

New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death

Abstract
1. Introduction
2. Vascular Aging and EC Senescence in Atherosclerosis
2.1. What Is Vascular Aging?
2.1.1. Vascular Aging
2.1.2. Morphological, Biochemical, Mechanical, and Metabolic Features of Senescent ECs
2.1.3. Cellular Senescence in Atherosclerosis
2.2. Mechanism of Endothelial Cell Senescence
2.2.1. Oxidative Stress
2.2.2. Inflammation
2.2.3. Genomic Instability
2.2.4. Metabolite-Sensing Longevity Pathways
Sirtuins
AMPK
mTOR
Klotho and FGF21
2.2.5. Activated Renin-Angiotensin System (RAS)
2.2.6. Uric Acid
2.2.7. Polyploidization
2.3. Senolysis—Therapeutic Targeting of Senescent ECs
2.3.1. Pharmacological Senolytic Approaches
2.3.2. Genetical Senolytic Approaches
2.3.3. Challenges of Senolysis
3. Vascular EC Death in Atherosclerosis
3.1. EC Death and Atherosclerosis
3.2. Autophagy—Protective and Detrimental
3.3. Apoptosis
3.4. Necroptosis
3.5. Pyroptosis
3.6. Ferroptosis
3.7. NETosis
3.8. Complexity and Limitations of Targeting EC Death in Atherosclerosis
4. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar] [CrossRef] [PubMed]
- Lesniewski, L.A.; Connell, M.L.; Durrant, J.R.; Folian, B.J.; Anderson, M.C.; Donato, A.J.; Seals, D.R. B6D2F1 Mice are a suitable model of oxidative stress-mediated impaired endothelium-dependent dilation with aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2009, 64, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takeshita, K.; Shimokawa, T.; Yi, H.; Isobe, K.; Loskutoff, D.J.; Saito, H. Plasminogen activator inhibitor-1 is a major stress-regulated gene: Implications for stress-induced thrombosis in aged individuals. Proc. Natl. Acad. Sci. USA 2002, 99, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Pelegrí, C.; Canudas, A.M.; del Valle, J.; Casadesus, G.; Smith, M.A.; Camins, A.; Pallàs, M.; Vilaplana, J. Increased permeability of blood-brain barrier on the hippocampus of a murine model of senescence. Mech. Ageing Dev. 2007, 128, 522–528. [Google Scholar] [CrossRef]
- Zhuo, Y.; Li, S.H.; Chen, M.S.; Wu, J.; Kinkaid, H.Y.; Fazel, S.; Weisel, R.D.; Li, R.K. Aging impairs the angiogenic response to ischemic injury and the activity of implanted cells: Combined consequences for cell therapy in older recipients. J. Thorac. Cardiovasc. Surg. 2010, 139, 1286–1294.e2. [Google Scholar] [CrossRef]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef]
- Hohensinner, P.J.; Kaun, C.; Buchberger, E.; Ebenbauer, B.; Demyanets, S.; Huk, I.; Eppel, W.; Maurer, G.; Huber, K.; Wojta, J. Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim. Et Biophys. Acta 2016, 1863, 360–367. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; DeMarco, V.G.; Martinez-Lemus, L.A.; Meininger, G.A.; Sowers, J.R. Vascular stiffness in insulin resistance and obesity. Front. Physiol. 2015, 6, 231. [Google Scholar] [CrossRef]
- Martinet, W.; Schrijvers, D.M.; De Meyer, G.R. Pharmacological modulation of cell death in atherosclerosis: A promising approach towards plaque stabilization? Br. J. Pharmacol. 2011, 164, 1–13. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell. Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [PubMed]
- Nishio, K.; Inoue, A. Senescence-associated alterations of cytoskeleton: Extraordinary production of vimentin that anchors cytoplasmic p53 in senescent human fibroblasts. Histochem. Cell Biol. 2005, 123, 263–273. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: Terminology for TOR-driven aging. Aging 2012, 4, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Brauer, E.; Lange, T.; Keller, D.; Görlitz, S.; Cho, S.; Keye, J.; Gossen, M.; Petersen, A.; Kornak, U. Dissecting the influence of cellular senescence on cell mechanics and extracellular matrix formation in vitro. Aging Cell 2023, 22, e13744. [Google Scholar] [CrossRef]
- Grandy, C.; Port, F.; Radzinski, M.; Singh, K.; Erz, D.; Pfeil, J.; Reichmann, D.; Gottschalk, K.E. Remodeling of the focal adhesion complex by hydrogen-peroxide-induced senescence. Sci. Rep. 2023, 13, 9735. [Google Scholar] [CrossRef]
- Shin, E.Y.; Park, J.H.; You, S.T.; Lee, C.S.; Won, S.Y.; Park, J.J.; Kim, H.B.; Shim, J.; Soung, N.K.; Lee, O.J.; et al. Integrin-mediated adhesions in regulation of cellular senescence. Sci. Adv. 2020, 6, eaay3909. [Google Scholar] [CrossRef]
- Phillip, J.M.; Aifuwa, I.; Walston, J.; Wirtz, D. The Mechanobiology of Aging. Annu. Rev. Biomed. Eng. 2015, 17, 113–141. [Google Scholar] [CrossRef]
- Chala, N.; Moimas, S.; Giampietro, C.; Zhang, X.; Zambelli, T.; Exarchos, V.; Nazari-Shafti, T.Z.; Poulikakos, D.; Ferrari, A. Mechanical Fingerprint of Senescence in Endothelial Cells. Nano Lett. 2021, 21, 4911–4920. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T. Metabolic regulation of endothelial senescence. Front. Cardiovasc. Med. 2023, 10, 1232681. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef]
- Unterluggauer, H.; Mazurek, S.; Lener, B.; Hütter, E.; Eigenbrodt, E.; Zwerschke, W.; Jansen-Dürr, P. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology 2008, 9, 247–259. [Google Scholar] [CrossRef]
- Peyton, K.J.; Liu, X.M.; Yu, Y.; Yates, B.; Behnammanesh, G.; Durante, W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 2018, 156, 204–214. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Culley, M.K.; Chan, S.Y. Endothelial Senescence: A New Age in Pulmonary Hypertension. Circ. Res. 2022, 130, 928–941. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Passos, J.F.; Kirkwood, T.B.L. On the evolution of cellular senescence. Aging Cell 2020, 19, e13270. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef]
- Idda, M.L.; McClusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent cell markers with age in human tissues. Aging 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Herrmann, J.; Babic, M.; Tölle, M.; Eckardt, K.U.; van der Giet, M.; Schuchardt, M. A Novel Protocol for Detection of Senescence and Calcification Markers by Fluorescence Microscopy. Int. J. Mol. Sci. 2020, 21, 3475. [Google Scholar] [CrossRef]
