
Neurohumoral Activation in Heart Failure

Abstract
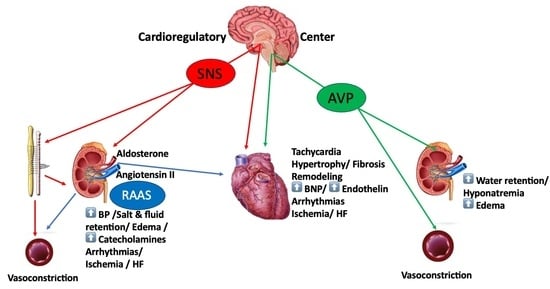
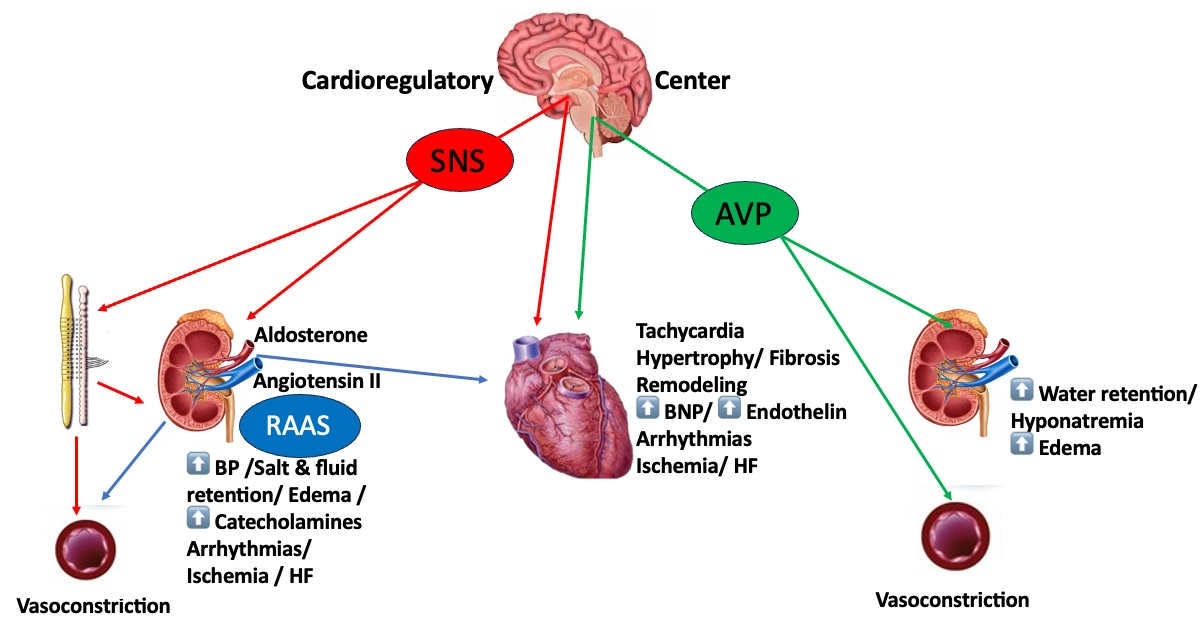
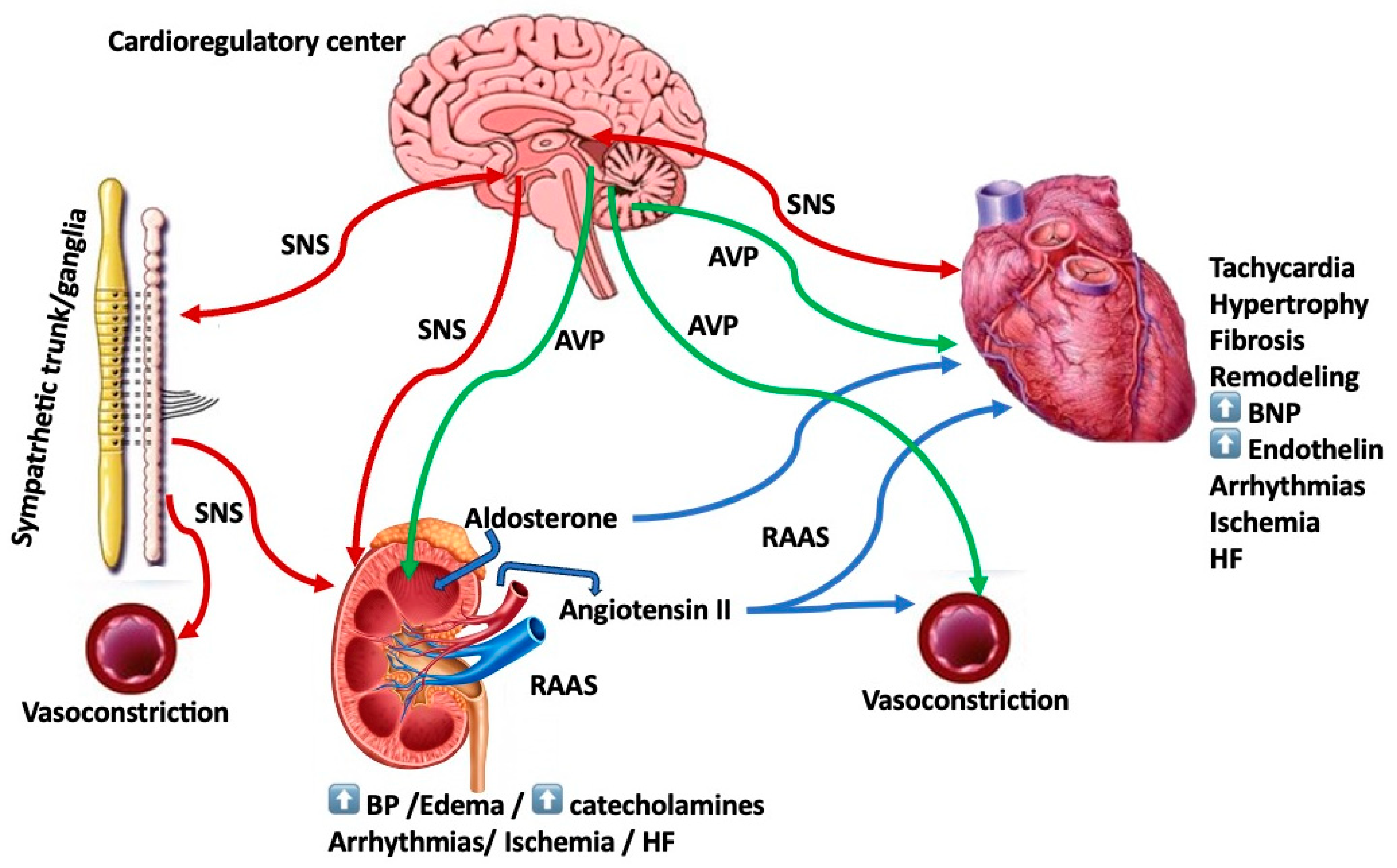
1. Introduction
2. Sympathetic Nervous System (SNS)
3. Arginine Vasopressin (AVP)
4. Renin–Angiotensin–Aldosterone System (RAAS)
5. Counterregulatory Systems
6. Cardiac Remodeling
7. Countering RAAS
8. Heart Failure with Preserved Ejection Fraction
9. Neurohormonal Biomarkers
10. Diuretics/Ultrafiltration
11. SGLT2 Inhibitors
12. Neurohumoral Blockade in Various Clinical Settings/Scenarios
13. Salt Intake and Heart Failure/The Role of Neurohormonal Activity
14. Combination Therapies
15. Natriuretic Peptides as Therapeutic Agents
16. Cardiac Arrhythmias and Sudden Cardiac Death/Arrhythmogenesis in Heart Failure
17. Updated Guideline-Directed Therapies for HF
18. Conclusions
19. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Grosman-Rimon, L.; Billia, F.; Wright, E.; Carasso, S.; Elbaz-Greener, G.; Kachel, E.; Rao, V.; Cherney, D. Neurohormones, inflammatory mediators, and cardiovascular injury in the setting of heart failure. Heart Fail. Rev. 2020, 25, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Li, A.; McNamara, J.; Dos Remedios, C.; Lal, S. Pathogenesis and pathophysiology of heart failure with reduced ejection fraction: Translation to human studies. Heart Fail. Rev. 2019, 24, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Nägele, M.P.; Barthelmes, J.; Kreysing, L.; Haider, T.; Nebunu, D.; Ruschitzka, F.; Sudano, I.; Flammer, A.J. Endocrine hormone imbalance in heart failure with reduced ejection fraction: A cross-sectional study. Health Sci. Rep. 2022, 5, e880. [Google Scholar] [CrossRef]
- Lip, G.Y.; Heinzel, F.R.; Gaita, F.; Juanatey, J.R.; Le Heuzey, J.Y.; Potpara, T.; Svendsen, J.H.; Vos, M.A.; Anker, S.D.; Coats, A.J.; et al. European Heart Rhythm Association/Heart Failure Association joint consensus document on arrhythmias in heart failure, endorsed by the Heart Rhythm Society and the Asia Pacific Heart Rhythm Society. Europace 2016, 18, 12–36. [Google Scholar] [CrossRef]
- Manolis, A.S.; Varriale, P.; Nobile, J. Short-term hemodynamic effects of intravenous methyldopa in patients with congestive heart failure. Pharmacotherapy 1987, 7, 216–222. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A.; Melita, H. Neprilysin Inhibitors: Filling a Gap in Heart Failure Management, Albeit Amidst Controversy and at a Significant Cost. Am. J. Cardiovasc. Drugs 2019, 19, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.S.; Benedict, C.; Johnstone, D.E.; Kirlin, P.C.; Nicklas, J.; Liang, C.S.; Kubo, S.H.; Rudin-Toretsky, E.; Yusuf, S. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 1990, 82, 1724–1729. [Google Scholar] [CrossRef]
- Leite, M.; Sampaio, F.; Saraiva, F.A.; Diaz, S.O.; Barros, A.S.; Fontes-Carvalho, R. The impact of heart failure therapy in patients with mildly reduced ejection fraction: A network meta-analysis. ESC Heart Fail. 2023, 10, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.A.; Manolis, T.A.; Manolis, A.S. Cardiovascular Safety of Antihyperglycemic Agents: “Do Good or Do No Harm”. Drugs 2018, 78, 1567–1592. [Google Scholar] [CrossRef]
- Manolis, A.A.; Manolis, T.A.; Apostolopoulos, E.J.; Apostolaki, N.E.; Melita, H.; Manolis, A.S. The role of the autonomic nervous system in cardiac arrhythmias: The neuro-cardiac axis, more foe than friend? Trends Cardiovasc. Med. 2021, 31, 290–302. [Google Scholar] [CrossRef]
- Rector, T.S.; Olivari, M.T.; Levine, T.B.; Francis, G.S.; Cohn, J.N. Predicting survival for an individual with congestive heart failure using the plasma norepinephrine concentration. Am. Heart J. 1987, 114, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Neve, K.A.; Molinoff, P.B. Effects of chronic administration of agonists and antagonists on the density of beta-adrenergic receptors. Am. J. Cardiol. 1986, 57, 17f–22f. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Ginsburg, R.; Minobe, W.; Cubicciotti, R.S.; Sageman, W.S.; Lurie, K.; Billingham, M.E.; Harrison, D.C.; Stinson, E.B. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 1982, 307, 205–211. [Google Scholar] [CrossRef]
- Heinsimer, J.A.; Lefkowitz, R.J. The beta-adrenergic receptor in heart failure. Hosp. Pract. 1983, 18, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Imperato-McGinley, J.; Gautier, T.; Ehlers, K.; Zullo, M.A.; Goldstein, D.S.; Vaughan, E.D., Jr. Reversibility of catecholamine-induced dilated cardiomyopathy in a child with a pheochromocytoma. N. Engl. J. Med. 1987, 316, 793–797. [Google Scholar] [CrossRef]
- Francis, G.S.; Goldsmith, S.R.; Levine, T.B.; Olivari, M.T.; Cohn, J.N. The neurohumoral axis in congestive heart failure. Ann. Intern. Med. 1984, 101, 370–377. [Google Scholar] [CrossRef]
- Grosman-Rimon, L.; Wright, E.; Sabovich, S.; Rimon, J.; Gleitman, S.; Sudarsky, D.; Lubovich, A.; Gabizon, I.; Lalonde, S.D.; Tsuk, S.; et al. Relationships among norepinephrine levels, exercise capacity, and chronotropic responses in heart failure patients. Heart Fail. Rev. 2023, 28, 35–45. [Google Scholar] [CrossRef]
- Manolis, A.A.; Manolis, T.A.; Melita, H.; Manolis, A.S. Takotsubo Syndrome and Sudden Cardiac Death. Angiology 2023, 74, 105–128. [Google Scholar] [CrossRef]
- Anninos, H.; Manolis, A.S. Takotsubo syndrome: A brief review in light of new evidence. Rhythmos 2016, 11, 90–95. [Google Scholar]
- Medina de Chazal, H.; Del Buono, M.G.; Keyser-Marcus, L.; Ma, L.; Moeller, F.G.; Berrocal, D.; Abbate, A. Stress Cardiomyopathy Diagnosis and Treatment: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1955–1971. [Google Scholar] [CrossRef]
- Koulouris, S.; Pastromas, S.; Sakellariou, D.; Kratimenos, T.; Piperopoulos, P.; Manolis, A.S. Takotsubo cardiomyopathy: The “broken heart” syndrome. Hell. J. Cardiol. 2010, 51, 451–457. [Google Scholar]
- Manolis, A.S.; Manolis, A.A.; Manolis, T.A.; Melita, H. Acute coronary syndromes in patients with angiographically normal or near normal (non-obstructive) coronary arteries. Trends Cardiovasc. Med. 2018, 28, 541–551. [Google Scholar] [CrossRef]
