
The Role of Genetics in the Management of Heart Failure Patients


 ,
,  and
and
Abstract
:1. Introduction
2. Advancements and Applications of Genetics in Cardiovascular Disease
- Assessing transmission modes: discerning the mode of cardiomyopathy transmission is critical, with a need to determine whether it is monogenic or polygenic [19].
- Establishing genetic counseling and support: genetic counseling, complemented by psychological support, is essential [23]. In certain cases, initiation during childhood might be warranted.
- Predicting adverse events: clinical management decisions often hinge on the ability to predict potential adverse outcomes linked to a specific disease.
- Providing prognostic information: offering insights about the disease’s potential severity and progression trajectory is pivotal for both the patient and their family [20].
- Personalizing therapy: treatment strategies often need tailoring based on the specific mutations identified or the potential application of gene therapy methods [24].
2.1. Assessment of Pathogenic Genetic Variants
- TGP (targeted gene panel): this method examines genes linked to a specific phenotype. However, its scope is primarily confined to selected genes known for their variants, necessitating ongoing updates. The process entails designing a gene panel correlated with a distinct disease and conducting parallel sequencing. It is commonly employed as the primary diagnostic test for probands [31,39].
- WES: this method is adept at diagnosing probands presenting with diverse disorders, including pediatric and syndromic cardiomyopathies. It encompasses all genes with the objective of sequencing the complete exome. Through an integrated process, data corresponding to the entire exome can be produced, eliminating the need for additional analyses when updated information becomes available [40,41].
- Whole-genome sequencing (WGS): this technique sequences the complete genome, offering diagnostic insights for probands with varied disorders and detailed data on pharmacokinetic variants. While comprehensive, WGS comes with a greater expense and necessitates intricate data analysis. If panel sequencing yields negative results, both WES and WGS stand as viable subsequent diagnostic options [42].
2.1.1. Sequencing Modalities
- Presequencing panels (targeted resequencing): this approach enables the simultaneous analysis of multiple patients by selectively enriching specific genomic regions prior to sequencing.
- In silico panels (targeted data analysis): this method is applied after exome sequencing and focuses on genes directly associated with the disease under investigation, generally allowing for the analysis of a restricted number of samples in each run [44].
- -
- Cost: The difference in cost is influenced by the reagents used. The use of TGP is more economical if the number of samples per run is optimized. In economic terms, WGS is the most expensive, and although WES is pricier than panels, it can be advantageous depending on the type of study to be performed [46].
- -
- Purpose: Typically, NGS techniques are employed for diseases with high genetic heterogeneity or Mendelian-based genetic diseases (or those suspected to be genetic) where the causative genes remain unidentified. For diseases with established genetic etiology, either custom-designed panels, WES, or WGS can be utilized [45].
- -