- Evangelou, K.; Gorgoulis, V.G. Sudan Black B, The Specific Histochemical Stain for Lipofuscin: A Novel Method to Detect Senescent Cells. Methods Mol. Biol. 2017, 1534, 111–119. [Google Scholar]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Couchie, D.; Vaisman, B.; Abderrazak, A.; Mahmood, D.F.D.; Hamza, M.M.; Canesi, F.; Diderot, V.; El Hadri, K.; Nègre-Salvayre, A.; Le Page, A.; et al. Human Plasma Thioredoxin-80 Increases With Age and in Apoe(-/-) Mice Induces Inflammation, Angiogenesis, and Atherosclerosis. Circulation 2017, 136, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Gao, H.; Yang, R.; Guo, K.; Miao, D. Pyrroloquinoline Quinone Prevents Estrogen Deficiency-Induced Osteoporosis by Inhibiting Oxidative Stress and Osteocyte Senescence. Int. J. Biol. Sci. 2019, 15, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Viehmann, M.; Adams, V.; Rabald, K.; Mangner, N.; Höllriegel, R.; Lurz, P.; Erbs, S.; Linke, A.; Kirsch, K.; et al. Chronic heart failure and aging—Effects of exercise training on endothelial function and mechanisms of endothelial regeneration: Results from the Leipzig Exercise Intervention in Chronic heart failure and Aging (LEICA) study. Eur. J. Prev. Cardiol. 2016, 23, 349–358. [Google Scholar] [CrossRef]
- Mwasongwe, S.; Gao, Y.; Griswold, M.; Wilson, J.G.; Aviv, A.; Reiner, A.P.; Raffield, L.M. Leukocyte telomere length and cardiovascular disease in African Americans: The Jackson Heart Study. Atherosclerosis 2017, 266, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Tranah, G.J.; Katzman, S.M.; Lauterjung, K.; Yaffe, K.; Manini, T.M.; Kritchevsky, S.; Newman, A.B.; Harris, T.B.; Cummings, S.R. Mitochondrial DNA m.3243A > G heteroplasmy affects multiple aging phenotypes and risk of mortality. Sci. Rep. 2018, 8, 11887. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- O’Neill, E.; Mosley, M.; Cornelissen, B. Imaging DNA damage response by γH2AX in vivo predicts treatment response to Lutetium-177 radioligand therapy and suggests senescence as a therapeutically desirable outcome. Theranostics 2023, 13, 1302–1310. [Google Scholar] [CrossRef]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Krouwer, V.J.; Hekking, L.H.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12. [Google Scholar] [CrossRef]
- Libby, P. Seeing and Sampling the Surface of the Atherosclerotic Plaque: Red or White Can Make Blue. Arter. Thromb. Vasc. Biol. 2016, 36, 2275–2277. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, R.; Yu, M.; Sausen, G.; Bichsel, C.A.; Corbo, C.; Folco, E.J.; Lee, G.Y.; Liu, Y.; Tesmenitsky, Y.; Shvartz, E.; et al. Targeted delivery of protein arginine deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc. Res. 2021, 117, 2652–2663. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arter. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Susser, L.I.; Rayner, K.J. Through the layers: How macrophages drive atherosclerosis across the vessel wall. J. Clin. Investig. 2022, 132, e157011. [Google Scholar] [CrossRef]
- Eshghjoo, S.; Kim, D.M.; Jayaraman, A.; Sun, Y.; Alaniz, R.C. Macrophage Polarization in Atherosclerosis. Genes 2022, 13, 756. [Google Scholar] [CrossRef]
- Vellasamy, D.M.; Lee, S.J.; Goh, K.W.; Goh, B.H.; Tang, Y.Q.; Ming, L.C.; Yap, W.H. Targeting Immune Senescence in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 13059. [Google Scholar] [CrossRef]
- Adachi, Y.; Ueda, K.; Takimoto, E. Perivascular adipose tissue in vascular pathologies-a novel therapeutic target for atherosclerotic disease? Front. Cardiovasc. Med. 2023, 10, 1151717. [Google Scholar] [CrossRef]
- Takaoka, M.; Nagata, D.; Kihara, S.; Shimomura, I.; Kimura, Y.; Tabata, Y.; Saito, Y.; Nagai, R.; Sata, M. Periadventitial adipose tissue plays a critical role in vascular remodeling. Circ. Res. 2009, 105, 906–911. [Google Scholar] [CrossRef]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef]
- Lüscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20 (Suppl. 2), Ii-3–Ii-10. [Google Scholar] [CrossRef]
- Novella, S.; Dantas, A.P.; Segarra, G.; Novensa, L.; Heras, M.; Hermenegildo, C.; Medina, P. Aging enhances contraction to thromboxane A2 in aorta from female senescence-accelerated mice. Age 2013, 35, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Westby, C.M.; Weil, B.R.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Endothelin-1 vasoconstriction and the age-related decline in endothelium-dependent vasodilatation in men. Clin. Sci. 2011, 120, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.E.; Kim, E.N.; Kim, M.Y.; Lim, J.H.; Jang, I.A.; Ban, T.H.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Choi, B.S. Age-Associated Changes in the Vascular Renin-Angiotensin System in Mice. Oxidative Med. Cell. Longev. 2016, 2016, 6731093. [Google Scholar] [CrossRef]
- Csiszar, A.; Wang, M.; Lakatta, E.G.; Ungvari, Z. Inflammation and endothelial dysfunction during aging: Role of NF-kappaB. J. Appl. Physiol. 2008, 105, 1333–1341. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Szarka, A.; Bánhegyi, G.; Sümegi, B. Mitochondria, oxidative stress and aging. Orvosi Hetil. 2014, 155, 447–452. [Google Scholar] [CrossRef]
- Lee, H.Y.; Zeeshan, H.M.A.; Kim, H.R.; Chae, H.J. Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic. Biol. Med. 2017, 113, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Lener, B.; Kozieł, R.; Pircher, H.; Hütter, E.; Greussing, R.; Herndler-Brandstetter, D.; Hermann, M.; Unterluggauer, H.; Jansen-Dürr, P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem. J. 2009, 423, 363–374. [Google Scholar] [CrossRef]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. CB 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Wolin, M.S.; Gupte, S.A.; Neo, B.H.; Gao, Q.; Ahmad, M. Oxidant-redox regulation of pulmonary vascular responses to hypoxia and nitric oxide-cGMP signaling. Cardiol. Rev. 2010, 18, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Park, H.C.; Patschan, S.; Brodsky, S.V.; Gealikman, O.; Kuo, M.C.; Li, H.; Addabbo, F.; Zhang, F.; Nasjletti, A.; et al. Premature vascular senescence in metabolic syndrome: Could it be prevented and reversed by a selenorganic antioxidant and peroxynitrite scavenger ebselen? Drug Discov. Today Ther. Strateg. 2007, 4, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Straface, E.; Pietraforte, D.; Gambardella, L.; Vona, R.; Maccaglia, A.; Minetti, M.; Malorni, W. Peroxynitrite induces senescence and apoptosis of red blood cells through the activation of aspartyl and cysteinyl proteases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 416–418. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef]
- Csiszar, A.; Ungvari, Z.; Edwards, J.G.; Kaminski, P.; Wolin, M.S.; Koller, A.; Kaley, G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 2002, 90, 1159–1166. [Google Scholar] [CrossRef]