- Y-Hassan, S.; Sörensson, P.; Ekenbäck, C.; Lundin, M.; Agewall, S.; Brolin, E.B.; Caidahl, K.; Cederlund, K.; Collste, O.; Daniel, M.; et al. Plasma catecholamine levels in the acute and subacute stages of takotsubo syndrome: Results from the Stockholm myocardial infarction with normal coronaries 2 study. Clin. Cardiol. 2021, 44, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Pelliccia, F.; Kaski, J.C.; Crea, F.; Camici, P.G. Pathophysiology of Takotsubo Syndrome. Circulation 2017, 135, 2426–2441. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.R.; Wolfe, B.B.; Harden, T.K.; Molinoff, P.B. Supersensitivity in rat cerebral cortex: Pre- and postsynaptic effects of 6-hydroxydopamine at noradrenergic synapses. Mol. Pharmacol. 1977, 13, 1170–1180. [Google Scholar] [PubMed]
- Glaubiger, G.; Lefkowitz, R.J. Elevated beta-adrenergic receptor number after chronic propranolol treatment. Biochem. Biophys. Res. Commun. 1977, 78, 720–725. [Google Scholar] [CrossRef]
- Swedberg, K.; Hjalmarson, A.; Waagstein, F.; Wallentin, I. Beneficial effects of long-term beta-blockade in congestive cardiomyopathy. Br. Heart J. 1980, 44, 117–133. [Google Scholar] [CrossRef]
- Engelmeier, R.S.; O’Connell, J.B.; Walsh, R.; Rad, N.; Scanlon, P.J.; Gunnar, R.M. Improvement in symptoms and exercise tolerance by metoprolol in patients with dilated cardiomyopathy: A double-blind, randomized, placebo-controlled trial. Circulation 1985, 72, 536–546. [Google Scholar] [CrossRef]
- Krum, H.; Mohacsi, P.; Katus, H.A.; Tendera, M.; Rouleau, J.L.; Fowler, M.B.; Coats, A.J.; Roecker, E.B.; Packer, M. Are beta-blockers needed in patients receiving spironolactone for severe chronic heart failure? An analysis of the COPERNICUS study. Am. Heart J. 2006, 151, 55–61. [Google Scholar] [CrossRef]
- Packer, M.; Fowler, M.B.; Roecker, E.B.; Coats, A.J.; Katus, H.A.; Krum, H.; Mohacsi, P.; Rouleau, J.L.; Tendera, M.; Staiger, C.; et al. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: Results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 2002, 106, 2194–2199. [Google Scholar] [CrossRef]
- Krukemyer, J.J. Use of beta-adrenergic blocking agents in congestive heart failure. Clin. Pharm. 1990, 9, 853–863. [Google Scholar]
- Urbach, J.; Goldsmith, S.R. Vasopressin antagonism in heart failure: A review of the hemodynamic studies and major clinical trials. Ther. Adv. Cardiovasc. Dis. 2021, 15, 1753944720977741. [Google Scholar] [CrossRef] [PubMed]
- Iovino, M.; Iacoviello, M.; De Pergola, G.; Licchelli, B.; Iovino, E.; Guastamacchia, E.; Giagulli, V.A.; Triggiani, V. Vasopressin in Heart Failure. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, S.R. Arginine vasopressin antagonism in heart failure: Current status and possible new directions. J. Cardiol. 2019, 74, 49–52. [Google Scholar] [CrossRef]
- Ali, F.; Guglin, M.; Vaitkevicius, P.; Ghali, J.K. Therapeutic potential of vasopressin receptor antagonists. Drugs 2007, 67, 847–858. [Google Scholar] [CrossRef]
- Oghlakian, G.; Klapholz, M. Vasopressin and vasopressin receptor antagonists in heart failure. Cardiol. Rev. 2009, 17, 10–15. [Google Scholar] [CrossRef]
- Udelson, J.E.; Orlandi, C.; Ouyang, J.; Krasa, H.; Zimmer, C.A.; Frivold, G.; Haught, W.H.; Meymandi, S.; Macarie, C.; Raef, D.; et al. Acute hemodynamic effects of tolvaptan, a vasopressin V2 receptor blocker, in patients with symptomatic heart failure and systolic dysfunction: An international, multicenter, randomized, placebo-controlled trial. J. Am. Coll. Cardiol. 2008, 52, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Costello-Boerrigter, L.C.; Smith, W.B.; Boerrigter, G.; Ouyang, J.; Zimmer, C.A.; Orlandi, C.; Burnett, J.C., Jr. Vasopressin-2-receptor antagonism augments water excretion without changes in renal hemodynamics or sodium and potassium excretion in human heart failure. Am. J. Physiol. Renal. Physiol. 2006, 290, F273–F278. [Google Scholar] [CrossRef]
- Urso, C.; Brucculeri, S.; Caimi, G. Employment of vasopressin receptor antagonists in management of hyponatraemia and volume overload in some clinical conditions. J. Clin. Pharm. Ther. 2015, 40, 376–385. [Google Scholar] [CrossRef]
- Bhandari, S.; Peri, A.; Cranston, I.; McCool, R.; Shaw, A.; Glanville, J.; Petrakova, L.; O’Reilly, K. A systematic review of known interventions for the treatment of chronic nonhypovolaemic hypotonic hyponatraemia and a meta-analysis of the vaptans. Clin. Endocrinol. 2017, 86, 761–771. [Google Scholar] [CrossRef]
- Rossi, J.; Bayram, M.; Udelson, J.E.; Lloyd-Jones, D.; Adams, K.F.; Oconnor, C.M.; Stough, W.G.; Ouyang, J.; Shin, D.D.; Orlandi, C.; et al. Improvement in hyponatremia during hospitalization for worsening heart failure is associated with improved outcomes: Insights from the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in CHF) trial. Acute Card Care 2007, 9, 82–86. [Google Scholar] [CrossRef]
- Hauptman, P.J.; Burnett, J.; Gheorghiade, M.; Grinfeld, L.; Konstam, M.A.; Kostic, D.; Krasa, H.B.; Maggioni, A.; Ouyang, J.; Swedberg, K.; et al. Clinical course of patients with hyponatremia and decompensated systolic heart failure and the effect of vasopressin receptor antagonism with tolvaptan. J. Card. Fail. 2013, 19, 390–397. [Google Scholar] [CrossRef]
- Konstam, M.A.; Gheorghiade, M.; Burnett, J.C., Jr.; Grinfeld, L.; Maggioni, A.P.; Swedberg, K.; Udelson, J.E.; Zannad, F.; Cook, T.; Ouyang, J.; et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: The EVEREST Outcome Trial. JAMA 2007, 297, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.J.; Yang, J.; Yang, J.; Fan, Z.X. Arginine vasopressin antagonist tolvaptan in the treatment of heart failure: A meta-analysis of randomized controlled trials. Int. J. Clin. Exp. Med. 2015, 8, 22117–22128. [Google Scholar] [PubMed]
- Álvarez-Zaballos, S.; Martínez-Sellés, M. Angiotensin-Converting Enzyme and Heart Failure. Front. Biosci. 2023, 28, 150. [Google Scholar] [CrossRef]