- Sensitivity: Sensitivity largely depends on the coverage of the sequences under investigation, that is, the number of reads for specific DNA sections and the overlap extent between these reads. A greater number of reads for a specific region translates to higher sensitivity for that DNA segment [45]. During panel analysis, a reduced genome proportion under investigation leads to enhanced coverage and sensitivity. Thus, if a disease is believed to result from a mosaic genomic alteration, NGS panel analysis offers higher sensitivity compared to WES.
- -
- IFs and VUSs: The probability of identifying IFs and VUSs depends on the genome proportion both analyzed and queried. Analysis based on panels has a reduced association with IFs, since the investigated sequences are directly relevant to the clinical presentation of the proband. In general, a broader sequence analysis correlates with a higher number of IFs and VUSs. Notably, VUSs can also appear in targeted analyses of specific gene panels [45].
- -
- Data storage: The extent of the genome analyzed directly influences the volume of data generated. Consequently, suitable platforms for data storage are essential, particularly for analyses yielding substantial data, such as those conducted via WES and, more prominently, WGS [45].
2.1.2. Identification and Interpretation of Pathogenic Genetic Variants in Clinical Diagnostics
- Their allele frequency within the general population;
2.2. Variants of Uncertain Significance (VUS)
2.3. Classification, Implications, and Clinical Utility of Genetic Variants in Cardiovascular Disease
2.4. Diagnosis and Counseling
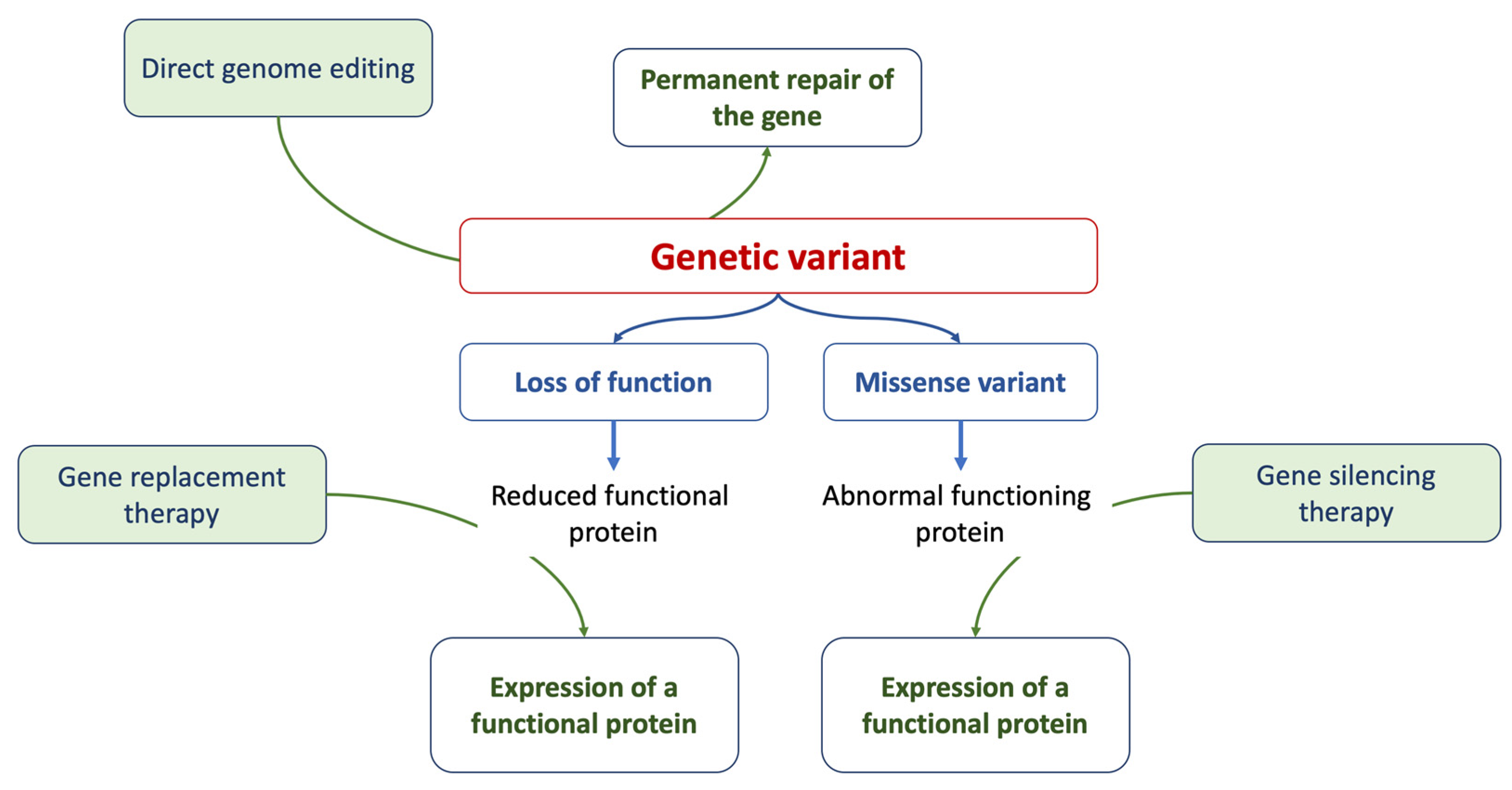
2.5. Gene Therapy
3. Genetics, Cardiomyopathies, and Heart Failure
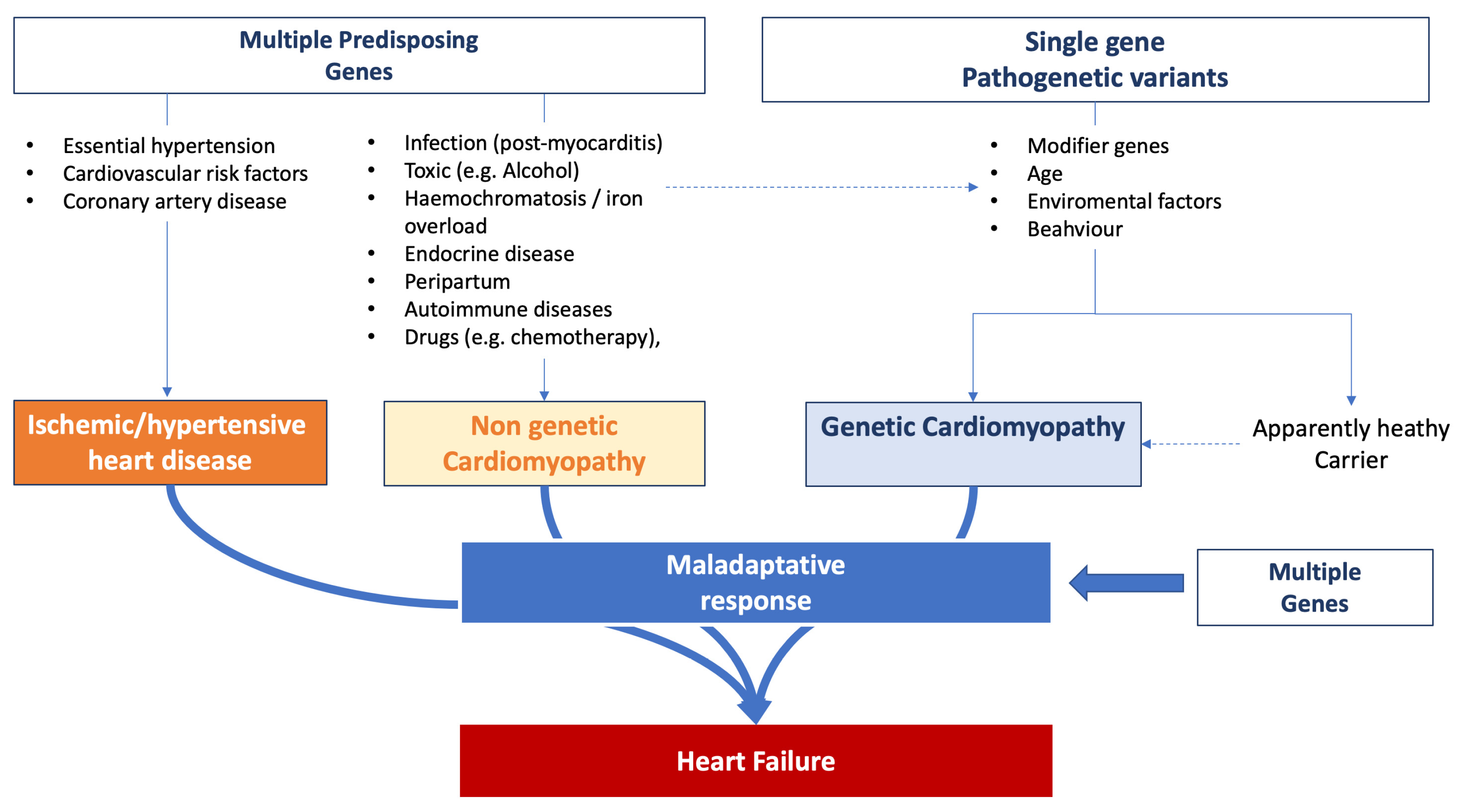
3.1. Genetic Factors Predisposing to Heart Failure
3.2. Genetics in Assessing the Risk of Heart Failure Progression
3.3. Arrhythmic Risk Stratification
3.4. Implications of Genetic Variants in Clinical Decisions
4. Heart Failure and Genetics: Beyond Cardiomyopathies
5. Current Guidelines and Recommendations
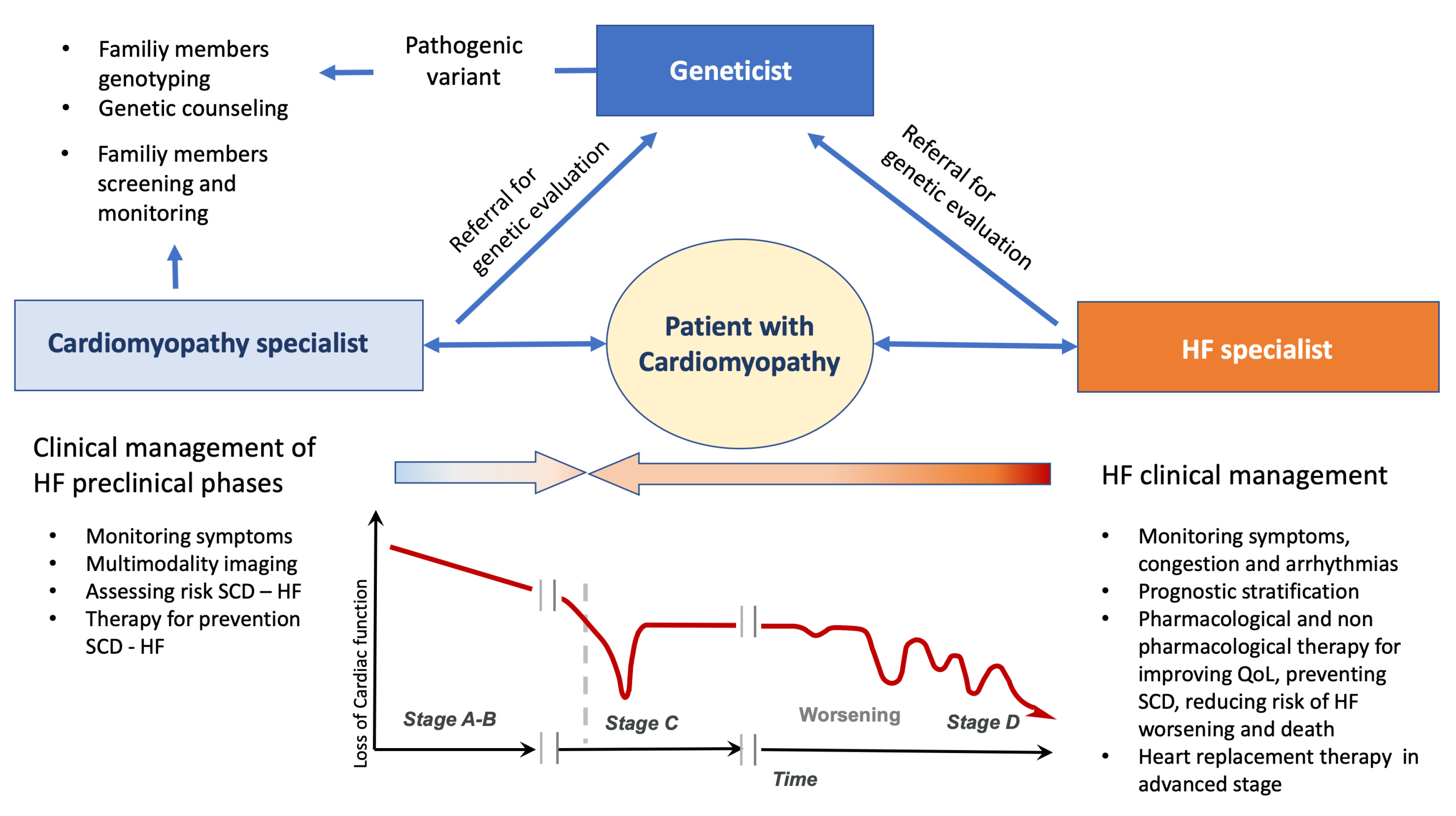
6. Genetics and Heart Failure in Clinical Practice
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shah, R.A.; Asatryan, B.; Sharaf Dabbagh, G.; Aung, N.; Khanji, M.Y.; Lopes, L.R.; Van Duijvenboden, S.; Holmes, A.; Muser, D.; Landstrom, A.P.; et al. Genotype-first approach I. Frequency, penetrance, and variable expressivity of dilated cardiomyopathy-associated putative pathogenic gene variants in UK Biobank participants. Circulation 2022, 146, 110–124. [Google Scholar] [CrossRef]
- De Marvao, A.; McGurk, K.A.; Zheng, S.L.; Thanaj, M.; Bai, W.; Duan, J.; Biffi, C.; Mazzarotto, F.; Statton, B.; Dawes, T.J.W.; et al. Phenotypic expression and outcomes in individuals with rare genetic variants of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 1097–1110. [Google Scholar] [CrossRef]
- McGurk, K.A.; Zheng, S.L.; Henry, A.; Josephs, K.; Edwards, M.; De Marvao, A.; Whiffin, N.; Roberts, A.; Lumbers, T.R.; O’Regan, D.P.; et al. Correspondence on “ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG)” by Miller et al. Genet. Med. 2022, 24, 744–746. [Google Scholar] [CrossRef]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.N.; et al. Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; Van Der Kooi, A.J.; Van Tintelen, J.P.; Van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal definition and classification of heart failure: A report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J. Card Fail. 2021, 27, 387–413. [Google Scholar]