- Cernadas, M.R.; Sánchez de Miguel, L.; García-Durán, M.; González-Fernández, F.; Millás, I.; Montón, M.; Rodrigo, J.; Rico, L.; Fernández, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286. [Google Scholar] [CrossRef]
- Cau, S.B.; Carneiro, F.S.; Tostes, R.C. Differential modulation of nitric oxide synthases in aging: Therapeutic opportunities. Front. Physiol. 2012, 3, 218. [Google Scholar] [CrossRef]
- Ungvari, Z.; Csiszar, A.; Kaley, G. Vascular inflammation in aging. Herz 2004, 29, 733–740. [Google Scholar] [CrossRef]
- Chou, T.C.; Yen, M.H.; Li, C.Y.; Ding, Y.A. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension 1998, 31, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Bortolotti, M.; Bolognesi, A.; Polito, L. Pro-Aging Effects of Xanthine Oxidoreductase Products. Antioxidants 2020, 9, 839. [Google Scholar] [CrossRef] [PubMed]
- Jankov, R.P.; Kantores, C.; Pan, J.; Belik, J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L233–L245. [Google Scholar] [CrossRef] [PubMed]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef]
- Shiina, K.; Tomiyama, H.; Tanaka, A.; Yoshida, H.; Eguchi, K.; Kario, K.; Kato, T.; Teragawa, H.; Toyoda, S.; Ohishi, M.; et al. Differential effect of a xanthine oxidase inhibitor on arterial stiffness and carotid atherosclerosis: A subanalysis of the PRIZE study. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2022, 45, 602–611. [Google Scholar] [CrossRef]
- Ameziane El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr. Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef]
- Bencivenga, L.; De Souto Barreto, P.; Rolland, Y.; Hanon, O.; Vidal, J.S.; Cestac, P.; Vellas, B.; Rouch, L. Blood pressure variability: A potential marker of aging. Ageing Res. Rev. 2022, 80, 101677. [Google Scholar] [CrossRef]
- Nagane, M.; Yasui, H.; Kuppusamy, P.; Yamashita, T.; Inanami, O. DNA damage response in vascular endothelial senescence: Implication for radiation-induced cardiovascular diseases. J. Radiat. Res. 2021, 62, 564–573. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Et Biophys. Acta. Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Xie, T.; Xu, Y.; Ji, L.; Sui, X.; Zhang, A.; Zhang, Y.; Chen, J. Heme Oxygenase 1/Peroxisome Proliferator-Activated Receptor Gamma Pathway Protects Intimal Hyperplasia and Mitigates Arteriovenous Fistula Dysfunction by Regulating Oxidative Stress and Inflammatory Response. Cardiovasc. Ther. 2022, 2022, 7576388. [Google Scholar] [CrossRef]
- Zhu, S.L.; Wang, M.L.; He, Y.T.; Guo, S.W.; Li, T.T.; Peng, W.J.; Luo, D. Capsaicin ameliorates intermittent high glucose-mediated endothelial senescence via the TRPV1/SIRT1 pathway. Phytomed. Int. J. Phytother. Phytopharm. 2022, 100, 154081. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Li, Q.; Kou, X.; Qin, X.; Li, Z.; Li, J.; Chen, C. BMP-4 impedes endothelial cell migration in neointimal hyperplasia via FoXO-3 specific modulation of reactive oxygen species. Atherosclerosis 2022, 351, 9–17. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.M.; Li, Y.; Kinzenbaw, D.A.; Sigmund, C.D.; Faraci, F.M. Endothelial PPARγ (Peroxisome Proliferator-Activated Receptor-γ) Is Essential for Preventing Endothelial Dysfunction with Aging. Hypertension 2018, 72, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Chen, Z.Y.; Xia, J.B.; Zheng, L.; Zhao, H.; Pi, L.Q.; Park, K.S.; Kim, S.K.; Lee, K.J.; Cai, D.Q. FoxO3a suppresses the senescence of cardiac microvascular endothelial cells by regulating the ROS-mediated cell cycle. J. Mol. Cell. Cardiol. 2015, 81, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Nyúl-Tóth, Á.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. GeroScience 2019, 41, 727–738. [Google Scholar] [CrossRef]
- Bresciani, G.; da Cruz, I.B.; González-Gallego, J. Manganese superoxide dismutase and oxidative stress modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar]
- Gebicka, L.; Krych-Madej, J. The role of catalases in the prevention/promotion of oxidative stress. J. Inorg. Biochem. 2019, 197, 110699. [Google Scholar] [CrossRef]
- Chu, Y.; Lan, R.S.; Huang, R.; Feng, H.; Kumar, R.; Dayal, S.; Chan, K.S.; Dai, D.F. Glutathione peroxidase-1 overexpression reduces oxidative stress, and improves pathology and proteome remodeling in the kidneys of old mice. Aging Cell 2020, 19, e13154. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef]
- Chung, H.Y.; Sung, B.; Jung, K.J.; Zou, Y.; Yu, B.P. The molecular inflammatory process in aging. Antioxid. Redox Signal. 2006, 8, 572–581. [Google Scholar] [CrossRef]
- Scuteri, A.; Orru, M.; Morrell, C.; Piras, M.G.; Taub, D.; Schlessinger, D.; Uda, M.; Lakatta, E.G. Independent and additive effects of cytokine patterns and the metabolic syndrome on arterial aging in the SardiNIA Study. Atherosclerosis 2011, 215, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Miles, E.A.; Rees, D.; Banerjee, T.; Cazzola, R.; Lewis, S.; Wood, R.; Oates, R.; Tallant, A.; Cestaro, B.; Yaqoob, P.; et al. Age-related increases in circulating inflammatory markers in men are independent of BMI, blood pressure and blood lipid concentrations. Atherosclerosis 2008, 196, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Walker, A.E.; Kaplon, R.E.; Pierce, G.L.; Nowlan, M.J.; Seals, D.R. Prevention of age-related endothelial dysfunction by habitual aerobic exercise in healthy humans: Possible role of nuclear factor κB. Clin. Sci. 2014, 127, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Lesniewski, L.A.; Durrant, J.R.; Connell, M.L.; Folian, B.J.; Donato, A.J.; Seals, D.R. Salicylate treatment improves age-associated vascular endothelial dysfunction: Potential role of nuclear factor kappaB and forkhead Box O phosphorylation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Lechel, A.; Satyanarayana, A.; Ju, Z.; Plentz, R.R.; Schaetzlein, S.; Rudolph, C.; Wilkens, L.; Wiemann, S.U.; Saretzki, G.; Malek, N.P.; et al. The cellular level of telomere dysfunction determines induction of senescence or apoptosis in vivo. EMBO Rep. 2005, 6, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Ikeda, K.; Urata, R.; Yamazaki, E.; Emoto, N.; Matoba, S. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci. Rep. 2021, 11, 14608. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Harrington, A.; Yang, X.; Friesel, R.E.; Liaw, L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 2010, 48, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.I.; Liu, Y.; Tucker, J.R.; Islam, M.T.; Machin, D.R.; Abdeahad, H.; Thomas, T.G.; Bramwell, R.C.; Lesniewski, L.A.; Donato, A.J. Endothelial cell telomere dysfunction induces senescence and results in vascular and metabolic impairments. Aging Cell 2023, 22, e13875. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Henson, G.D.; Morgan, R.G.; Enz, R.A.; Walker, A.E.; Lesniewski, L.A. TNF-α impairs endothelial function in adipose tissue resistance arteries of mice with diet-induced obesity. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H672–H679. [Google Scholar] [CrossRef][Green Version]