- Wang, Y.; Seto, S.W.; Golledge, J. Angiotensin II, sympathetic nerve activity and chronic heart failure. Heart Fail. Rev. 2014, 19, 187–198. [Google Scholar] [CrossRef]
- Mirzoyev, Z.; Anavekar, N.S.; Chen, H.H. Renal and humoral pathophysiological actions of angiotensin II in congestive heart failure. Timely Top. Med. Cardiovasc. Dis. 2005, 9, E9. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef]
- Balakumar, P.; Handa, S.; Alqahtani, A.; Alqahtani, T.; Khan, N.A.; LakshmiRaj, R.S.; Thangathirupathi, A.; Sundram, K.; Shenoy, V. Unraveling the Differentially Articulated Axes of the Century-Old Renin-Angiotensin-Aldosterone System: Potential Therapeutic Implications. Cardiovasc. Toxicol. 2022, 22, 246–253. [Google Scholar] [CrossRef]
- Tsutsui, H.; Albert, N.M.; Coats, A.J.S.; Anker, S.D.; Bayes-Genis, A.; Butler, J.; Chioncel, O.; Defilippi, C.R.; Drazner, M.H.; Felker, G.M.; et al. Natriuretic Peptides: Role in the Diagnosis and Management of Heart Failure: A Scientific Statement From the Heart Failure Association of the European Society of Cardiology, Heart Failure Society of America and Japanese Heart Failure Society. J. Card. Fail. 2023, 29, 787–804. [Google Scholar] [CrossRef]
- Díez, J. Chronic heart failure as a state of reduced effectiveness of the natriuretic peptide system: Implications for therapy. Eur. J. Heart Fail. 2017, 19, 167–176. [Google Scholar] [CrossRef]
- Cruden, N.L.; Fox, K.A.; Ludlam, C.A.; Johnston, N.R.; Newby, D.E. Neutral endopeptidase inhibition augments vascular actions of bradykinin in patients treated with angiotensin-converting enzyme inhibition. Hypertension 2004, 44, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling--concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Anavekar, N.; Skali, H.; McMurray, J.J.; Swedberg, K.; Yusuf, S.; Granger, C.B.; Michelson, E.L.; Wang, D.; Pocock, S.; et al. Influence of ejection fraction on cardiovascular outcomes in a broad spectrum of heart failure patients. Circulation 2005, 112, 3738–3744. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.R.; Daubert, C.; Abraham, W.T.; Ghio, S.; St John Sutton, M.; Hudnall, J.H.; Cerkvenik, J.; Linde, C. The effect of reverse remodeling on long-term survival in mildly symptomatic patients with heart failure receiving cardiac resynchronization therapy: Results of the REVERSE study. Heart Rhythm 2015, 12, 524–530. [Google Scholar] [CrossRef]
- Pascual-Figal, D.; Bayés-Genis, A.; Beltrán-Troncoso, P.; Caravaca-Pérez, P.; Conde-Martel, A.; Crespo-Leiro, M.G.; Delgado, J.F.; Díez, J.; Formiga, F.; Manito, N. Sacubitril-Valsartan, Clinical Benefits and Related Mechanisms of Action in Heart Failure with Reduced Ejection Fraction. A Review. Front. Cardiovasc. Med. 2021, 8, 754499. [Google Scholar] [CrossRef]
- St John Sutton, M.; Ghio, S.; Plappert, T.; Tavazzi, L.; Scelsi, L.; Daubert, C.; Abraham, W.T.; Gold, M.R.; Hassager, C.; Herre, J.M.; et al. Cardiac resynchronization induces major structural and functional reverse remodeling in patients with New York Heart Association class I/II heart failure. Circulation 2009, 120, 1858–1865. [Google Scholar] [CrossRef]
- Martignani, C.; Diemberger, I.; Nanni, C.; Biffi, M.; Ziacchi, M.; Boschi, S.; Corzani, A.; Fanti, S.; Sambuceti, G.; Boriani, G. Cardiac resynchronization therapy and cardiac sympathetic function. Eur. J. Clin. Investig. 2015, 45, 792–799. [Google Scholar] [CrossRef]
- Tarquini, R.; Guerra, C.T.; Porciani, M.C.; Michelucci, A.; Padeletti, M.; Ricciardi, G.; Chiostri, M.; Jelic, S.; Padeletti, L. Effects of cardiac resynchronization therapy on systemic inflammation and neurohormonal pathways in heart failure. Cardiol. J. 2009, 16, 545–552. [Google Scholar]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, 1757–1780. [Google Scholar] [CrossRef]
- Tai, C.; Gan, T.; Zou, L.; Sun, Y.; Zhang, Y.; Chen, W.; Li, J.; Zhang, J.; Xu, Y.; Lu, H.; et al. Effect of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on cardiovascular events in patients with heart failure: A meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2017, 17, 257. [Google Scholar] [CrossRef]
- Cohen-Segev, R.; Nativ, O.; Kinaneh, S.; Aronson, D.; Kabala, A.; Hamoud, S.; Karram, T.; Abassi, Z. Effects of Angiotensin 1–7 and Mas Receptor Agonist on Renal System in a Rat Model of Heart Failure. Int. J. Mol. Sci. 2023, 24, 11470. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.X.; Hu, J.; Cheng, J.; Liu, C.; Wei, X. The function of the ACE2/Ang(1-7)/Mas receptor axis of the renin-angiotensin system in myocardial ischemia reperfusion injury. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 1852–1859. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Dhalla, N.S. Adaptive and maladaptive roles of different angiotensin receptors in the development of cardiac hypertrophy and heart failure. Can. J. Physiol. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Berry, C.; Murphy, N.F.; De Vito, G.; Galloway, S.; Seed, A.; Fisher, C.; Sattar, N.; Vallance, P.; Hillis, W.S.; McMurray, J. Effects of aldosterone receptor blockade in patients with mild-moderate heart failure taking a beta-blocker. Eur. J. Heart Fail. 2007, 9, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Krum, H.; Shi, H.; Pitt, B.; McMurray, J.; Swedberg, K.; van Veldhuisen, D.J.; Vincent, J.; Pocock, S.; Zannad, F. Clinical benefit of eplerenone in patients with mild symptoms of systolic heart failure already receiving optimal best practice background drug therapy: Analysis of the EMPHASIS-HF study. Circ. Heart Fail. 2013, 6, 711–718. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Rossello, X.; Eschalier, R.; McMurray, J.J.V.; Pocock, S.; Girerd, N.; Rossignol, P.; Pitt, B.; Zannad, F. MRAs in Elderly HF Patients: Individual Patient-Data Meta-Analysis of RALES, EMPHASIS-HF, and TOPCAT. JACC Heart Fail. 2019, 7, 1012–1021. [Google Scholar] [CrossRef]
- Manolis, A.A.; Manolis, T.A.; Melita, H.; Manolis, A.S. Eplerenone Versus Spironolactone in Resistant Hypertension: An Efficacy and/or Cost or Just a Men’s Issue? Curr. Hypertens. Rep. 2019, 21, 22. [Google Scholar] [CrossRef]
