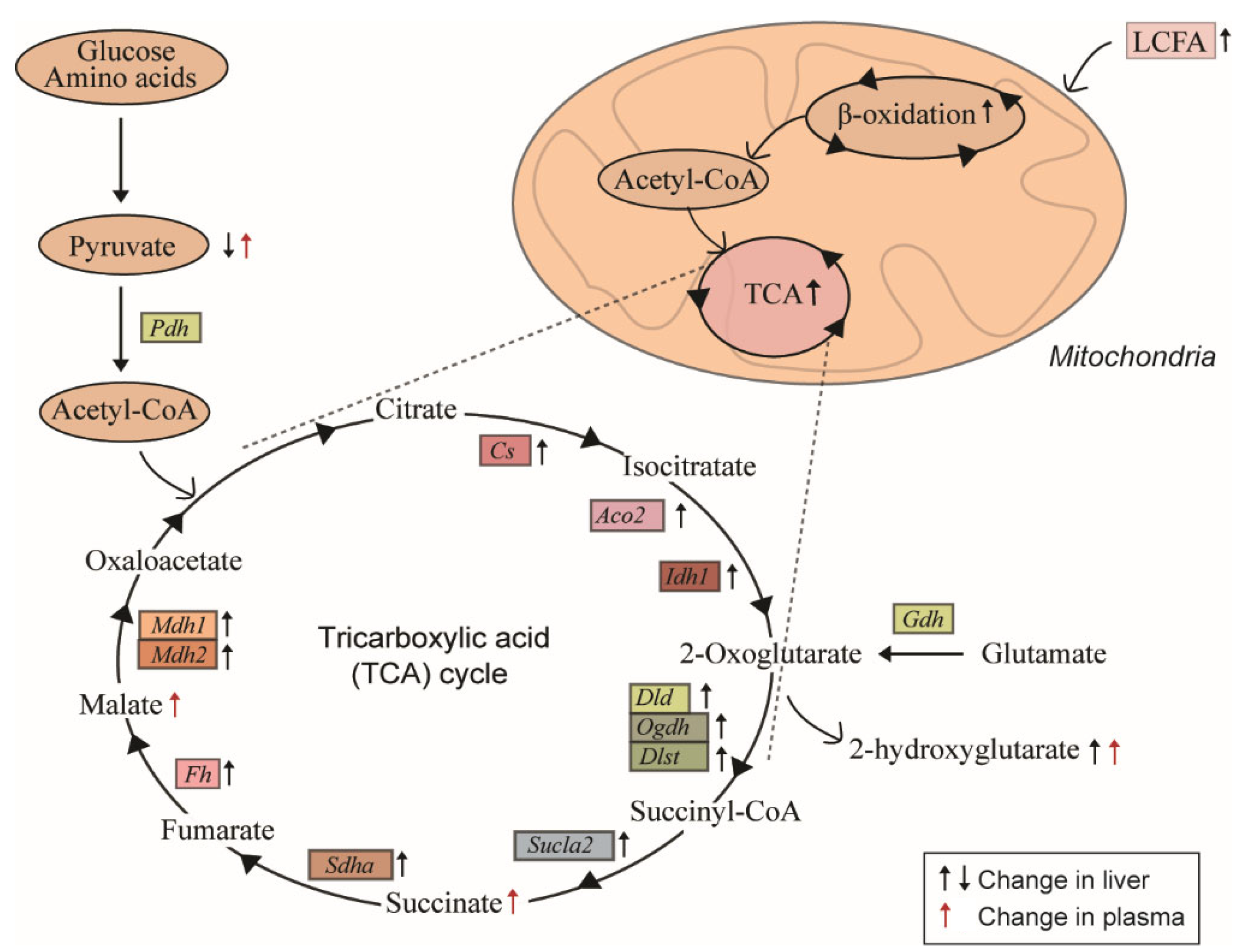
2.3. Induction of Hepatic Fatty Acid Oxidation Alters Liver and Plasma Levels of TCA Cycle Intermediates and the Expression of Related Genes
2.4. Effects on the Mitochondrial Solute Carrier (SLC) 25 Transporters
2.5. The 3-Thia Fatty Acid Administration Alters TCA-Related Enzyme Activity and Expression
3. Discussion
4. Materials and Methods
4.1. Animal Study
4.2. Plasma Malonyl-CoA
4.3. Plasma Acetylcarnitine, Palmitoylcarnitine, Beta-Hydroxybutyrate, and Acetoacetate
4.4. Plasma and Liver Pyruvate, Lactate, and TCA Cycle Intermediates
4.5. Mitochondrial FA Oxidation, Free Fatty Acids, NAD, and Enzyme Activities
4.6. Hepatic Gene Expression Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Ramanathan, R.; Ali, A.H.; Silva, G.; Murri, M.; Ramanathan, R.; Ali, A.H.; Ibdah, J.A.; Memorial, H.S.T. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Inigo, M.; Deja, S.; Burgess, S.C. Ins and Outs of the TCA Cycle: The Central Role of Anaplerosis. Ann. Rev. Nutr. 2021, 41, 19–47. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Abek, A.D.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef]
- Corkey, B.E.; Deeney, J.T. The Redox Communication Network as a Regulator of Metabolism. Front. Physiol. 2020, 11, 567796. [Google Scholar] [CrossRef]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef]
- Nocito, L.; Kleckner, A.S.; Yoo, E.J.; Jones, A.R.; Liesa, M.; Corkey, B.E. The extracellular redox state modulates mitochondrial function, gluconeogenesis, and glycogen synthesis in murine hepatocytes. PLoS ONE 2015, 10, e0122818. [Google Scholar] [CrossRef]
- Selen, E.S.; Rodriguez, S.; Cavagnini, K.S.; Kim, H.B.; Na, C.H.; Wolfgang, M.J. Requirement of hepatic pyruvate carboxylase during fasting, high fat, and ketogenic diet. J. Biol. Chem. 2022, 298, 102648. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Saha, A.K.; Vavvas, D.; Witters, L.A. Malonyl-CoA, fuel sensing, and insulin resistance. Am. J. Physiol.-Endocrinol. Metab. 1999, 276, E1–E18. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, C.; Bjørndal, B.; Rossmann, C.R.; Svardal, A.; Hallström, S.; Berge, R.K. A fatty acid analogue targeting mitochondria exerts a plasma triacylglycerol lowering effect in rats with impaired carnitine biosynthesis. PLoS ONE 2018, 13, e0194978. [Google Scholar] [CrossRef]
- Lindquist, C.; Bjorndal, B.; Bakke, H.G.; Slettom, G.; Karoliussen, M.; Rustan, A.C.; Hege Thoresen, G.; Skorve, J.; Nygard, O.K.; Berge, R.K. A mitochondria-targeted fatty acid analogue influences hepatic glucose metabolism and reduces the plasma insulin/glucose ratio in male Wistar rats. PLoS ONE 2019, 14, e0222558. [Google Scholar] [CrossRef]