- Waddell-Smith, K.E.; Donoghue, T.; Oates, S.; Graham, A.; Crawford, J.; Stiles, M.K.; Aitken, A.; Skinner, J.R. Inpatient detection of cardiac-inherited disease: The impact of improving family history taking. Open Heart 2016, 3, e000329. [Google Scholar] [CrossRef]
- Ingles, J.; Lind, J.M.; Phongsavan, P.; Semsarian, C. Psychosocial impact of specialized cardiac genetic clinics for hypertrophic cardiomyopathy. Genet. Med. 2008, 10, 117–120. [Google Scholar] [CrossRef]
- Reuter, C.; Grove, M.E.; Orland, K.; Spoonamore, K.; Caleshu, C. Clinical Cardiovascular Genetic Counselors Take a Leading Role in Team-based Variant Classification. J. Genet. Couns. 2018, 27, 751–760. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq Transatlantic Network of Excellence to Cure Phospholamban-Induced Cardiomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Grote Beverborg, N.; Später, D.; Knöl, R.; Hidalgo, A.; Yeh, S.T.; Elbeck, Z.; Silljé, H.H.W.; Eijgenraam, T.R.; Siga, H.; Zurek, M.; et al. Phospholamban antisense oligonucleotides improve cardiac function in murine cardiomyopathy. Nat. Commun. 2021, 12, 5180. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Motsinger-Reif, A.A.; Jorgenson, E.; Relling, M.V.; Kroetz, D.L.; Weinshilboum, R.; Cox, N.J.; Roden, D.M. Genome-wide association studies in pharmacogenomics: Successes and lessons. Pharmacogenet. Genom. 2013, 23, 383–394. [Google Scholar] [CrossRef]
- Huertas-Vazquez, A.; Nelson, C.P.; Sinsheimer, J.S.; Reinier, K.; Uy-Evanado, A.; Teodorescu, C.; Ayala, J.; Hall, A.S.; Gunson, K.; Jui, J.; et al. Cumulative effects of common genetic variants on risk of sudden cardiac death. Int. J. Cardiol. Heart Vasc. 2015, 7, 88–91. [Google Scholar] [CrossRef]
- Hajek, C.; Guo, X.; Yao, J.; Hai, Y.; Johnson, W.C.; Frazier-Wood, A.C.; Post, W.S.; Psaty, B.M.; Taylor, K.D.; Rotter, J.I. Coronary heart disease genetic risk score predicts cardiovascular disease risk in men, not women. Circ. Genom. Precis Med. 2018, 11, e002324. [Google Scholar] [CrossRef]
- Subas, T.; Luiten, R.; Hanson-Kahn, A.; Wheeler, M.; Caleshu, C. Evolving decisions: Perspectives of active and athletic individuals with inherited heart disease who exercise against recommendations. J. Genet. Couns. 2019, 28, 119–129. [Google Scholar] [CrossRef]
- Day, S.M. Exercise in hypertrophic cardiomyopathy. J. Cardiovasc. Transl. Res. 2009, 2, 407–414. [Google Scholar] [CrossRef]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef]
- Van Velzen, H.G.; Schinkel, A.F.L.; Baart, S.J.; Oldenburg, R.A.; Frohn-Mulder, I.M.E.; van Slegtenhorst, M.A.; Michels, M. Outcomes of contemporary family screening in hypertrophic cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001896. [Google Scholar] [CrossRef]
- Gimeno, J.R.; Lacunza, J.; García-Alberola, A.; Cerdán, M.C.; Oliva, M.J.; García-Molina, E.; López-Ruiz, M.; Castro, F.; González-Carrillo, J.; de la Morena, G.; et al. Penetrance and risk profile in inherited cardiac diseases studied in a dedicated screening clinic. Am. J. Cardiol. 2009, 104, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; McGaughran, J.; Scuffham, P.A.; Atherton, J.; Semsarian, C. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart 2012, 98, 625–630. [Google Scholar] [CrossRef]