- Rossignol, P.; Dobre, D.; McMurray, J.J.; Swedberg, K.; Krum, H.; van Veldhuisen, D.J.; Shi, H.; Messig, M.; Vincent, J.; Girerd, N.; et al. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: Results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ. Heart Fail. 2014, 7, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Girerd, N.; Collier, T.; Pocock, S.; Krum, H.; McMurray, J.J.; Swedberg, K.; Van Veldhuisen, D.J.; Vincent, J.; Pitt, B.; Zannad, F. Clinical benefits of eplerenone in patients with systolic heart failure and mild symptoms when initiated shortly after hospital discharge: Analysis from the EMPHASIS-HF trial. Eur. Heart J. 2015, 36, 2310–2317. [Google Scholar] [CrossRef]
- Bozkurt, B.; Nair, A.P.; Misra, A.; Scott, C.Z.; Mahar, J.H.; Fedson, S. Neprilysin Inhibitors in Heart Failure: The Science, Mechanism of Action, Clinical Studies, and Unanswered Questions. JACC Basic Transl. Sci. 2023, 8, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Perrone-Filardi, P.; Paolillo, S.; Agostoni, P.; Basile, C.; Basso, C.; Barillà, F.; Correale, M.; Curcio, A.; Mancone, M.; Merlo, M.; et al. Renin-angiotensin-aldosterone system inhibition in patients affected by heart failure: Efficacy, mechanistic effects and practical use of sacubitril/valsartan. Position Paper of the Italian Society of Cardiology. Eur. J. Intern. Med. 2022, 102, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, P.; Coelho, C.; Costa-Oliveira, C.; Rocha, S. The Effect of Sacubitril-Valsartan on Ventricular Arrhythmia Burden in Patients with Heart Failure with Reduced Ejection Fraction. Cureus 2023, 15, e34508. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, V.; Gentile, F.; Ghionzoli, N.; Chiriacò, M.; Panichella, G.; Aimo, A.; Vergaro, G.; Giannoni, A.; Passino, C.; Emdin, M. Pathophysiological Rationale and Clinical Evidence for Neurohormonal Modulation in Heart Failure with Preserved Ejection Fraction. Card. Fail. Rev. 2023, 9, e09. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Docherty, K.F.; Claggett, B.L.; Jhund, P.S.; de Boer, R.A.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. SGLT-2 inhibitors in patients with heart failure: A comprehensive meta-analysis of five randomised controlled trials. Lancet 2022, 400, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Litwin, S.E.; East, C.A. Assessing clinical and biomarker characteristics to optimize the benefits of sacubitril/valsartan in heart failure. Front. Cardiovasc. Med. 2022, 9, 1058998. [Google Scholar] [CrossRef]
- Minatoguchi, S. Heart failure and its treatment from the perspective of sympathetic nerve activity. J. Cardiol. 2022, 79, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Gaggin, H.K.; Januzzi, J.L., Jr. Biomarkers and diagnostics in heart failure. Biochim. Et Biophys. Acta 2013, 1832, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Schelbert, E.B.; Bonnett, L.J.; Lewis, G.A.; Lagan, J.; Orsborne, C.; Brown, P.F.; Black, N.; Naish, J.H.; Williams, S.G.; et al. Growth differentiation factor-15 in patients with or at risk of heart failure but before first hospitalisation. Heart 2023. [Google Scholar] [CrossRef]
- Spoto, S.; Argemi, J.; Di Costanzo, R.; Gavira Gomez, J.J.; Salterain Gonzales, N.; Basili, S.; Cangemi, R.; Abbate, A.; Locorriere, L.; Masini, F.; et al. Mid-Regional Pro-Adrenomedullin and N-Terminal Pro-B-Type Natriuretic Peptide Measurement: A Multimarker Approach to Diagnosis and Prognosis in Acute Heart Failure. J. Pers. Med. 2023, 13, 1155. [Google Scholar] [CrossRef]
- Zhang, X.; Wan, Y.; Karunathilaka, N.; Chan, W.; Kostner, K.; Hartel, G.; Coats, A.J.S.; Atherton, J.J.; Punyadeera, C. Prognostic utility of serum NT-proBNP (fragments 1-76aa and 13-71aa) and galectin-3 in predicting death and re-hospitalisation due to cardiovascular events in patients with heart failure. Heart Vessel. 2023. [Google Scholar] [CrossRef]
- Dmour, B.A.; Costache, A.D.; Dmour, A.; Huzum, B.; Duca, Ș.T.; Chetran, A.; Miftode, R.; Afrăsânie, I.; Tuchiluș, C.; Cianga, C.M.; et al. Could Endothelin-1 Be a Promising Neurohormonal Biomarker in Acute Heart Failure? Diagnostics 2023, 13, 2277. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.; Burger, A.J. Neurohumoral activation and ventricular arrhythmias in patients with decompensated congestive heart failure: Role of endothelin. Pacing Clin. Electrophysiol. 2003, 26, 703–710. [Google Scholar] [CrossRef]
- Aspromonte, N.; Gulizia, M.M.; Clerico, A.; Di Tano, G.; Emdin, M.; Feola, M.; Iacoviello, M.; Latini, R.; Mortara, A.; Valle, R.; et al. ANMCO/ELAS/SIBioC Consensus Document: Biomarkers in heart failure. Eur. Heart J. Suppl. 2017, 19, D102–D112. [Google Scholar] [CrossRef] [PubMed]
- Clerico, A.; Giannoni, A.; Vittorini, S.; Passino, C. Thirty years of the heart as an endocrine organ: Physiological role and clinical utility of cardiac natriuretic hormones. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H12–H20. [Google Scholar] [CrossRef] [PubMed]
- Sangaralingham, S.J.; Kuhn, M.; Cannone, V.; Chen, H.H.; Burnett, J.C. Natriuretic peptide pathways in heart failure: Further therapeutic possibilities. Cardiovasc. Res. 2023, 118, 3416–3433. [Google Scholar] [CrossRef]
- Bencivenga, L.; Palaia, M.E.; Sepe, I.; Gambino, G.; Komici, K.; Cannavo, A.; Femminella, G.D.; Rengo, G. Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker? Cells 2021, 10, 457. [Google Scholar] [CrossRef]
- Ibrahim, N.E.; Januzzi, J.L., Jr. Established and Emerging Roles of Biomarkers in Heart Failure. Circ. Res. 2018, 123, 614–629. [Google Scholar] [CrossRef]