- Lindquist, C.; Bjørndal, B.; Rossmann, C.R.; Tusubira, D.; Svardal, A.; Røsland, G.V.; Tronstad, K.J.; Hallström, S.; Berge, R.K. Increased hepatic mitochondrial FA oxidation reduces plasma and liver TG levels and is associated with regulation of UCPs and APOC-III in rats. J. Lipid Res. 2017, 58, 1362–1373. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef]
- Krycer, J.R.; Quek, L.E.; Francis, D.; Fazakerley, D.J.; Elkington, S.D.; Diaz-Vegas, A.; Cooke, K.C.; Weiss, F.C.; Duan, X.; Kurdyukov, S.; et al. Lactate production is a prioritized feature of adipocyte metabolism. J. Biol. Chem. 2020, 295, 83–98. [Google Scholar] [CrossRef]
- Stanko, R.T.; Arch, J.E. Inhibition of regain in body weight and fat with addition of 3-carbon compounds to the diet with hyperenergetic refeeding after weight reduction. Int. J. Obes. 1996, 20, 925–930. [Google Scholar]
- Stanko, R.T.; Reynolds, H.R.; Hoyson, R.; Janosky, J.E.; Wolf, R. Pyruvate supplementation of a low-cholesterol, low-fat diet: Effects on plasma lipid concentrations and body composition in hyperlipidemic patients. Am. J. Clin. Nutr. 1994, 59, 423–427. [Google Scholar] [CrossRef]
- Stanko, R.T.; Robertson, R.J.; Spina, R.J.; Reilly, J.J.; Greenawalt, K.D.; Goss, F.L. Enhancement of arm exercise endurance capacity with dihydroxyacetone and pyruvate. J. Appl. Physiol. 1990, 68, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Kalman, D.; Colker, C.M.; Wilets, I.; Roufs, J.B.; Antonio, J. The effects of pyruvate supplementation on body composition in overweight individuals. Nutrition 1999, 15, 337–340. [Google Scholar] [CrossRef]
- Stanko, R.T.; Tietze, D.L.; Arch, J.E. Body composition, energy utilization, and nitrogen metabolism with a severely restricted diet supplemented with dihydroxyacetone and pyruvate. Am. J. Clin. Nutr. 1992, 55, 771–776. [Google Scholar] [CrossRef]
- Nakamichi, N.; Kambe, Y.; Oikawa, H.; Ogura, M.; Takano, K.; Tamaki, K.; Inoue, M.; Hinoi, E.; Yoneda, Y. Protection by exogenous pyruvate through a mechanism related to monocarboxylate transporters against cell death induced by hydrogen peroxide in cultured rat cortical neurons. J. Neurochem. 2005, 93, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Glowinski, J.; Prémont, J. Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J. Neurosci. 1997, 17, 9060–9067. [Google Scholar] [CrossRef]
- Patgiri, A.; Skinner, O.S.; Miyazaki, Y.; Schleifer, G.; Marutani, E.; Shah, H.; Sharma, R.; Goodman, R.P.; To, T.L.; Robert Bao, X.; et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD+ imbalance. Nat. Biotechnol. 2020, 38, 309–313. [Google Scholar] [CrossRef]