- Stiles, M.K.; Wilde, A.A.M.; Abrams, D.J.; Ackerman, M.J.; Albert, C.M.; Behr, E.R.; Chugh, S.S.; Cornel, M.C.; Gardner, K.; Ingles, J.; et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm 2021, 18, e1–e50. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, C.; Olivotto, I. The importance of sex differences in patients with hypertrophic cardiomyopathy–Tailoring management and future perspectives. Am. J. Med. Sci. 2020, 360, 433–434. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, epidemiology, and global burden of cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef]
- Conrad, D.F.; Keebler, J.E.; DePristo, M.A.; Lindsay, S.J.; Zhang, Y.; Casals, F.; Idaghdour, Y.; Hartl, C.L.; Torroja, C.; Garimella, K.V.; et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011, 43, 712–714. [Google Scholar]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef]
- Visser, M.; Dooijes, D.; van der Smagt, J.J.; van der Heijden, J.F.; Doevendans, P.A.; Loh, P.; Asselbergs, F.W.; Hassink, R.J. Next-generation sequencing of a large gene panel in patients initially diagnosed with idiopathic ventricular fibrillation. Heart Rhythm 2017, 14, 1035–1040. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A prospective study of sudden cardiac death among children and young adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef]
- Michels, V.V.; Moll, P.P.; Miller, F.A.; Tajik, A.J.; Chu, J.S.; Driscoll, D.J.; Burnett, J.C.; Rodeheffer, R.J.; Chesebro, J.H.; Tazelaar, H.D. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1992, 326, 77–82. [Google Scholar] [CrossRef]
- Whiffin, N.; Karczewski, K.J.; Zhang, X.; Chothani, S.; Smith, M.J.; Evans, D.G.; Roberts, A.M.; Quaife, N.M.; Schafer, S.; Rackham, O.; et al. Characterizing the loss-of-function impact of 5’ untranslated region variants in 15,708 individuals. Nat. Commun. 2020, 11, 2523. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Crotti, L.; George, A.L., Jr. Modifier genes for sudden cardiac death. Eur. Heart J. 2018, 39, 3925–3931. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Hanchard, N.A.; Umana, L.A.; D’Alessandro, L.; Azamian, M.; Poopola, M.; Morris, S.A.; Fernbach, S.; Lalani, S.R.; Towbin, J.A.; Zender, G.A.; et al. Assessment of large copy number variants in patients with apparently isolated congenital left-sided cardiac lesions reveals clinically relevant genomic events. Am. J. Med. Genet. A 2017, 173, 2176–2188. [Google Scholar] [CrossRef] [PubMed]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Lieve, K.V.V.; Rohatgi, R.K.; Koca, F.; Tester, D.J.; van der Werf, C.; Martijn Bos, J.; Wilde, A.A.M.; Ackerman, M.J. Assessment and validation of a phenotype-enhanced variant classification framework to promote or demote RYR2 missense variants of uncertain significance. Circ. Genom. Precis. Med. 2019, 12, e002510. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Green, R.C. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014, 370, 2418–2425. [Google Scholar] [CrossRef]
- Seleman, M.; Hoyos-Bachiloglu, R.; Geha, R.S.; Chou, J. Uses of Next-Generation Sequencing Technologies for the Diagnosis of Primary Immunodeficiencies. Front. Immunol. 2017, 8, 847. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 15 September 2023).
- Available online: https://www.omim.org (accessed on 15 September 2023).
- Available online: http://genetics.bwh.harvard.edu/pph2/ (accessed on 15 September 2023).
- Available online: https://www.mutationtaster.org (accessed on 15 September 2023).