- Lin, D.C.; Diamandis, E.P.; Januzzi, J.L., Jr.; Maisel, A.; Jaffe, A.S.; Clerico, A. Natriuretic peptides in heart failure. Clin. Chem. 2014, 60, 1040–1046. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Fonarow, G.C.; Peacock, W.F.; Phillips, C.O.; Givertz, M.M.; Lopatin, M. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. J. Am. Coll. Cardiol. 2007, 49, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Sourour, N.; Riveland, E.; Naesgaard, P.; Kjekshus, H.; Larsen, A.I.; Omland, T.; Røsjø, H.; Myhre, P.L. N-terminal pro-B-type natriuretic peptide for prediction of ventricular arrhythmias: Data from the SMASH study. Clin. Cardiol. 2023, 46, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Prausmüller, S.; Arfsten, H.; Spinka, G.; Freitag, C.; Bartko, P.E.; Goliasch, G.; Strunk, G.; Pavo, N.; Hülsmann, M. Plasma Neprilysin Displays No Relevant Association with Neurohumoral Activation in Chronic HFrEF. J. Am. Heart Assoc. 2020, 9, e015071. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Emdin, M.; Iervasi, A.; Zyw, L.; Gabutti, A.; Poletti, R.; Mammini, C.; Giannoni, A.; Fontana, M.; Passino, C. Prognostic value of plasma renin activity in heart failure. Am. J. Cardiol. 2011, 108, 246–251. [Google Scholar] [CrossRef]
- Zalewska, E.; Kmieć, P.; Sworczak, K. Role of Catestatin in the Cardiovascular System and Metabolic Disorders. Front. Cardiovasc. Med. 2022, 9, 909480. [Google Scholar] [CrossRef]
- Bozic, J.; Kumric, M.; Ticinovic Kurir, T.; Urlic, H.; Martinovic, D.; Vilovic, M.; Tomasovic Mrcela, N.; Borovac, J.A. Catestatin as a Biomarker of Cardiovascular Diseases: A Clinical Perspective. Biomedicines 2021, 9, 1757. [Google Scholar] [CrossRef]
- Mahapatra, N.R. Catestatin is a novel endogenous peptide that regulates cardiac function and blood pressure. Cardiovasc. Res. 2008, 80, 330–338. [Google Scholar] [CrossRef]
- Mahata, S.K.; Kiranmayi, M.; Mahapatra, N.R. Catestatin: A Master Regulator of Cardiovascular Functions. Curr. Med. Chem. 2018, 25, 1352–1374. [Google Scholar] [CrossRef]
- Mullens, W.; Verbrugge, F.H.; Nijst, P.; Tang, W.H.W. Renal sodium avidity in heart failure: From pathophysiology to treatment strategies. Eur. Heart J. 2017, 38, 1872–1882. [Google Scholar] [CrossRef]
- von Lueder, T.G.; Atar, D.; Krum, H. Diuretic use in heart failure and outcomes. Clin. Pharmacol. Ther. 2013, 94, 490–498. [Google Scholar] [CrossRef]
- Verbrugge, F.H. Editor’s Choice-Diuretic resistance in acute heart failure. Eur. Heart J. Acute Cardiovasc. Care 2018, 7, 379–389. [Google Scholar] [CrossRef]
- De Vecchis, R.; Ciccarelli, A.; Pucciarelli, A. Unloading therapy by intravenous diuretic in chronic heart failure: A double-edged weapon? J. Cardiovasc. Med. 2010, 11, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Talha, K.M.; Anker, S.D.; Butler, J. SGLT-2 Inhibitors in Heart Failure: A Review of Current Evidence. Int. J. Heart Fail. 2023, 5, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.S.; Siddiqi, T.J.; Anker, S.D.; Bakris, G.L.; Bhatt, D.L.; Filippatos, G.; Fonarow, G.C.; Greene, S.J.; Januzzi, J.L., Jr.; Khan, M.S.; et al. Effect of SGLT2 Inhibitors on Cardiovascular Outcomes Across Various Patient Populations. J. Am. Coll. Cardiol. 2023, 81, 2377–2387. [Google Scholar] [CrossRef]
- Jhund, P.S. Improving heart failure outcomes with sodium-glucose cotransporter 2 inhibitors in different patient groups. Diabetes Obes. Metab. 2023, 25 (Suppl. 3), 26–32. [Google Scholar] [CrossRef]
- Manolis, A.A.; Manolis, T.A.; Melita, H.; Manolis, A.S. Sodium-glucose cotransporter type 2 inhibitors and cardiac arrhythmias. Trends Cardiovasc. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Biegus, J.; Fudim, M.; Salah, H.M.; Heerspink, H.J.L.; Voors, A.A.; Ponikowski, P. Sodium-glucose cotransporter-2 inhibitors in heart failure: Potential decongestive mechanisms and current clinical studies. Eur. J. Heart Fail. 2023. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Rao, V.S.; Ivey-Miranda, J.; Fleming, J.; Mahoney, D.; Maulion, C.; Suda, N.; Siwakoti, K.; Ahmad, T.; Jacoby, D.; et al. Empagliflozin in Heart Failure: Diuretic and Cardiorenal Effects. Circulation 2020, 142, 1028–1039. [Google Scholar] [CrossRef]
- Packer, M. Molecular, Cellular, and Clinical Evidence That Sodium-Glucose Cotransporter 2 Inhibitors Act as Neurohormonal Antagonists When Used for the Treatment of Chronic Heart Failure. J. Am. Heart Assoc. 2020, 9, e016270. [Google Scholar] [CrossRef]
- Gronda, E.; Jessup, M.; Iacoviello, M.; Palazzuoli, A.; Napoli, C. Glucose Metabolism in the Kidney: Neurohormonal Activation and Heart Failure Development. J. Am. Heart Assoc. 2020, 9, e018889. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Borges, J.I.; Cora, N.; Sizova, A. Sympatholytic Mechanisms for the Beneficial Cardiovascular Effects of SGLT2 Inhibitors: A Research Hypothesis for Dapagliflozin’s Effects in the Adrenal Gland. Int. J. Mol. Sci. 2021, 22, 7684. [Google Scholar] [CrossRef] [PubMed]
- von Lueder, T.G.; Kotecha, D.; Atar, D.; Hopper, I. Neurohormonal Blockade in Heart Failure. Card. Fail. Rev. 2017, 3, 19–24. [Google Scholar] [CrossRef]
- Silva, J.E.; Melo, N.; Ferreira, A.I.; Silva, C.; Oliveira, D.; Lume, M.J.; Pereira, J.; Araújo, J.P.; Lourenço, P. Prognostic impact of neurohormonal modulation in very old patients with chronic heart failure. Age Ageing 2022, 51, afac076. [Google Scholar] [CrossRef] [PubMed]
- Barywani, S.B.; Ergatoudes, C.; Schaufelberger, M.; Petzold, M.; Fu, M.L. Does the target dose of neurohormonal blockade matter for outcome in Systolic heart failure in octogenarians? Int. J. Cardiol. 2015, 187, 666–672. [Google Scholar] [CrossRef]
- Gilstrap, L.; Solomon, N.; Chiswell, K.; James O’Malley, A.; Skinner, J.S.; Fonarow, G.C.; Bhatt, D.L.; Yancy, C.W.; Devore, A.D. The Association between Beta-blocker and Renin-Angiotensin System Inhibitor Use after Heart Failure with Reduced Ejection Fraction Hospitalization and Outcomes in Older Patients. J. Card. Fail. 2023, 29, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Brunner-La Rocca, H.P.; Knackstedt, C.; Eurlings, L.; Rolny, V.; Krause, F.; Pfisterer, M.E.; Tobler, D.; Rickenbacher, P.; Maeder, M.T. Impact of worsening renal function related to medication in heart failure. Eur. J. Heart Fail. 2015, 17, 159–168. [Google Scholar] [CrossRef]