- Siess, E.A.; Brocks, D.G.; Wieland, O.H. Subcellular distribution of key metabolites in isolated liver cells from fasted rats. FEBS Lett. 1976, 69, 265–271. [Google Scholar] [CrossRef]
- Berge, R.K.; Tronstad, K.J.; Berge, K.; Rost, T.H.; Wergedahl, H.; Gudbrandsen, O.A.; Skorve, J. The metabolic syndrome and the hepatic fatty acid drainage hypothesis. Biochimie 2005, 87, 15–20. [Google Scholar] [CrossRef]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Miki, S.; Araki, H.; Takahashi, K.; Kotani, H. Identification of dicarboxylate carrier Slc25a10 as malate transporter in de Novo fatty acid synthesis. J. Biol. Chem. 2005, 280, 32434–32441. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagne, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lanks, C.W.; Tong, L. Molecular mechanism for the regulation of human mitochondrial NAD(P)+-Dependent malic enzyme by ATP and umarate. Structure 2002, 10, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Liu, D.; Tang, J.; Dong, L.; Liu, Y.; Tao, Z.; Wan, J.; Gao, D.; Wang, L.; Sun, H.; et al. Malic enzyme 1 induces epithelial–mesenchymal transition and indicates poor prognosis in hepatocellular carcinoma. Tumor Biol. 2015, 36, 6211–6221. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Ryter, S.W.; Hong, P.K.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M.K. Mechanisms of Cell Death in Oxidative Stress. Antioxid. Redox Signal. 2006, 9, 49–89. [Google Scholar] [CrossRef]
- Vigerust, N.F.; Cacabelos, D.; Burri, L.; Berge, K.; Wergedahl, H.; Christensen, B.; Portero-Otin, M.; Viste, A.; Pamplona, R.; Berge, R.K.; et al. Fish oil and 3-thia fatty acid have additive effects on lipid metabolism but antagonistic effects on oxidative damage when fed to rats for 50 weeks. J. Nutr. Biochem. 2012, 23, 1384–1393. [Google Scholar] [CrossRef]
- Raspé, E.; Madsen, L.; Lefebvre, A.M.; Leitersdorf, I.; Gelman, L.; Peinado-Onsurbe, J.; Dallongeville, J.; Fruchart, J.C.; Berge, R.; Staels, B. Modulation of rat liver apolipoprotein gene expression and serum lipid levels by tetradecylthioacetic acid (TTA) via PPARα activation. J. Lipid Res. 1999, 40, 2099–2110. [Google Scholar] [CrossRef]
- Løvås, K.; Røst, T.H.; Skorve, J.; Ulvik, R.J.; Gudbrandsen, O.A.; Bohov, P.; Wensaas, A.J.; Rustan, A.C.; Berge, R.K.; Husebye, E.S. Tetradecylthioacetic acid attenuates dyslipidaemia in male patients with type 2 diabetes mellitus, possibly by dual PPAR-α/δ activation and increased mitochondrial fatty acid oxidation. Diabetes Obes. Metab. 