- Available online: https://www.jcvi.org/research/provean (accessed on 15 September 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Doh, C.Y.; Kampourakis, T.; Campbell, K.S.; Stelzer, J.E. Basic science methods for the characterization of variants of uncertain significance in hypertrophic cardiomyopathy. Front. Cardiovasc. Med. 2023, 10, 1238515. [Google Scholar] [CrossRef]
- Oulas, A.; Minadakis, G.; Zachariou, M.; Spyrou, G.M. Selecting variants of unknown significance through network-based gene-association significantly improves risk prediction for disease-control cohorts. Sci. Rep. 2019, 9, 3266. [Google Scholar] [CrossRef]
- Zhang, X.; Walsh, R.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Govind, R.; Li, N.; Ahmad, M.; Mazzarotto, F.; et al. Disease-specific variant pathogenicity prediction significantly improves variant interpretation in inherited cardiac conditions. Genet. Med. 2021, 23, 69–79. [Google Scholar] [CrossRef]
- Barbosa, P.; Ribeiro, M.; Carmo-Fonseca, M.; Fonseca, A. Clinical significance of genetic variation in hypertrophic cardiomyopathy: Comparison of computational tools to prioritize missense variants. Front. Cardiovasc. Med. 2022, 9, 975478. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.K.; Bartels, K.; Hathaway, J.; Burns, C.; Yeates, L.; Semsarian, C.; Krahn, A.D.; Virani, A.; Ingles, J. Perceptions of genetic variant reclassification in patients with inherited cardiac disease. Eur. J. Hum. Genet. 2019, 27, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Yeates, L.; O’Brien, L.; McGaughran, J.; Scuffham, P.A.; Atherton, J.; Semsarian, C. Genetic testing for inherited heart diseases: Longitudinal impact on health-related quality of life. Genet. Med 2012, 14, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Breithardt, G.; Howard, A.J.; Julian, D.G.; Rehnqvist Ahlberg, N. Task Force Report: The legal implications of medical guidelines—A Task Force of the European Society of Cardiology. Eur. Heart J. 1999, 20, 1152–1157. [Google Scholar] [CrossRef]
- Lambert, S.A.; Gil, L.; Jupp, S.; Ritchie, S.C.; Xu, Y.; Buniello, A.; McMahon, A.; Abraham, G.; Chapman, M.; Parkinson, H.; et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 2021, 53, 420–425. [Google Scholar] [CrossRef]
- Lahrouchi, N.; Tadros, R.; Crotti, L.; Mizusawa, Y.; Postema, P.G.; Beekman, L.; Walsh, R.; Hasegawa, K.; Barc, J.; Ernsting, M.; et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation 2020, 142, 324–338. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. HCMR Investigators. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef]
- Tadros, R.; Tan, H.L.; ESCAPE-NET Investigators; El Mathari, S.; Kors, J.A.; Postema, P.G.; Lahrouchi, N.; Beekman, L.; Radivojkov-Blagojevic, M.; Amin, A.S.; et al. Predicting cardiac electrical response to sodiumchannel blockade and Brugada syndrome using polygenic risk scores. Eur. Heart J. 2019, 40, 3097–3107. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Dotzler, S.M.; Tester, D.J.; Giudicessi, J.R.; Bos, J.M.; Speziale, A.D.; Vollenweider, J.M.; Ackerman, M.J. Corrected QT interval-polygenic risk score and its contribution to type 1, type 2, and type 3 long-QT syndrome in probands and genotype-positive family members. Circ. Genom. Precis. Med. 2020, 13, e002922. [Google Scholar] [CrossRef]
- Connolly, S.J.; Hallstrom, A.P.; Cappato, R.; Schron, E.B.; Kuck, K.H.; Zipes, D.P.; Greene, H.L.; Boczor, S.; Domanski, M.; Follmann, D.; et al. Meta-analysis of the implantable cardioverter defibrillator secondary prevention trials. Eur. Heart J. 2000, 21, 2071–2078. [Google Scholar] [CrossRef]
- Chivulescu, M.; Lie, Ø.H.; Popescu, B.A.; Skulstad, H.; Edvardsen, T.; Jurcut, R.O.; Haugaa, K.H. High penetrance and similar disease progression in probands and in family members with arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Cardim, N.; Freitas, A.; Brito, D. From hypertrophic cardiomyopathy centers to inherited cardiovascular disease centers in Europe. A small or a major step? A position paper from the Nucleus of the Working Group on Myocardial and Pericardial Diseases of the Portuguese Society of Cardiology. Rev. Port. Cardiol. 2011, 30, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Barriales-Villa, R.; Gimeno-Blanes, J.R.; Zorio-Grima, E.; Ripoll-Vera, T.; Evangelista-Masip, A.; Moya-Mitjans, A.; Serratosa-Fernández, L.; Albert-Brotons, D.C.; García-Pinilla, J.M.; García-Pavía, P. Plan of action for inherited cardiovascular diseases: Synthesis of recommendations and action algorithms. Rev. Esp. Cardiol. 2016, 69, 300–309. [Google Scholar] [CrossRef]
- Edwards, A.; Gray, J.; Clarke, A.; Dundon, J.; Elwyn, G.; Gaff, C.; Hood, K.; Iredale, R.; Sivell, S.; Shaw, C.; et al. Interventions to improve risk communication in clinical genetics: Systematic review. Patient Educ. Couns. 2008, 71, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Whyte, S.; Green, A.; McAllister, M.; Shipman, H. Family communication in inherited cardiovascular conditions in Ireland. J. Genet. Couns. 2016, 25, 1317–1326. [Google Scholar] [CrossRef]
- Daly, M.B.; Montgomery, S.; Bingler, R.; Ruth, K. Communicating genetic test results within the family: Is it lost in translation? A survey of relatives in the randomized six-step study. Fam. Cancer 2016, 15, 697–706. [Google Scholar] [CrossRef]
- Kaphingst, K.A.; Blanchard, M.; Milam, L.; Pokharel, M.; Elrick, A.; Goodman, M.S. Relationships between health literacy and genomics-related knowledge, self-efficacy, perceived importance, and communication in a medically underserved population. J. Health Commun. 2016, 21, 58–68. [Google Scholar] [CrossRef]