- Csiszar, A.; Labinskyy, N.; Smith, K.; Rivera, A.; Orosz, Z.; Ungvari, Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am. J. Pathol. 2007, 170, 388–398. [Google Scholar] [CrossRef]
- Csiszar, A.; Ungvari, Z.; Koller, A.; Edwards, J.G.; Kaley, G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genom. 2004, 17, 21–30. [Google Scholar] [CrossRef]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [PubMed]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging 2017, 9, 2411–2435. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Ungvari, Z.; Koller, A.; Edwards, J.G.; Kaley, G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1183–1185. [Google Scholar] [CrossRef]
- Liu, Z.J.; Tan, Y.; Beecham, G.W.; Seo, D.M.; Tian, R.; Li, Y.; Vazquez-Padron, R.I.; Pericak-Vance, M.; Vance, J.M.; Goldschmidt-Clermont, P.J.; et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: Implication of Notch signaling in atherosclerosis. Atherosclerosis 2012, 225, 296–303. [Google Scholar] [CrossRef]
- Liang, J.; Ke, X.; Yang, R.; Wang, X.; Du, Z.; Hu, C. Notch pathway activation mediated the senescence of endothelial progenitor cells in hypercholesterolemic mice. J. Bioenerg. Biomembr. 2020, 52, 431–440. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef]
- Xu, Q.; Mojiri, A.; Boulahouache, L.; Morales, E.; Walther, B.K.; Cooke, J.P. Vascular senescence in progeria: Role of endothelial dysfunction. Eur. Heart J. Open 2022, 2, oeac047. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef]
- Park, J.W.; Park, J.E.; Kim, S.R.; Sim, M.K.; Kang, C.M.; Kim, K.S. Metformin alleviates ionizing radiation-induced senescence by restoring BARD1-mediated DNA repair in human aortic endothelial cells. Exp. Gerontol. 2022, 160, 111706. [Google Scholar] [CrossRef]
- Shah, P.; Hobson, C.M.; Cheng, S.; Colville, M.J.; Paszek, M.J.; Superfine, R.; Lammerding, J. Nuclear Deformation Causes DNA Damage by Increasing Replication Stress. Curr. Biol. CB 2021, 31, 753–765.e6. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.; Hu, Y.; Hainaut, H.; Xu, Q. Mechanical stress-induced DNA damage and rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Mahmoudi, M. The role of DNA damage and repair in atherosclerosis: A review. J. Mol. Cell. Cardiol. 2015, 86, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Kurz, C.; Hakimi, M.; Kloor, M.; Grond-Ginsbach, C.; Gross-Weissmann, M.L.; Böckler, D.; von Knebel Doeberitz, M.; Dihlmann, S. Coding Microsatellite Frameshift Mutations Accumulate in Atherosclerotic Carotid Artery Lesions: Evaluation of 26 Cases and Literature Review. Mol. Med. 2015, 21, 479–486. [Google Scholar] [CrossRef]
- Chang, E.; Harley, C.B. Telomere length and replicative aging in human vascular tissues. Proc. Natl. Acad. Sci. USA 1995, 92, 11190–11194. [Google Scholar] [CrossRef]
- Fuster, J.J.; Andrés, V. Telomere biology and cardiovascular disease. Circ. Res. 2006, 99, 1167–1180. [Google Scholar] [CrossRef]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2019, 26, e12487. [Google Scholar] [CrossRef]
- Kotla, S.; Vu, H.T.; Ko, K.A.; Wang, Y.; Imanishi, M.; Heo, K.S.; Fujii, Y.; Thomas, T.N.; Gi, Y.J.; Mazhar, H.; et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight 2019, 4, e124867. [Google Scholar] [CrossRef]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Bhaggoe, U.M.; Leijten, F.; et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012, 126, 468–478. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Sirtuins in atherosclerosis: Guardians of healthspan and therapeutic targets. Nat. Rev. Cardiol. 2022, 19, 668–683. [Google Scholar] [CrossRef] [PubMed]
- Kaldirim, M.; Lang, A.; Pfeiler, S.; Fiegenbaum, P.; Kelm, M.; Bönner, F.; Gerdes, N. Modulation of mTOR Signaling in Cardiovascular Disease to Target Acute and Chronic Inflammation. Front. Cardiovasc. Med. 2022, 9, 907348. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Kume, S.; Takeda-Watanabe, A.; Kanasaki, K.; Koya, D. Sirtuins and renal diseases: Relationship with aging and diabetic nephropathy. Clin. Sci. 2013, 124, 153–164. [Google Scholar] [CrossRef]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 2011, 589, 4545–4554. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef]
- Stein, S.; Schäfer, N.; Breitenstein, A.; Besler, C.; Winnik, S.; Lohmann, C.; Heinrich, K.; Brokopp, C.E.; Handschin, C.; Landmesser, U.; et al. SIRT1 reduces endothelial activation without affecting vascular function in Apoe-/-mice. Aging 2010, 2, 353–360. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.Y.; Yun, M.; Jeong, J.; Park, E.R.; Shin, H.J.; Woo, S.R.; Jung, J.K.; Kim, Y.M.; Park, J.J.; Kim, J.; et al. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1α (HIF-1α) via direct interactions during hypoxia. Biochem. Biophys. Res. Commun. 2015, 462, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A.; et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014, 6, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M., Jr.; Sindler, A.L.; Seals, D.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef]
- Curry, A.M.; White, D.S.; Donu, D.; Cen, Y. Human Sirtuin Regulators: The “Success” Stories. Front. Physiol. 2021, 12, 752117. [Google Scholar] [CrossRef]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Kilic, U.; Gok, O.; Bacaksiz, A.; Izmirli, M.; Elibol-Can, B.; Uysal, O. SIRT1 gene polymorphisms affect the protein expression in cardiovascular diseases. PLoS ONE 2014, 9, e90428. [Google Scholar] [CrossRef]
- Nasiri, M.; Rauf, M.; Kamfiroozie, H.; Zibaeenezhad, M.J.; Jamali, Z. SIRT1 gene polymorphisms associated with decreased risk of atherosclerotic coronary artery disease. Gene 2018, 672, 16–20. [Google Scholar] [CrossRef]