2009, 11, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Dyrøy, E.; Yndestad, A.; Ueland, T.; Halvorsen, B.; Damås, J.K.; Aukrust, P.; Berge, R.K. Antiinflammatory effects of tetradecylthioacetic acid involve both peroxisome proliferator-activated receptor α-dependent and -independent pathways. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Castelein, H.; Gulick, T.; Declercq, P.E.; Mannaerts, G.P.; Moore, D.D.; Baes, M.I. The peroxisome proliferator activated receptor regulates malic enzyme gene expression. J. Biol. Chem. 1994, 269, 26754–26758. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, G.; Tirujillo, C.; Chang, V.; Blanco, L.; Joseph, S.B.; Bassilian, S.; Saad, M.F.; Tontonoz, P.; Lee, W.N.P.; et al. Peroxisome proliferator-activated receptor α (PPARα) influences: Substrate utilization for hepatic glucose production. J. Biol. Chem. 2002, 277, 50237–50244. [Google Scholar] [CrossRef] [PubMed]
- Lysne, V.; Strand, E.; Svingen, G.F.T.; Bjørndal, B.; Pedersen, E.R.; Midttun, Ø.; Olsen, T.; Ueland, P.M.; Berge, R.K.; Nygård, O. Peroxisome Proliferator-Activated Receptor Activation is Associated with Altered Plasma One-Carbon Metabolites and B-Vitamin Status in Rats. Nutrients 2016, 8, 26. [Google Scholar] [CrossRef]
- Pettersen, I.K.N.; Tusubira, D.; Ashrafi, H.; Dyrstad, S.E.; Hansen, L.; Liu, X.Z.; Nilsson, L.I.H.; Løvsletten, N.G.; Berge, K.; Wergedahl, H.; et al. Upregulated PDK4 expression is a sensitive marker of increased fatty acid oxidation. Mitochondrion 2019, 49, 97–110. [Google Scholar] [CrossRef]
- Vik, R.; Busnelli, M.; Parolini, C.; Bjørndal, B.; Holm, S.; Bohov, P.; Halvorsen, B.; Brattelid, T.; Manzini, S.; Ganzetti, G.S.; et al. An Immunomodulating Fatty Acid Analogue Targeting Mitochondria Exerts Anti-Atherosclerotic Effect beyond Plasma Cholesterol-Lowering Activity in apoE−/− Mice. PLoS ONE 2013, 8, e81963. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Keshari, K.R. Metabolic analysis as a driver for discovery, diagnosis and therapy. Cell 2022, 185, 2678–2689. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Bjørndal, B.; Alterås, E.K.; Lindquist, C.; Svardal, A.; Skorve, J.; Berge, R.K. Associations between fatty acid oxidation, hepatic mitochondrial function, and plasma acylcarnitine levels in mice. Nutr. Metab. 2018, 15, 10. [Google Scholar] [CrossRef]
- Froyland, L.; Asiedu, D.K.; Vaagenes, H.; Garras, A.; Lie, O.; Totland, G.K.; Berge, R.K. Tetradecylthioacetic acid incorporated into very low density lipoprotein: Changes in the fatty acid composition and reduced plasma lipids in cholesterol-fed hamsters. J. Lipid Res. 1995, 36, 2529–2540. [Google Scholar] [CrossRef] [PubMed]