- de Boer, R.A.; Heymans, S.; Backs, J.; Carrier, L.; Coats, A.J.S.; Dimmeler, S.; Eschenhagen, T.; Filippatos, G.; Gepstein, L.; Hulot, J.S.; et al. Targeted therapies in genetic dilated and hypertrophic cardiomyopathies: From molecular mechanisms to therapeutic targets. A position paper from the Heart Failure Association (HFA) and the Working Group on Myocardial Function of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2022, 24, 406–420. [Google Scholar]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Fomin, A.; Gärtner, A.; Cyganek, L.; Tiburcy, M.; Tuleta, I.; Wellers, L.; Folsche, L.; Hobbach, A.J.; von Frieling-Salewsky, M.; Unger, A.; et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci. Transl. Med. 2021, 13, eabd3079. [Google Scholar] [CrossRef]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.; Casini, S.; Davis, R.P.; D’Aniello, C.; Haas, J.; Ward-van Oostwaard, D.; Tertoolen, L.G.; Jung, C.B.; Elliott, D.A.; Welling, A.; et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013, 32, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horváth, A.; Di Mauro, V.; Koivumäki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Krämer, E.; et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Moretti, A.; Fonteyne, L.; Giesert, F.; Hoppmann, P.; Meier, A.B.; Bozoglu, T.; Baehr, A.; Schneider, C.M.; Sinnecker, D.; Klett, K.; et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020, 26, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef]
- Yu, G.; Chakrabarti, S.; Tischenko, M.; Chen, A.L.; Wang, Z.; Cho, H.; French, B.A.; Naga Prasad, S.V.; Chen, Q.; Wang, Q.K. Gene therapy targeting protein trafficking regulator MOG1 in mouse models of Brugada syndrome, arrhythmias, and mild cardiomyopathy. Sci. Transl. Med. 2022, 14, eabf3136. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT05302271?cond=Cardiomyopathies&intr=gene%20therapy&rank=1 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT00494195?cond=Cardiomyopathies&intr=gene%20therapy&rank=4 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT05885412?cond=Cardiomyopathies&intr=gene%20therapy&page=2&rank=11 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT01344798?cond=Cardiomyopathies&intr=gene%20therapy&page=1&rank=7 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT02346422?cond=Cardiomyopathies&intr=gene%20therapy&page=1&rank=9 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT05445323?cond=Cardiomyopathies&intr=gene%20therapy&rank=3 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT01966887?cond=Cardiomyopathies&intr=gene%20therapy&rank=2 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT03439514?cond=Cardiomyopathies&intr=gene%20therapy&page=2&rank=12 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT05836259?cond=Cardiomyopathies&intr=gene%20therapy&page=2&rank=13 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT00279539?cond=Cardiomyopathies&intr=gene%20therapy&page=2&rank=20 (accessed on 15 September 2023).
- Available online: https://clinicaltrials.gov/study/NCT03882437?cond=Cardiomyopathies&intr=gene%20therapy&page=3&rank=21 (accessed on 15 September 2023).
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef]
- Hada, Y.; Sakamoto, T.; Amano, K.; Yamaguchi, T.; Takenaka, K.; Takahashi, H.; Takikawa, R.; Hasegawa, I.; Takahashi, T.; Suzuki, J. Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening. Am. J. Cardiol. 1987, 59, 183–184. [Google Scholar] [CrossRef]
- Maro, E.E.; Janabi, M.; Kaushik, R. Clinical and echocardiographic study of hypertrophic cardiomyopathy in Tanzania. Trop. Doct. 2006, 36, 225–227. [Google Scholar] [CrossRef]
- Anan, R.; Shono, H.; Kisanuki, A.; Arima, S.; Nakao, S.; Tanaka, H. Patients with familial hypertrophic cardiomyopathy caused by a Phe110Ile missense mutation in the cardiac troponin T gene have variable cardiac morphologies and a favorable prognosis. Circulation 1998, 98, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Appleton, C.P.; Gillebert, T.C.; Marino, P.N.; Oh, J.K.; Smiseth, O.A.; Waggoner, A.D.; Flachskampf, F.A.; Pellikka, P.A.; Evangelisa, A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. Eur. J. Echocardiogr. 2009, 10, 165–193. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Codd, M.B.; Sugrue, D.D.; Gersh, B.J.; Melton, L.J., III. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989, 80, 564–572. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Meyners, W. Prevalence of right ventricular dysplasiacardiomyopathy in a non-referral hospital. Int. J. Cardiol. 2004, 97, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Migliore, F.; Zorzi, A.; Michieli, P.; Marra Perazzolo, M.; Siciliano, M.; Rigato, I.; Bauce, B.; Basso, C.; Toazza, D.; Schiavon, M.; et al. Prevalence of cardiomyopathy in Italian asymptomatic children with electrocardiographic T-wave inversion at preparticipation screening. Circulation 2012, 125, 529–538. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the clinical genome resource framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.; Wiesfeld, A.C.; Jongbloed, J.D.; van den Wijngaard, A.; Kuks, J.B.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009, 6, 1574–1583. [Google Scholar] [CrossRef]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Pencina, M.J.; Benjamin, E.J.; Wang, T.J.; Levy, D.; O’Donnell, C.J.; Nam, B.H.; Larson, M.G.; D’Agostino, R.B.; Vasan, R.S. Association of parental heart failure with risk of heart failure in offspring. N. Engl. J. Med. 2006, 355, 138–147. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Narula, J. Cardiolaminopathies: Weighing in on the Concept of Genotype-First Screening. J. Am. Coll. Cardiol. 2022, 80, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules 2019, 9, 878. [Google Scholar] [CrossRef]