- Kedenko, L.; Lamina, C.; Kedenko, I.; Kollerits, B.; Kiesslich, T.; Iglseder, B.; Kronenberg, F.; Paulweber, B. Genetic polymorphisms at SIRT1 and FOXO1 are associated with carotid atherosclerosis in the SAPHIR cohort. BMC Med. Genet. 2014, 15, 112. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, L.; Chen, S.; Liu, X.; Li, H.; Lu, X.; Yang, X.; Huang, J.; Gu, D. Association between the SIRT1 mRNA expression and acute coronary syndrome. J. Atheroscler. Thromb. 2015, 22, 165–182. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Li, T.; Liu, M.; Wang, Y.; Ma, H.; Mu, N.; Wang, H. SIRT6 in Senescence and Aging-Related Cardiovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 641315. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, P.; Ge, J.; Li, H. SIRT6 in Aging, Metabolism, Inflammation and Cardiovascular Diseases. Aging Dis. 2022, 13, 1787–1822. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yin, M.; Koroleva, M.; Mastrangelo, M.A.; Zhang, W.; Bai, P.; Little, P.J.; Jin, Z.G. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging 2016, 8, 1064–1082. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W.; et al. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury Through Targeting Nkx3.2-GATA5 Signaling. Circ. Res. 2019, 124, 1448–1461. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, J.; Huang, X.; Li, Z.; Liu, P. Deletion of sirtuin 6 accelerates endothelial dysfunction and atherosclerosis in apolipoprotein E-deficient mice. Transl. Res. J. Lab. Clin. Med. 2016, 172, 18–29.e2. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, C.; Hu, L.; Gao, E.; Li, C.; Wang, H.; Sun, D. Sirt6 stabilizes atherosclerosis plaques by promoting macrophage autophagy and reducing contact with endothelial cells. Biochem. Cell Biol. = Biochim. Et Biol. Cell. 2020, 98, 120–129. [Google Scholar] [CrossRef]
- Cardus, A.; Uryga, A.K.; Walters, G.; Erusalimsky, J.D. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc. Res. 2013, 97, 571–579. [Google Scholar] [CrossRef]
- Lee, O.H.; Woo, Y.M.; Moon, S.; Lee, J.; Park, H.; Jang, H.; Park, Y.Y.; Bae, S.K.; Park, K.H.; Heo, J.H.; et al. Sirtuin 6 deficiency induces endothelial cell senescence via downregulation of forkhead box M1 expression. Aging 2020, 12, 20946–20967. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Nie, H.; Wu, D.; Ying, W. SIRT2 plays a significant role in maintaining the survival and energy metabolism of PIEC endothelial cells. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 120–127. [Google Scholar]
- Chen, T.; Ma, C.; Fan, G.; Liu, H.; Lin, X.; Li, J.; Li, N.; Wang, S.; Zeng, M.; Zhang, Y.; et al. SIRT3 protects endothelial cells from high glucose-induced senescence and dysfunction via the p53 pathway. Life Sci. 2021, 264, 118724. [Google Scholar] [CrossRef]
- Sun, L.; Dang, W. SIRT7 slows down stem cell aging by preserving heterochromatin: A perspective on the new discovery. Protein Cell 2020, 11, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, J.; Yang, X.; Lai, P.; Mou, Y.; Deng, J.; Li, X.; Wang, H.; Liu, X.; Zhou, L.; et al. H2O2 down-regulates SIRT7’s protective role of endothelial premature dysfunction via microRNA-335-5p. Biosci. Rep. 2022, 42, BSR20211775. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Du, K. DHHC17 Is a New Regulator of AMPK Signaling. Mol. Cell. Biol. 2022, 42, e0013122. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zhou, M.; Chen, C.; Wu, X.; Wang, X. Role of AMPK mediated pathways in autophagy and aging. Biochimie 2022, 195, 100–113. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e6. [Google Scholar] [CrossRef]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef]
- Mori, H.; Inoki, K.; Münzberg, H.; Opland, D.; Faouzi, M.; Villanueva, E.C.; Ikenoue, T.; Kwiatkowski, D.; MacDougald, O.A.; Myers, M.G., Jr.; et al. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab. 2009, 9, 362–374. [Google Scholar] [CrossRef]
- Levine, Y.C.; Li, G.K.; Michel, T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway. J. Biol. Chem. 2007, 282, 20351–20364. [Google Scholar] [CrossRef]
- Lesniewski, L.A.; Zigler, M.C.; Durrant, J.R.; Donato, A.J.; Seals, D.R. Sustained activation of AMPK ameliorates age-associated vascular endothelial dysfunction via a nitric oxide-independent mechanism. Mech. Ageing Dev. 2012, 133, 368–371. [Google Scholar] [CrossRef]
- Yang, R.; Fang, W.; Liang, J.; Lin, C.; Wu, S.; Yan, S.; Hu, C.; Ke, X. Apelin/APJ axis improves angiotensin II-induced endothelial cell senescence through AMPK/SIRT1 signaling pathway. Arch. Med. Sci. AMS 2018, 14, 725–734. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, J.; Fu, M.; Dong, R.; Yang, Y.; Luo, J.; Hu, S.; Li, W.; Xu, X.; Tu, L. Dipeptidyl peptidase-4 inhibition improves endothelial senescence by activating AMPK/SIRT1/Nrf2 signaling pathway. Biochem. Pharmacol. 2020, 177, 113951. [Google Scholar] [CrossRef]
- Lyu, T.J.; Zhang, Z.X.; Chen, J.; Liu, Z.J. Ginsenoside Rg1 ameliorates apoptosis, senescence and oxidative stress in ox-LDL-induced vascular endothelial cells via the AMPK/SIRT3/p53 signaling pathway. Exp. Ther. Med. 2022, 24, 545. [Google Scholar] [CrossRef]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. TEM 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: Relevance in age-associated vascular dysfunction. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2018, 1864, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Sharp, Z.D.; Strong, R. The role of mTOR signaling in controlling mammalian life span: What a fungicide teaches us about longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 580–589. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef]
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 2017, 16, 17–26. [Google Scholar] [CrossRef]
- Lin, A.L.; Zheng, W.; Halloran, J.J.; Burbank, R.R.; Hussong, S.A.; Hart, M.J.; Javors, M.; Shih, Y.Y.; Muir, E.; Solano Fonseca, R.; et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 1412–1421. [Google Scholar] [CrossRef]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, A.G.; Yepuri, G.; Carvas, J.M.; Stein, S.; Matter, C.M.; Scerri, I.; Ruffieux, J.; Montani, J.P.; Ming, X.F.; Yang, Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: Inhibition by resveratrol. PLoS ONE 2011, 6, e19237. [Google Scholar] [CrossRef] [PubMed]
- Sabini, E.; O’Mahony, A.; Caturegli, P. MyMD-1 Improves Health Span and Prolongs Life Span in Old Mice: A Noninferiority Study to Rapamycin. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2023, 78, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Yang, H.W.; Hong, H.L.; Luo, W.W.; Dai, C.M.; Chen, X.Y.; Wang, L.P.; Li, Q.; Li, Z.Q.; Liu, P.Q.; Li, Z.M. mTORC2 facilitates endothelial cell senescence by suppressing Nrf2 expression via the Akt/GSK-3β/C/EBPα signaling pathway. Acta Pharmacol. Sin. 2018, 39, 1837–1846. [Google Scholar] [CrossRef]