- Ceperuelo-Mallafré, V.; Reverté, L.; Peraire, J.; Madeira, A.; Maymó-Masip, E.; López-Dupla, M.; Gutierrez-Valencia, A.; Ruiz-Mateos, E.; Buzón, M.J.; Jorba, R.; et al. Circulating pyruvate is a potent prognostic marker for critical COVID-19 outcomes. Front. Immunol. 2022, 13, 912579. [Google Scholar] [CrossRef] [PubMed]
- Skorve, J.; Asiedu, D.; Rustan, A.C.; Drevon, C.A.; Al-Shurbaji, A.; Berge, R.K. Regulation of fatty acid oxidation and triglyceride and phospholipid metabolism by hypolipidemic sulfur-substituted fatty acid analogues. J. Lipid Res. 1990, 31, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Vernez, L.; Wenk, M.; Krähenbühl, S. Determination of carnitine and acylcarnitines in plasma by high-performance liquid chromatography/electrospray ionization ion trap tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 1233–1238. [Google Scholar] [CrossRef]
- Tan, B.; Lu, Z.; Dong, S.; Zhao, G.; Kuo, M.S. Derivatization of the tricarboxylic acid intermediates with O-benzylhydroxylamine for liquid chromatography-tandem mass spectrometry detection. Anal. Biochem. 2014, 465, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Røst, L.M.; Brekke Thorfinnsdottir, L.; Kumar, K.; Fuchino, K.; Langørgen, I.E.; Bartosova, Z.; Kristiansen, K.A.; Bruheim, P. Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites 2020, 10, 74. [Google Scholar] [CrossRef]
- Willumsen, N.; Hexeberg, S.; Skorve, J.; Lundquist, M.; Berge, R.K. Docosahexaenoic acid shows no triglyceride-lowering effects but increases the peroxisomal fatty acid oxidation in liver of rats. J. Lipid Res. 1993, 34, 13–22. [Google Scholar] [CrossRef]
- Srere, P.A.; Brazil, H.; Gonen, L.; Takahashi, M. The Citrate Condensing Enzyme of Pigeon Breast Muscle and Moth Flight Muscle. Acta Chem. Scand. 1963, 17, S129–S134. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kolattukudy, P.E. Malonyl-CoA decarboxylase from the uropygial gland of waterfowl: Purification, properties, immunological comparison, and role in regulating the synthesis of multimethyl-branched fatty acids. Arch. Biochem. Biophys. 1978, 170, 585–597. [Google Scholar] [CrossRef]
- Clinkenbeard, K.D.; Reed, W.D.; Mooney, R.A.; Lane, M.D. Intracellular localization of the 3-hydroxy-3-methylglutaryl coenzme A cycle enzymes in liver. Separate cytoplasmic and mitochondrial 3-hydroxy-3-methylglutaryl coenzyme A generating systems for cholesterogenesis and ketogenesis. J. Biol. Chem. 1975, 250, 3108–3116. [Google Scholar] [CrossRef]