- Yousaf, M.; Khan, W.A.; Shahzad, K.; Khan, H.N.; Ali, B.; Hussain, M.; Awan, F.R.; Mustafa, H.; Sheikh, F.N. Genetic Association of Beta-Myosin Heavy-Chain Gene (MYH7) with Cardiac Dysfunction. Genes 2022, 13, 1554. [Google Scholar] [CrossRef]
- Tudurachi, B.S.; Zăvoi, A.; Leonte, A.; Țăpoi, L.; Ureche, C.; Bîrgoan, S.G.; Chiuariu, T.; Anghel, L.; Radu, R.; Sascău, R.A.; et al. An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 10510. [Google Scholar] [CrossRef]
- Velicki, L.; Jakovljevic, D.G.; Preveden, A.; Golubovic, M.; Bjelobrk, M.; Ilic, A.; Stojsic, S.; Barlocco, F.; Tefelmeier, M.; Okwose, N.; et al. Genetic determinants of clinical phenotype in hypertrophic cardiomyopathy. BMC Cardiovasc. Disord. 2020, 20, 516. [Google Scholar] [CrossRef]
- Herrera-Rodríguez, D.L.; Totomoch-Serra, A.; Rosas-Madrigal, S.; Luna-Limón, C.; Marroquín-Ramírez, D.; Carnevale, A.; Rosendo-Gutiérrez, R.; Villarreal-Molina, M.T.; Márquez-Murillo, M.F. Genes frequently associated with sudden death in primary hypertrophic cardiomyopathy. Arch. Cardiol. Mex. 2020, 90, 58–68. [Google Scholar] [CrossRef]
- Christensen, A.H.; Platonov, P.G.; Jensen, H.K.; Chivulescu, M.; Svensson, A.; Dahlberg, P.; Madsen, T.; Frederiksen, T.C.; Heliö, T.; Lie, Ø.H.; et al. Genotype-phenotype correlation in arrhythmogenic right ventricular cardiomyopathy-risk of arrhythmias and heart failure. J. Med. Genet. 2022, 59, 858–864. [Google Scholar] [CrossRef]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. A 2016, 170, 3241–3248. [Google Scholar] [CrossRef]
- Kaviarasan, V.; Mohammed, V.; Veerabathiran, R. Genetic predisposition study of heart failure and its association with cardiomyopathy. Egypt. Heart J. 2022, 74, 5. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Olivotto, I.; Maron, B.J.; Prasad, S.K.; Cecchi, F.; Udelson, J.E.; Camici, P.G. The case for myocardial ischemia in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Cahill, T.J.; Ashrafian, H.; Watkins, H. Genetic cardiomyopathies causing heart failure. Circ. Res. 2013, 113, 660–675. [Google Scholar] [CrossRef]
- Janjusevic, M.; Fluca, A.L.; Ferro, F.; Gagno, G.; D’Alessandra, Y.; Beltrami, A.P.; Sinagra, G.; Aleksova, A. Traditional and Emerging Biomarkers in Asymptomatic Left Ventricular Dysfunction-Promising Non-Coding RNAs and Exosomes as Biomarkers in Early Phases of Cardiac Damage. Int. J. Mol. Sci. 2021, 22, 4937. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef]
- Verstraelen, T.E.; van Lint, F.H.M.; Bosman, L.P.; de Brouwer, R.; Proost, M.V.; Abeln, B.G.S.; Taha, K.; Zwinderman, A.H.; Dickhoff, C.; Oomen, T.; et al. Prediction of ventricular arrhythmia in phospholamban p.Arg14del mutation carriersreaching the frontiers of individual risk prediction. Eur. Heart J. 2021, 42, 2842–2850. [Google Scholar] [CrossRef]
- Available online: https://lmna-risk-vta.fr (accessed on 15 September 2023).
- Mirelis, J.G.; Escobar-Lopez, L.; Ochoa, J.P.; Espinosa, M.Á.; Villacorta, E.; Navarro, M.; Casas, G.; Mora-Ayestarán, N.; Barriales-Villa, R.; Mogollón-Jiménez, M.V.; et al. Combination of late gadolinium enhancement and genotype improves prediction of prognosis in non-ischaemic dilated cardiomyopathy. Eur. J. Heart Fail. 2022, 24, 1183–1196. [Google Scholar]
- Ader, F.; De Groote, P.; Réant, P.; Rooryck-Thambo, C.; Dupin-Deguine, D.; Rambaud, C.; Khraiche, D.; Perret, C.; Pruny, J.F.; Mathieu-Dramard, M.; et al. FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations. Clin. Genet. 2019, 96, 317–329. [Google Scholar] [CrossRef]
- de Frutos, F.; Ochoa, J.; Navarro-Peñalver, M.; Peñalver, M.; Baas, A.; Bjerre, J.V.; Zorio, E.; Méndez, I.; Lorca, R.; Verdonschot, J.A.J.; et al. Natural History of MYH7-Related Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 80, 1447–1461. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, J.R.; Tomé-Esteban, M.; Lofiego, C.; Hurtado, J.; Pantazis, A.; Mist, B.; Lambiase, P.; McKenna, W.J.; Elliott, P.M. Exercise-induced ventricular arrhythmias and risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2009, 30, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://qxmd.com/calculate/calculator_303/hcm-risk-scd (accessed on 15 September 2023).
- Chan, R.H.; Maron, B.J.; Olivotto, I.; Pencina, M.J.; Azzenza, G.E.; Haas, T.; Lesser, J.R.; Gruner, C.; Crean, A.M.; Rakowski, H.; et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation 2014, 130, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.; Murray, B.; te Riele, A.S.; van den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/ cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, S.E.; Cho, M.C. Clinical Implication of Genetic Testing in Dilated Cardiomyopathy. Int. J. Heart Fail. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Bondue, A.; Arbustini, E.; Bianco, A.; Ciccarelli, M.; Dawson, D.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef]
- Escobar-Lopez, L.; Ochoa, J.P.; Mirelis, J.G.; Espinosa, M.Á.; Navarro, M.; Gallego-Delgado, M.; Barriales-Villa, R.; Robles-Mezcua, A.; Basurte-Elorz, M.T.; Gutiérrez García-Moreno, L.; et al. Association of Genetic Variants with Outcomes in patients with nonischemic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 1682–1699. [Google Scholar] [CrossRef]
- Kamiya, C.A.; Yoshimatsu, J.; Ikeda, T. Peripartum Cardiomyopathy From a Genetic Perspective. Circ. J. 2016, 80, 1684–1688. [Google Scholar] [CrossRef]
- Cooper, L.T., Jr.; Čiháková, D. Do Genes Influence Susceptibility to Myocarditis? JACC Basic. Transl. Sci. 2021, 6, 593–594. [Google Scholar] [CrossRef]