- Zhang, K.S.; Schecker, J.; Krull, A.; Riechert, E.; Jürgensen, L.; Kamuf-Schenk, V.; Burghaus, J.; Kiper, L.; Cao Ho, T.; Wöltje, K.; et al. PRAS40 suppresses atherogenesis through inhibition of mTORC1-dependent pro-inflammatory signaling in endothelial cells. Sci. Rep. 2019, 9, 16787. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Ferri, C.M.; Martín-Núñez, E.; Pérez-Delgado, N.; González-Luis, A.; Mora-Fernández, C.; Navarro-González, J.F. Klotho as a biomarker of subclinical atherosclerosis in patients with moderate to severe chronic kidney disease. Sci. Rep. 2021, 11, 15877. [Google Scholar] [CrossRef] [PubMed]
- Castelblanco, E.; Hernández, M.; Alonso, N.; Ribes-Betriu, A.; Real, J.; Granado-Casas, M.; Rossell, J.; Rojo-López, M.I.; Dusso, A.S.; Julve, J.; et al. Association of α-klotho with subclinical carotid atherosclerosis in subjects with type 1 diabetes mellitus. Cardiovasc. Diabetol. 2022, 21, 207. [Google Scholar] [CrossRef] [PubMed]
- Keles, N.; Dogan, B.; Kalcik, M.; Caliskan, M.; Keles, N.N.; Aksu, F.; Bulut, M.; Kostek, O.; Isbilen, B.; Yilmaz, Y.; et al. Is serum Klotho protective against atherosclerosis in patients with type 1 diabetes mellitus? J. Diabetes Complicat. 2016, 30, 126–132. [Google Scholar] [CrossRef]
- Yu, L.; Kang, L.; Ren, X.Z.; Diao, Z.L.; Liu, W.H. Circulating α-Klotho Levels in Hemodialysis Patients and Their Relationship to Atherosclerosis. Kidney Blood Press. Res. 2018, 43, 1174–1182. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Donate-Correa, J.; Muros de Fuentes, M.; Pérez-Hernández, H.; Martínez-Sanz, R.; Mora-Fernández, C. Reduced Klotho is associated with the presence and severity of coronary artery disease. Heart (Br. Card. Soc.) 2014, 100, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; San Hipólito-Luengo, Á.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef]
- Buendía, P.; Carracedo, J.; Soriano, S.; Madueño, J.A.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Klotho Prevents NFκB Translocation and Protects Endothelial Cell from Senescence Induced by Uremia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1198–1209. [Google Scholar] [CrossRef]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef]
- Mao, Q.; Deng, M.; Zhao, J.; Zhou, D.; Tong, W.; Xu, S.; Zhao, X. Klotho ameliorates angiotension-II-induced endothelial senescence via restoration of autophagy by inhibiting Wnt3a/GSK-3β/mTOR signaling: A potential mechanism involved in prognostic performance of Klotho in coronary atherosclerotic disease. Mech. Ageing Dev. 2023, 211, 111789. [Google Scholar] [CrossRef]
- Kuro, O.M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Nar, G.; Cetin, S.S.; Nar, R.; Kilic, O.; Furkan, O.M.; Gunver, G.; Ilyas, S.C. Is serum fibroblast growth factor 21 associated with the severity or presence of coronary artery disease? J. Med. Biochem. 2022, 41, 162–167. [Google Scholar] [CrossRef]
- Lin, Z.; Pan, X.; Wu, F.; Ye, D.; Zhang, Y.; Wang, Y.; Jin, L.; Lian, Q.; Huang, Y.; Ding, H.; et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation 2015, 131, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.P.; Chen, C.Y.; Lin, T.W.; Kuo, C.S.; Huang, H.L.; Huang, P.H.; Lin, S.J. Fibroblast growth factor 21 reverses high-fat diet-induced impairment of vascular function via the anti-oxidative pathway in Apoe knockout mice. J. Cell. Mol. Med. 2022, 26, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Li, N.; He, Z.; Zeng, X.; Nan, Y.; Chen, J.; Miao, P.; Ying, Y.; Lin, W.; Zhao, X.; et al. Fibroblast growth factor 21 Ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKα signaling pathway. Cell Death Dis. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Zheng, Q.; Chen, J.; Tan, X.; Li, Q.; Ding, L.; Zhang, R.; Lin, X. FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp. Cell Res. 2020, 393, 112108. [Google Scholar] [CrossRef]
- Chen, J.J.; Tao, J.; Zhang, X.L.; Xia, L.Z.; Zeng, J.F.; Zhang, H.; Wei, D.H.; Lv, Y.C.; Li, G.H.; Wang, Z. Inhibition of the ox-LDL-Induced Pyroptosis by FGF21 of Human Umbilical Vein Endothelial Cells Through the TET2-UQCRC1-ROS Pathway. DNA Cell Biol. 2020, 39, 661–670. [Google Scholar] [CrossRef]
- Yan, X.; Gou, Z.; Li, Y.; Wang, Y.; Zhu, J.; Xu, G.; Zhang, Q. Fibroblast growth factor 21 inhibits atherosclerosis in Apoe-/- mice by ameliorating Fas-mediated apoptosis. Lipids Health Dis. 2018, 17, 203. [Google Scholar] [CrossRef]
- Yan, J.; Wang, J.; Huang, H.; Huang, Y.; Mi, T.; Zhang, C.; Zhang, L. Fibroblast growth factor 21 delayed endothelial replicative senescence and protected cells from H(2)O(2)-induced premature senescence through SIRT1. Am. J. Transl. Res. 2017, 9, 4492–4501. [Google Scholar]
- Wang, X.M.; Song, S.S.; Xiao, H.; Gao, P.; Li, X.J.; Si, L.Y. Fibroblast growth factor 21 protects against high glucose induced cellular damage and dysfunction of endothelial nitric-oxide synthase in endothelial cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 34, 658–671. [Google Scholar] [CrossRef]
- Lauder, L.; Mahfoud, F.; Azizi, M.; Bhatt, D.L.; Ewen, S.; Kario, K.; Parati, G.; Rossignol, P.; Schlaich, M.P.; Teo, K.K.; et al. Hypertension management in patients with cardiovascular comorbidities. Eur. Heart J. 2023, 44, 2066–2077. [Google Scholar] [CrossRef]
- de Cavanagh, E.M.; Inserra, F.; Ferder, M.; Ferder, L. From mitochondria to disease: Role of the renin-angiotensin system. Am. J. Nephrol. 2007, 27, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Haslem, L.; Hays, J.M.; Hays, F.A. p66Shc in Cardiovascular Pathology. Cells 2022, 11, 1855. [Google Scholar] [CrossRef] [PubMed]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Investig. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Zhang, K.; Zhou, B.; Zhang, L. Association study of angiotensin II type 1 receptor: A1166C (rs5186) polymorphism with coronary heart disease using systematic meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2013, 14, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Mo, Y.P.; He, Y.S.; Yang, F.; Xu, Y.; Li, C.C.; Wang, J.; Reng, H.M.; Long, L. Correlation between renin-angiotensin system gene polymorphisms and essential hypertension in the Chinese Yi ethnic group. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2015, 16, 975–981. [Google Scholar] [CrossRef]