- Miura, M.; Sugimura, K.; Sakata, Y.; Miyata, S.; Tadaki, S.; Yamauchi, T.; Onose, T.; Tsuji, K.; Abe, R.; Oikawa, T.; et al. Prognostic Impact of Loop Diuretics in Patients with Chronic Heart Failure-Effects of Addition of Renin-Angiotensin-Aldosterone System Inhibitors and β-Blockers. Circ. J. 2016, 80, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Chatur, S.; Claggett, B.L.; McCausland, F.R.; Rouleau, J.; Zile, M.R.; Packer, M.; Pfeffer, M.A.; Lefkowitz, M.; McMurray, J.J.V.; Solomon, S.D.; et al. Variation in Renal Function Following Transition to Sacubitril/Valsartan in Patients with Heart Failure. J. Am. Coll. Cardiol. 2023, 81, 1443–1455. [Google Scholar] [CrossRef]
- Grupper, A.; Zhao, Y.M.; Sajgalik, P.; Joyce, L.D.; Park, S.J.; Pereira, N.L.; Stulak, J.M.; Burnett, J.C., Jr.; Edwards, B.S.; Daly, R.C.; et al. Effect of Neurohormonal Blockade Drug Therapy on Outcomes and Left Ventricular Function and Structure after Left Ventricular Assist Device Implantation. Am. J. Cardiol. 2016, 117, 1765–1770. [Google Scholar] [CrossRef]
- Imamura, T.; Mehta, P.; Nguyen, A.; Chung, B.; Narang, N.; Rodgers, D.; Raikhelkar, J.; Smith, B.; Song, T.; Ota, T.; et al. Neurohormonal Blockade during Left Ventricular Assist Device Support. Asaio J. 2020, 66, 881–885. [Google Scholar] [CrossRef]
- McCullough, M.; Caraballo, C.; Ravindra, N.G.; Miller, P.E.; Mezzacappa, C.; Levin, A.; Gruen, J.; Rodwin, B.; Reinhardt, S.; van Dijk, D.; et al. Neurohormonal Blockade and Clinical Outcomes in Patients with Heart Failure Supported by Left Ventricular Assist Devices. JAMA Cardiol. 2020, 5, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S. Cardiac resynchronization therapy in congestive heart failure: Ready for prime time? Heart Rhythm 2004, 1, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Hürlimann, D.; Starck, C.T.; Hindricks, G.; Lüscher, T.F.; Ruschitzka, F.; Steffel, J. Treatment with higher dosages of heart failure medication is associated with improved outcome following cardiac resynchronization therapy. Eur. Heart J. 2014, 35, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Hogas, M.; Statescu, C.; Padurariu, M.; Ciobica, A.; Bilha, S.C.; Haisan, A.; Timofte, D.; Hogas, S. Salt, Not Always a Cardiovascular Enemy? A Mini-Review and Modern Perspective. Medicina 2022, 58, 1175. [Google Scholar] [CrossRef]
- Arafa, A.; Kokubo, Y.; Teramoto, M.; Kashima, R.; Shimamoto, K.; Nakao, Y.M.; Matsuo, M.; Yoshihara, F.; Izumi, C. Blood pressure per the 2017 ACC/AHA and 2018 ESC/ESH guidelines and heart failure risk: The Suita Study. Hypertens. Res. 2023, 46, 575–582. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2018, 71, e127–e248. [Google Scholar] [CrossRef]
- Rami, K. Aggressive salt and water restriction in acutely decompensated heart failure: Is it worth its weight in salt? Expert Rev. Cardiovasc. Ther. 2013, 11, 1125–1128. [Google Scholar] [CrossRef]
- O’Donnell, M.; Mente, A.; Alderman, M.H.; Brady, A.J.B.; Diaz, R.; Gupta, R.; López-Jaramillo, P.; Luft, F.C.; Lüscher, T.F.; Mancia, G.; et al. Salt and cardiovascular disease: Insufficient evidence to recommend low sodium intake. Eur. Heart J. 2020, 41, 3363–3373. [Google Scholar] [CrossRef]
- Manolis, A.S. You Are What You Eat, Hence Curtail Saturated and Trans Fats, Free Sugars and Salt. Rhythmos 2016, 11, 28–38. [Google Scholar]
- Manolis, A.S.; Sakellaris, N.; Pyrros, I. Salt controversy stirred by “PURE” but settled by “NUTRICODE”. Hosp. Chron. 2014, 9, 232–235. [Google Scholar]
- Manolis, A.A.; Manolis, T.A.; Melita, H.; Manolis, A.S. Features of a Balanced Healthy Diet with Cardiovascular and Other Benefits. Curr. Vasc. Pharmacol. 2023, 21, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.; Joseph, J. Sodium Intake and Heart Failure. Int. J. Mol. Sci. 2020, 21, 9474. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Huang, L.H.; Ku, C.H. Use of dietary sodium intervention effect on neurohormonal and fluid overload in heart failure patients: Review of select research based literature. Appl. Nurs. Res. 2018, 42, 17–21. [Google Scholar] [CrossRef]
- O’Donnell, M.; Mente, A.; Yusuf, S. Sodium intake and cardiovascular health. Circ. Res. 2015, 116, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Ouwerkerk, W.; van Veldhuisen, D.J.; Hillege, H.L.; Richards, A.M.; van der Meer, P.; Anand, I.S.; Lam, C.S.P.; Voors, A.A. A Systematic Review and Network Meta-Analysis of Pharmacological Treatment of Heart Failure with Reduced Ejection Fraction. JACC Heart Fail. 2022, 10, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.E.; Burnett, J.C., Jr.; Butler, J.; Camacho, A.; Felker, G.M.; Fiuzat, M.; O’Connor, C.; Solomon, S.D.; Vaduganathan, M.; Zile, M.R.; et al. Natriuretic Peptides as Inclusion Criteria in Clinical Trials: A JACC: Heart Failure Position Paper. JACC Heart Fail. 2020, 8, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Sackner-Bernstein, J.D.; Kowalski, M.; Fox, M.; Aaronson, K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: A pooled analysis of randomized controlled trials. JAMA 2005, 293, 1900–1905. [Google Scholar] [CrossRef]
- Sackner-Bernstein, J.D.; Skopicki, H.A.; Aaronson, K.D. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation 2005, 111, 1487–1491. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Starling, R.C.; Hernandez, A.F.; Armstrong, P.W.; Dickstein, K.; Hasselblad, V.; Heizer, G.M.; Komajda, M.; Massie, B.M.; McMurray, J.J.; et al. Effect of nesiritide in patients with acute decompensated heart failure. N. Engl. J. Med. 2011, 365, 32–43. [Google Scholar] [CrossRef]
- Mentz, R.J.; Felker, G.M.; Ahmad, T.; Peacock, W.F.; Pitt, B.; Fiuzat, M.; Maggioni, A.P.; Gheorghiade, M.; Ando, Y.; Pocock, S.J.; et al. Learning from recent trials and shaping the future of acute heart failure trials. Am. Heart J. 2013, 166, 629–635. [Google Scholar] [CrossRef]