- Fox, C.S.; Evans, J.C.; Larson, M.G.; Kannel, W.B.; Levy, D. Temporal trends in coronary heart disease mortality and sudden cardiac death from 1950 to 1999: The Framingham Heart Study. Circulation 2004, 110, 522–527. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Wilson, P.W.F. Framingham risk score and prediction of lifetime risk for coronary heart disease. Am. J. Cardiol. 2004, 94, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Skrzynia, C.; Berg, J.S.; Willis, M.S.; Jensen, B.C. Genetics and heart failure: A concise guide for the clinician. Curr. Cardiol. Rev. 2015, 11, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Czepluch, F.S.; Wollnik, B.; Hasenfuß, G. Genetic determinants of heart failure: Facts and numbers. ESC Heart Fail. 2018, 5, 211–217. [Google Scholar] [CrossRef]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Bordet, C.; Brice, S.; Maupain, C.; Gandjbakhch, E.; Isidor, B.; Palmyre, A.; Moerman, A.; Toutain, A.; Akloul, L.; Brehin, A.C.; et al. Psychosocial impact of predictive genetic testing in hereditary heart diseases: The PREDICT study. J. Clin. Med. 2020, 9, 1365. [Google Scholar] [CrossRef]
- Michie, S.; Marteau, T.M.; Bobrow, M. Genetic counselling: The psychological impact of meeting patients’ expectations. J. Med. Genet. 1997, 34, 237–241. [Google Scholar] [CrossRef]
- Charron, P.; Arad, M.; Arbustini, E.; Basso, C.; Bilinska, Z.; Elliott, P.; Helio, T.; Keren, A.; McKenna, W.J.; Monserrat, L.; et al. Genetic counselling and testing in cardiomyopathies: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2010, 31, 2715–2726. [Google Scholar] [CrossRef]
- Borry, P.; Evers-Kiebooms, G.; Cornel, M.C.; Clarke, A.; Dierickx, K. Public Professional Policy Committee (PPPC) of the European Society of Human Genetics (ESHG). Genetic testing in asymptomatic minors: Background considerations towards ESHG recommendations. Eur. J. Hum. Genet. 2009, 17, 711–719. [Google Scholar] [CrossRef]
- Marey, I.; Fressart, V.; Rambaud, C.; Fornes, P.; Martin, L.; Grotto, S.; Alembik, Y.; Gorka, H.; Millat, G.; Gandjbakhch, E.; et al. Clinical impact of post-mortem genetic testing in cardiac death and cardiomyopathy. Open Med. 2020, 15, 435–446. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Technique | Characteristic | Applications | Advantage | Disadvantages |
|---|---|---|---|---|
| WES | Sequencing of the entire exonic genome portion |
|
|
|
| WGS | Sequencing of the entire genome, both exonic and intronic |
|
|
|
| TGP | Panel of known genes that can be sequenced simultaneously Two types: |
|
|
|
| Targeted Data Analysis Panel constructed with genes known to be associated with a specific disease following a WES |
|
| ||
| Targeted ResequencingEnrichment of specific genomic regions before sequencing |
|
|
| Technique | Setting | Treatment | Gene | Phase Study | Ongoing Study ID |
|---|---|---|---|---|---|
| AAV | Friedreich’s ataxia DCM | AAVrh.10hFXN | Frataxin (FXN) | I | NCT05302271 [84] |
| AAV serotype 1 | LGMD2D | rAAV1.tMCK.hαSG | Alpha-sarcoglycan (haSG) | I | NCT00494195 [85] |
| AAV | ARVC | RP-A601 | Plakophilin-2a (PKP2a) | I | NCT05885412 [86] |
| AAV serotype 1 | Limb girdle muscular dystrophy type 2C | Gamma-sarcoglycan vector injection | SGCG | I | NCT01344798 [87] |
| AAV | Adv-HF | MYDICAR | SERCA2a | I/II | NCT02346422 [88] |
| AAV serotype 1 | CHF | AAV1-CMV-SERCA2a | SERCA2a | II | NCT01966887 [89] |
| AAV | Friedreich’s ataxia with evidence of cardiomyopathy | LX2006 (AAVrh.10hFXN) | hFXN | I/II | NCT05445323 [90] |
| Drug PF-07265803 | REALM-DCM | ARRY-371797 | Lamin A/C | III | NCT03439514 [91] |
| AAV | MYBPC3 nHCM | TN-201 | MYBPC3 | I | NCT05836259 [92] |
| Genetic (VEGF1) | CHF | VEGF1 | phVEGF165 | I | NCT00279539 [93] |
| AAV serotype 9 | Danon Disease | RP-A501 | LAMP2B | I | NCT03882437 [94] |
| Cardiomyopathy | Gene (Protein) | Arrhythmic Risk | Heart Failure Risk |
|---|---|---|---|
| DCM | LMNA (Lamin A/C) | X [110] | |
| TTN (Titin) truncating mutations | X [106,134] | ||
| RBM20 (RNA-binding motif protein 20) | X [134] | X [134] | |
| PLN (Phospholamban) | X [135] | ||
| FLNC (Filamin C) | X [135] | ||
| TMEM43 (Transmembrane protein 43) | X [106] | ||
| EMD (Emerin) | X [136] | ||
| DSG2 (Desmoglein 2) | X [106] | ||
| DSP (Desmoplakin) | X [106] | ||
| DES (Desmin) | X [106] | ||
| MYH7 (Beta-myosin heavy chain) | X [129] | ||
| HCM | MYBPC3 (Cardiac myosin binding protein C) | X [106] | X [113] |
| MYH7 (Beta-myosin heavy chain) | X [106] | X [129] | |
| RCM | TNNT2 (Troponin T2) | X [106] | |
| TNNI3 (Troponin T3) | X [108] | ||
| ARVC | DSC2 (Desmocollin 2) | X [116] | |
| DSG2 (Desmoglein 2) | X [116] | ||
| DSP (Desmoplakin) | X [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, G.; D’Ambrosio, M.F.; Correale, M.; Brunetti, N.D.; Santacroce, R.; Iacoviello, M.; Margaglione, M. The Role of Genetics in the Management of Heart Failure Patients. Int. J. Mol. Sci. 2023, 24, 15221. https://doi.org/10.3390/ijms242015221
Palmieri G, D’Ambrosio MF, Correale M, Brunetti ND, Santacroce R, Iacoviello M, Margaglione M. The Role of Genetics in the Management of Heart Failure Patients. International Journal of Molecular Sciences. 2023; 24(20):15221. https://doi.org/10.3390/ijms242015221
Chicago/Turabian StylePalmieri, Gianpaolo, Maria Francesca D’Ambrosio, Michele Correale, Natale Daniele Brunetti, Rosa Santacroce, Massimo Iacoviello, and Maurizio Margaglione. 2023. "The Role of Genetics in the Management of Heart Failure Patients" International Journal of Molecular Sciences 24, no. 20: 15221. https://doi.org/10.3390/ijms242015221
APA StylePalmieri, G., D’Ambrosio, M. F., Correale, M., Brunetti, N. D., Santacroce, R., Iacoviello, M., & Margaglione, M. (2023). The Role of Genetics in the Management of Heart Failure Patients. International Journal of Molecular Sciences, 24(20), 15221. https://doi.org/10.3390/ijms242015221