- Ivanova, Z.; Bjørndal, B.; Grigorova, N.; Roussenov, A.; Vachkova, E.; Berge, K.; Burri, L.; Berge, R.; Stanilova, S.; Milanova, A.; et al. Effect of fish and krill oil supplementation on glucose tolerance in rabbits with experimentally induced obesity. Eur. J. Nutr. 2015, 54, 1055–1067. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Control | 1-Triple TTA | p-Value | Control | 1-Triple TTA | p-Value |
|---|---|---|---|---|---|---|
| Liver (nmol/mg) | Plasma (µM) | |||||
| Oxaloacetate | 1.76 (1.18) | 1.37 (0.71) | 0.523 | 4.36 (2.27) | 10.7 (12.3) | 0.17 |
| Citrate | 0.150 (0.06) | 0.103 (0.02) | 0.183 | 234 (36.1) | 242 (33.0) | 0.70 |
| Isocitrate | N/A | N/A | N/A | 10.5 (6.74) | 14.6 (6.34) | 0.30 |
| 2-hydroxyglutarate | 0.002 (0.001) | 0.009 (0.003) | <0.001 | 0.76 (0.18) | 1.14 (0.35) | 0.024 |
| Succinate | 0.984 (0.393) | 1.49 (0.743) | 0.130 | 6.98 (1.58) | 19.5 (10.8) | 0.007 |
| Fumarate | 0.544 (0.230) | 0.433 (0.259) | 0.465 | 2.24 (0.72) | 3.0 (1.53) | 0.263 |
| Malate | 0.098 (0.055) | 0.120 (0.06) | 0.517 | 6.51 (2.18) | 10.4 (4.08) | 0.043 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dankel, S.N.; Kalleklev, T.-L.; Tungland, S.L.; Stafsnes, M.H.; Bruheim, P.; Aloysius, T.A.; Lindquist, C.; Skorve, J.; Nygård, O.K.; Madsen, L.; et al. Changes in Plasma Pyruvate and TCA Cycle Metabolites upon Increased Hepatic Fatty Acid Oxidation and Ketogenesis in Male Wistar Rats. Int. J. Mol. Sci. 2023, 24, 15536. https://doi.org/10.3390/ijms242115536
Dankel SN, Kalleklev T-L, Tungland SL, Stafsnes MH, Bruheim P, Aloysius TA, Lindquist C, Skorve J, Nygård OK, Madsen L, et al. Changes in Plasma Pyruvate and TCA Cycle Metabolites upon Increased Hepatic Fatty Acid Oxidation and Ketogenesis in Male Wistar Rats. International Journal of Molecular Sciences. 2023; 24(21):15536. https://doi.org/10.3390/ijms242115536
Chicago/Turabian StyleDankel, Simon Nitter, Tine-Lise Kalleklev, Siri Lunde Tungland, Marit Hallvardsdotter Stafsnes, Per Bruheim, Thomas Aquinas Aloysius, Carine Lindquist, Jon Skorve, Ottar Kjell Nygård, Lise Madsen, and et al. 2023. "Changes in Plasma Pyruvate and TCA Cycle Metabolites upon Increased Hepatic Fatty Acid Oxidation and Ketogenesis in Male Wistar Rats" International Journal of Molecular Sciences 24, no. 21: 15536. https://doi.org/10.3390/ijms242115536
APA StyleDankel, S. N., Kalleklev, T.-L., Tungland, S. L., Stafsnes, M. H., Bruheim, P., Aloysius, T. A., Lindquist, C., Skorve, J., Nygård, O. K., Madsen, L., Bjørndal, B., Sydnes, M. O., & Berge, R. K. (2023). Changes in Plasma Pyruvate and TCA Cycle Metabolites upon Increased Hepatic Fatty Acid Oxidation and Ketogenesis in Male Wistar Rats. International Journal of Molecular Sciences, 24(21), 15536. https://doi.org/10.3390/ijms242115536