- Packer, M.; O’Connor, C.; McMurray, J.J.V.; Wittes, J.; Abraham, W.T.; Anker, S.D.; Dickstein, K.; Filippatos, G.; Holcomb, R.; Krum, H.; et al. Effect of Ularitide on Cardiovascular Mortality in Acute Heart Failure. N. Engl. J. Med. 2017, 376, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.J.; Hernandez, A.F.; Sun, J.L.; Butler, J.; Armstrong, P.W.; Ezekowitz, J.A.; Zannad, F.; Ferreira, J.P.; Coles, A.; Metra, M.; et al. Relationship between Enrolling Country Income Level and Patient Profile, Protocol Completion, and Trial End Points. Circulation. Cardiovasc. Qual. Outcomes 2018, 11, e004783. [Google Scholar] [CrossRef] [PubMed]
- Cody, R.J.; Atlas, S.A.; Laragh, J.H.; Kubo, S.H.; Covit, A.B.; Ryman, K.S.; Shaknovich, A.; Pondolfino, K.; Clark, M.; Camargo, M.J.; et al. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J. Clin. Investig. 1986, 78, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakao, K.; Nishimura, K.; Sugawara, A.; Okumura, K.; Obata, K.; Sonoda, R.; Ban, T.; Yasue, H.; Imura, H. Clinical application of atrial natriuretic polypeptide in patients with congestive heart failure: Beneficial effects on left ventricular function. Circulation 1987, 76, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Nomura, F.; Kurobe, N.; Mori, Y.; Hikita, A.; Kawai, M.; Suwa, M.; Okutani, Y. Multicenter prospective investigation on efficacy and safety of carperitide as a first-line drug for acute heart failure syndrome with preserved blood pressure: COMPASS: Carperitide Effects Observed Through Monitoring Dyspnea in Acute Decompensated Heart Failure Study. Circ. J. 2008, 72, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Iwakami, N.; Nakai, M.; Nishimura, K.; Sumita, Y.; Mizuno, A.; Tsutsui, H.; Ogawa, H.; Anzai, T. Effect of intravenous carperitide versus nitrates as first-line vasodilators on in-hospital outcomes in hospitalized patients with acute heart failure: Insight from a nationwide claim-based database. Int. J. Cardiol. 2019, 280, 104–109. [Google Scholar] [CrossRef]
- Nogi, K.; Ueda, T.; Matsue, Y.; Nogi, M.; Ishihara, S.; Nakada, Y.; Kawakami, R.; Kagiyama, N.; Kitai, T.; Oishi, S.; et al. Effect of carperitide on the 1 year prognosis of patients with acute decompensated heart failure. ESC Heart Fail. 2022, 9, 1061–1070. [Google Scholar] [CrossRef]
- Kamiya, M.; Sato, N.; Matsuda, J.; Nozaki, A.; Akiya, M.; Sato, T.; Okazaki, H.; Takahashi, Y.; Shimizu, W. Predictors of responders for low-dose carperitide monotherapy in patients with acute heart failure. Heart Vessel. 2020, 35, 59–68. [Google Scholar] [CrossRef]
- Sharma, A.; Felker, G.M. Brain Natriuretic Peptide Treatment and Heart Failure Prevention: Reliving the Mistakes of the Past or Charting a New Course for the Future? JACC Heart Fail. 2016, 4, 548–550. [Google Scholar] [CrossRef]
- Chen, H.H.; Anstrom, K.J.; Givertz, M.M.; Stevenson, L.W.; Semigran, M.J.; Goldsmith, S.R.; Bart, B.A.; Bull, D.A.; Stehlik, J.; LeWinter, M.M.; et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: The ROSE acute heart failure randomized trial. JAMA 2013, 310, 2533–2543. [Google Scholar] [CrossRef]
- Yancy, C.W.; Krum, H.; Massie, B.M.; Silver, M.A.; Stevenson, L.W.; Cheng, M.; Kim, S.S.; Evans, R. Safety and efficacy of outpatient nesiritide in patients with advanced heart failure: Results of the Second Follow-Up Serial Infusions of Nesiritide (FUSION II) trial. Circ. Heart Fail. 2008, 1, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S. Sudden death risk stratification in non-ischemic dilated cardiomyopathy using old and new tools: A clinical challenge. Expert Rev. Cardiovasc. Ther. 2017, 15, 315–325. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, A.A.; Manolis, T.A.; Melita, H. Sudden death in heart failure with preserved ejection fraction and beyond: An elusive target. Heart Fail. Rev. 2019, 24, 847–866. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S.; Estes, I.N.A.M. Reversal of electrophysiologic effects of flecainide on the accessory pathway by isoproterenol in the Wolff-Parkinson-White syndrome. Am. J. Cardiol. 1989, 64, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S. Preventing sudden death in idiopathic dilated cardiomyopathy: A difficult to settle issue. Rhythmos 2017, 12, 1–6. [Google Scholar]
- Squire, I. Neurohormonal intervention to reduce sudden cardiac death in heart failure: What is the optimal pharmacologic strategy? Heart Fail. Rev. 2004, 9, 337–345. [Google Scholar] [CrossRef]
- Kiuchi, M.G.; Chen, S.; Paz, L.M.R.; Purerfellner, H. Renal sympathetic denervation guided by renal nerve stimulation to treat ventricular arrhythmia in CKD patients with ICD. Oncotarget 2017, 8, 37296–37307. [Google Scholar] [CrossRef]
- Kiuchi, M.G.; Nolde, J.M.; Villacorta, H.; Carnagarin, R.; Chan, J.J.S.; Lugo-Gavidia, L.M.; Ho, J.K.; Matthews, V.B.; Dwivedi, G.; Schlaich, M.P. New Approaches in the Management of Sudden Cardiac Death in Patients with Heart Failure-Targeting the Sympathetic Nervous System. Int. J. Mol. Sci. 2019, 20, 2430. [Google Scholar] [CrossRef]
- Vecchi, A.L.; Abete, R.; Marazzato, J.; Iacovoni, A.; Mortara, A.; De Ponti, R.; Senni, M. Ventricular arrhythmias and ARNI: Is it time to reappraise their management in the light of new evidence? Heart Fail. Rev. 2022, 27, 103–110. [Google Scholar] [CrossRef]
- Yamada, C.; Kuwahara, K.; Yamazaki, M.; Nakagawa, Y.; Nishikimi, T.; Kinoshita, H.; Kuwabara, Y.; Minami, T.; Yamada, Y.; Shibata, J.; et al. The renin-angiotensin system promotes arrhythmogenic substrates and lethal arrhythmias in mice with non-ischaemic cardiomyopathy. Cardiovasc. Res. 2016, 109, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Francia, P.; Palano, F.; Tocci, G.; Adduci, C.; Ricotta, A.; Semprini, L.; Caprinozzi, M.; Balla, C.; Volpe, M. Angiotensin receptor antagonists to prevent sudden death in heart failure: Does the dose matter? ISRN Cardiol. 2014, 2014, 652421. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, J.; Abriel, H. Neurohormonal regulation of cardiac ion channels in chronic heart failure. J. Cardiovasc. Pharmacol. 2009, 54, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Cutler, M.J.; Rosenbaum, D.S.; Dunlap, M.E. Structural and electrical remodeling as therapeutic targets in heart failure. J. Electrocardiol. 2007, 40, S1–S7. [Google Scholar] [CrossRef]