- Parchwani, D.N.; Patel, D.D.; Rawtani, J.; Yadav, D. Analysis of Association of Angiotensin II Type 1 Receptor Gene A1166C Gene Polymorphism with Essential Hypertension. Indian J. Clin. Biochem. IJCB 2018, 33, 53–60. [Google Scholar] [CrossRef]
- Xu, Y.; Bao, Q.; He, B.; Pan, Y.; Zhang, R.; Mao, X.; Tang, Z.; Qu, L.; Zhu, C.; Tian, F.; et al. Association of angiotensin I converting enzyme, angiotensin II type 1 receptor and angiotensin I converting enzyme 2 gene polymorphisms with the dyslipidemia in type 2 diabetic patients of Chinese Han origin. J. Endocrinol. Investig. 2012, 35, 378–383. [Google Scholar]
- Chandra, S.; Narang, R.; Sreenivas, V.; Bhatia, J.; Saluja, D.; Srivastava, K. Association of angiotensin II type 1 receptor (A1166C) gene polymorphism and its increased expression in essential hypertension: A case-control study. PLoS ONE 2014, 9, e101502. [Google Scholar] [CrossRef]
- Yi, W.; Chen, F.; Zhang, H.; Tang, P.; Yuan, M.; Wen, J.; Wang, S.; Cai, Z. Role of angiotensin II in aging. Front. Aging Neurosci. 2022, 14, 1002138. [Google Scholar] [CrossRef]
- Neves, M.F.; Cunha, A.R.; Cunha, M.R.; Gismondi, R.A.; Oigman, W. The Role of Renin-Angiotensin-Aldosterone System and Its New Components in Arterial Stiffness and Vascular Aging. High Blood Press. Cardiovasc. Prev. Off. J. Ital. Soc. Hypertens. 2018, 25, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, A.; Feely, J. Effect of angiotensin ii receptor blockade on arterial stiffness: Beyond blood pressure reduction. Am. J. Hypertens. 2002, 15, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Intengan, H.D.; Schiffrin, E.L. Reduction of resistance artery stiffness by treatment with the AT(1)-receptor antagonist losartan in essential hypertension. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2000, 1, 40–45. [Google Scholar] [CrossRef]
- Mahmud, A.; Feely, J. Arterial stiffness and the renin-angiotensin-aldosterone system. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2004, 5, 102–108. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Wang, B.X.; Ou, W.S.; Lv, Y.H.; Li, M.M.; Miao, Z.; Wang, S.X.; Fei, J.C.; Guo, T. Administration of losartan improves aortic arterial stiffness and reduces the occurrence of acute coronary syndrome in aged patients with essential hypertension. J. Cell. Biochem. 2019, 120, 5713–5721. [Google Scholar] [CrossRef] [PubMed]
- Basso, N.; Cini, R.; Pietrelli, A.; Ferder, L.; Terragno, N.A.; Inserra, F. Protective effect of long-term angiotensin II inhibition. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1351–H1358. [Google Scholar] [CrossRef] [PubMed]
- González Bosc, L.V.; Kurnjek, M.L.; Müller, A.; Terragno, N.A.; Basso, N. Effect of chronic angiotensin II inhibition on the nitric oxide synthase in the normal rat during aging. J. Hypertens. 2001, 19, 1403–1409. [Google Scholar] [CrossRef]
- González Bosc, L.; Kurnjek, M.L.; Müller, A.; Basso, N. Effect of chronic angiotensin II inhibition on the cardiovascular system of the normal rat. Am. J. Hypertens. 2000, 13, 1301–1307. [Google Scholar] [CrossRef]
- Xu, S.; Zhi, H.; Hou, X.; Jiang, B. Angiotensin II modulates interleukin-1β-induced inflammatory gene expression in vascular smooth muscle cells via interfering with ERK-NF-κB crosstalk. Biochem. Biophys. Res. Commun. 2011, 410, 543–548. [Google Scholar] [CrossRef]
- Zhang, G.H.; Chao, M.; Hui, L.H.; Xu, D.L.; Cai, W.L.; Zheng, J.; Gao, M.; Zhang, M.X.; Wang, J.; Lu, Q.H. Poly(ADP-ribose)polymerase 1 inhibition protects against age-dependent endothelial dysfunction. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1266–1274. [Google Scholar] [CrossRef]
- Nassif, M.E.; Windsor, S.L.; Borlaug, B.A.; Kitzman, D.W.; Shah, S.J.; Tang, F.; Khariton, Y.; Malik, A.O.; Khumri, T.; Umpierrez, G.; et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: A multicenter randomized trial. Nat. Med. 2021, 27, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Udell, J.A.; Jones, W.S.; Petrie, M.C.; Harrington, J.; Anker, S.D.; Bhatt, D.L.; Hernandez, A.F.; Butler, J. Sodium Glucose Cotransporter-2 Inhibition for Acute Myocardial Infarction: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2022, 79, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Belcastro, E.; Hasan, H.; Matsushita, K.; Marchandot, B.; Abbas, M.; Toti, F.; Auger, C.; Jesel, L.; Ohlmann, P.; et al. Angiotensin II-induced upregulation of SGLT1 and 2 contributes to human microparticle-stimulated endothelial senescence and dysfunction: Protective effect of gliflozins. Cardiovasc. Diabetol. 2021, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2020, 24, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Malhotra, K.; Sowers, J.; Aroor, A. Uric Acid—Key ingredient in the recipe for cardiorenal metabolic syndrome. Cardiorenal Med. 2013, 3, 208–220. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Whaley-Connell, A.T.; Sowers, J.R. Fructose and uric acid: Is there a role in endothelial function? Curr. Hypertens. Rep. 2014, 16, 434. [Google Scholar] [CrossRef]
- Canepa, M.; Viazzi, F.; Strait, J.B.; Ameri, P.; Pontremoli, R.; Brunelli, C.; Studenski, S.; Ferrucci, L.; Lakatta, E.G.; AlGhatrif, M. Longitudinal Association Between Serum Uric Acid and Arterial Stiffness: Results from the Baltimore Longitudinal Study of Aging. Hypertension 2017, 69, 228–235. [Google Scholar] [CrossRef]
- Puddu, P.; Puddu, G.M.; Cravero, E.; Vizioli, L.; Muscari, A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: Molecular mechanisms and clinical implications. J. Cardiol. 2012, 59, 235–242. [Google Scholar] [CrossRef]
- Kanbay, M.; Yilmaz, M.I.; Sonmez, A.; Turgut, F.; Saglam, M.; Cakir, E.; Yenicesu, M.; Covic, A.; Jalal, D.; Johnson, R.J. Serum uric acid level and endothelial dysfunction in patients with nondiabetic chronic kidney disease. Am. J. Nephrol. 2011, 33, 298–304. [Google Scholar] [CrossRef]