- Shugg, T.; Hudmon, A.; Overholser, B.R. Neurohormonal Regulation of I(Ks) in Heart Failure: Implications for Ventricular Arrhythmogenesis and Sudden Cardiac Death. J. Am. Heart Assoc. 2020, 9, e016900. [Google Scholar] [CrossRef]
- Kääb, S.; Dixon, J.; Duc, J.; Ashen, D.; Näbauer, M.; Beuckelmann, D.J.; Steinbeck, G.; McKinnon, D.; Tomaselli, G.F. Molecular basis of transient outward potassium current downregulation in human heart failure: A decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 1998, 98, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Silverman, N.; Sabbah, H.N.; Undrovinas, A.I. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur. J. Heart Fail. 2007, 9, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S.M.; Bers, D.M. Calcium cycling in heart failure: The arrhythmia connection. J. Cardiovasc. Electrophysiol. 2002, 13, 88–91. [Google Scholar] [CrossRef]
- Schillinger, W.; Schneider, H.; Minami, K.; Ferrari, R.; Hasenfuss, G. Importance of sympathetic activation for the expression of Na+-Ca2+ exchanger in end-stage failing human myocardium. Eur. Heart J. 2002, 23, 1118–1124. [Google Scholar] [CrossRef][Green Version]
- Jost, N.; Virág, L.; Bitay, M.; Takács, J.; Lengyel, C.; Biliczki, P.; Nagy, Z.; Bogáts, G.; Lathrop, D.A.; Papp, J.G.; et al. Restricting excessive cardiac action potential and QT prolongation: A vital role for IKs in human ventricular muscle. Circulation 2005, 112, 1392–1399. [Google Scholar] [CrossRef]
- Jost, N.; Papp, J.G.; Varró, A. Slow delayed rectifier potassium current (IKs) and the repolarization reserve. Ann. Noninvasive Electrocardiol. 2007, 12, 64–78. [Google Scholar] [CrossRef]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Stengl, M.; Ramakers, C.; Donker, D.W.; Nabar, A.; Rybin, A.V.; Spätjens, R.L.; van der Nagel, T.; Wodzig, W.K.; Sipido, K.R.; Antoons, G.; et al. Temporal patterns of electrical remodeling in canine ventricular hypertrophy: Focus on IKs downregulation and blunted beta-adrenergic activation. Cardiovasc. Res. 2006, 72, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Codina, P.; Zamora, E.; Levy, W.C.; Cediel, G.; Santiago-Vacas, E.; Domingo, M.; Ruiz-Cueto, M.; Casquete, D.; Sarrias, A.; Borrellas, A.; et al. Sudden Cardiac Death in Heart Failure: A 20-Year Perspective From a Mediterranean Cohort. J. Card. Fail. 2023, 29, 236–245. [Google Scholar] [CrossRef]
- Mujadzic, H.; Prousi, G.S.; Napier, R.; Siddique, S.; Zaman, N. The Impact of Angiotensin Receptor-Neprilysin Inhibitors on Arrhythmias in Patients with Heart Failure: A Systematic Review and Meta-analysis. J. Innov. Card. Rhythm Manag. 2022, 13, 5164–5175. [Google Scholar] [CrossRef]
- Sato, T.; Kouzu, H.; Yano, T.; Sakuma, I.; Furuhashi, M.; Tohse, N. Potential favorable action of sodium-glucose cotransporter-2 inhibitors on sudden cardiac death: A brief overview. Front. Cardiovasc. Med. 2023, 10, 1159953. [Google Scholar] [CrossRef] [PubMed]
- Oates, C.P.; Santos-Gallego, C.G.; Smith, A.; Basyal, B.; Moss, N.; Kawamura, I.; Musikantow, D.R.; Turagam, M.K.; Miller, M.A.; Whang, W.; et al. SGLT2 inhibitors reduce sudden cardiac death risk in heart failure: Meta-analysis of randomized clinical trials. J. Cardiovasc. Electrophysiol. 2023, 34, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.C.; Flather, M.D.; Wang, D. Systematic review of the impact of beta blockers on mortality and hospital admissions in heart failure. Eur. J. Heart Fail. 2001, 3, 351–357. [Google Scholar] [CrossRef]
- Garg, R.; Yusuf, S. Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. Collaborative Group on ACE Inhibitor Trials. JAMA 1995, 273, 1450–1456. [Google Scholar] [CrossRef]
- Pinilla-Vera, M.; Hahn, V.S.; Kass, D.A. Leveraging Signaling Pathways to Treat Heart Failure with Reduced Ejection Fraction. Circ. Res. 2019, 124, 1618–1632. [Google Scholar] [CrossRef]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. Calmodulin-dependent protein kinase II: Linking heart failure and arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef]
- Yuyun, M.F.; Kinlay, S.; Singh, J.P.; Joseph, J. Are arrhythmias the drivers of sudden cardiac death in heart failure with preserved ejection fraction? A review. ESC Heart Fail. 2023, 10, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Leyva, F.; Israel, C.W.; Singh, J. Declining Risk of Sudden Cardiac Death in Heart Failure: Fact or Myth? Circulation 2023, 147, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Leyva, F.; Zegard, A.; Patel, P.; Stegemann, B.; Marshall, H.; Ludman, P.; de Bono, J.; Boriani, G.; Qiu, T. Improved prognosis after cardiac resynchronization therapy over a decade. Europace 2023, 25, euad141. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, e263–e421. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| I. Activation of the Sympathetic Nervous System (SNS) |
|
| II. Activation of the Renin–Angiotensin–Aldosterone System (RAAS) * † |
|
| III. Activation of the Arginine Vasopressin (AVP) System |
|
| IV. Natriuretic Peptides (NPs) |
|
| V. Sodium-Glucose Co-Transporter-2 (SGLT2) Inhibitors |
|
| VI. Future Directions |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manolis, A.A.; Manolis, T.A.; Manolis, A.S. Neurohumoral Activation in Heart Failure. Int. J. Mol. Sci. 2023, 24, 15472. https://doi.org/10.3390/ijms242015472
Manolis AA, Manolis TA, Manolis AS. Neurohumoral Activation in Heart Failure. International Journal of Molecular Sciences. 2023; 24(20):15472. https://doi.org/10.3390/ijms242015472
Chicago/Turabian StyleManolis, Antonis A., Theodora A. Manolis, and Antonis S. Manolis. 2023. "Neurohumoral Activation in Heart Failure" International Journal of Molecular Sciences 24, no. 20: 15472. https://doi.org/10.3390/ijms242015472
APA StyleManolis, A. A., Manolis, T. A., & Manolis, A. S. (2023). Neurohumoral Activation in Heart Failure. International Journal of Molecular Sciences, 24(20), 15472. https://doi.org/10.3390/ijms242015472