- Tang, Z.; Cheng, L.T.; Li, H.Y.; Wang, T. Serum uric acid and endothelial dysfunction in continuous ambulatory peritoneal dialysis patients. Am. J. Nephrol. 2009, 29, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Maio, R.; Mallamaci, F.; Sesti, G.; Perticone, F. Uric acid and endothelial dysfunction in essential hypertension. J. Am. Soc. Nephrol. JASN 2006, 17, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Sincer, I.; Kurtoglu, E.; Calıskan, M.; Akkaya, E.; Vuruskan, E.; Küçükosmanoglu, M.; Çoşkun, F.Y.; Inci, M.F.; Zorlu, A. Significant correlation between uric acid levels and flow-mediated dilatation in patients with masked hypertension. Clin. Exp. Hypertens. 2014, 36, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, A.J.; Bruinsma, K.A. Uric acid is closely linked to vascular nitric oxide activity. Evidence for mechanism of association with cardiovascular disease. J. Am. Coll. Cardiol. 2001, 38, 1850–1858. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jia, G.; Habibi, J.; Sun, Z.; Ramirez-Perez, F.I.; Brady, B.; Chen, D.; Martinez-Lemus, L.A.; Manrique, C.; Nistala, R.; et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metab. Clin. Exp. 2017, 74, 32–40. [Google Scholar] [CrossRef]
- Lastra, G.; Manrique, C.; Jia, G.; Aroor, A.R.; Hayden, M.R.; Barron, B.J.; Niles, B.; Padilla, J.; Sowers, J.R. Xanthine oxidase inhibition protects against Western diet-induced aortic stiffness and impaired vasorelaxation in female mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R67–R77. [Google Scholar] [CrossRef]
- Yu, M.A.; Sánchez-Lozada, L.G.; Johnson, R.J.; Kang, D.H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Qi, W. Uric acid, as a double-edged sword, affects the activity of epidermal growth factor (EGF) on human umbilical vein endothelial cells by regulating aging process. Bioengineered 2022, 13, 3877–3895. [Google Scholar] [CrossRef]
- Rodríguez-Mañas, L.; El-Assar, M.; Vallejo, S.; López-Dóriga, P.; Solís, J.; Petidier, R.; Montes, M.; Nevado, J.; Castro, M.; Gómez-Guerrero, C.; et al. Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 2009, 8, 226–238. [Google Scholar] [CrossRef]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef]
- Repin, V.S.; Dolgov, V.V.; Zaikina, O.E.; Novikov, I.D.; Antonov, A.S.; Nikolaeva, M.A.; Smirnov, V.N. Heterogeneity of endothelium in human aorta. A quantitative analysis by scanning electron microscopy. Atherosclerosis 1984, 50, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; McCrann, D.J.; Nguyen, H.; St Hilaire, C.; DePinho, R.A.; Jones, M.R.; Ravid, K. Increased polyploidy in aortic vascular smooth muscle cells during aging is marked by cellular senescence. Aging Cell 2007, 6, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, O.; Fan, J.L.; Watanabe, T. Atherosclerosis- and age-related multinucleated variant endothelial cells in primary culture from human aorta. Am. J. Pathol. 1989, 135, 967–976. [Google Scholar] [PubMed]
- Devlin, A.M.; Davidson, A.O.; Gordon, J.F.; Campbell, A.M.; Morton, J.J.; Reid, J.L.; Dominiczak, A.F. Vascular smooth muscle polyploidy in genetic hypertension: The role of angiotensin II. J. Hum. Hypertens. 1995, 9, 497–500. [Google Scholar]
- Devlin, A.M.; Gordon, J.F.; Davidson, A.O.; Clark, J.S.; Hamilton, C.A.; Morton, J.J.; Campbell, A.M.; Reid, J.L.; Dominiczak, A.F. The effects of perindopril on vascular smooth muscle polyploidy in stroke-prone spontaneously hypertensive rats. J. Hypertens. 1995, 13, 211–218. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, J.G. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 2009, 8, 100–112. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, J.G. Polyploidy impairs human aortic endothelial cell function and is prevented by nicotinamide phosphoribosyltransferase. Am. J. Physiol. Cell Physiol. 2010, 298, C66–C74. [Google Scholar] [CrossRef]
- Pandit, S.K.; Westendorp, B.; Nantasanti, S.; van Liere, E.; Tooten, P.C.; Cornelissen, P.W.; Toussaint, M.J.; Lamers, W.H.; de Bruin, A. E2F8 is essential for polyploidization in mammalian cells. Nat. Cell Biol. 2012, 14, 1181–1191. [Google Scholar] [CrossRef]
- Prata, L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Macdonald, K.; Chan, S.W.; Boyle, J.J.; Weissberg, P.L. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ. Res. 1998, 82, 704–712. [Google Scholar] [CrossRef]
- Suda, M.; Paul, K.H.; Minamino, T.; Miller, J.D.; Lerman, A.; Ellison-Hughes, G.M.; Tchkonia, T.; Kirkland, J.L. Senescent Cells: A Therapeutic Target in Cardiovascular Diseases. Cells 2023, 12, 1296. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Y.; Carr, C.; Dhandapani, K.M.; Brann, D.W. Senolytic therapy is neuroprotective and improves functional outcome long-term after traumatic brain injury in mice. Front. Neurosci. 2023, 17, 1227705. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab 2021, 33, 818–832.e7. [Google Scholar] [CrossRef]
- Lérida-Viso, A.; Estepa-Fernández, A.; Morellá-Aucejo, Á.; Lozano-Torres, B.; Alfonso, M.; Blandez, J.F.; Bisbal, V.; Sepúlveda, P.; García-Fernández, A.; Orzáez, M. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol. Res. 2022, 183, 106356. [Google Scholar] [CrossRef]
- Jia, K.; Dai, Y.; Liu, A.; Li, X.; Wu, L.; Lu, L.; Bao, Y.; Jin, Q. Senolytic Agent Navitoclax Inhibits Angiotensin II-Induced Heart Failure in Mice. J. Cardiovasc. Pharmacol. 2020, 76, 452–460. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech. Ageing Dev. 2022, 208, 111740. [Google Scholar] [PubMed]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Kaistha, A.; Uryga, A.K.; Oc, S.; Foote, K.; Shah, A.; Finigan, A.; Figg, N.; Dobnikar, L.; Jørgensen, H.; et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc. Res. 2022, 118, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Karnewar, S.; Karnewar, V.; Shankman, L.S.; Owens, G.K. Treatment of advanced atherosclerotic mice with the senolytic agent ABT-263 is associated with reduced indices of plaque stability and increased mortality. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [PubMed]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martínez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 2020, 19, e13133. [Google Scholar] [CrossRef]
- González-Gualda, E.; Pàez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., 3rd; González-López, C.; Ou, H.L.; Mirón-Barroso, S.; Zhang, Z.; Lérida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef]
- van der Feen, D.E.; Bossers, G.P.L.; Hagdorn, Q.A.J.; Moonen, J.R.; Kurakula, K.; Szulcek, R.; Chappell, J.; Vallania, F.; Donato, M.; Kok, K.; et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 2020, 12, eaaw4974. [Google Scholar] [PubMed]
- Ramadhiani, R.; Ikeda, K.; Miyagawa, K.; Ryanto, G.R.T.; Tamada, N.; Suzuki, Y.; Kirita, Y.; Matoba, S.; Hirata, K.I.; Emoto, N. Endothelial cell senescence exacerbates pulmonary hypertension by inducing juxtacrine Notch signaling in smooth muscle cells. iScience 2023, 26, 106662. [Google Scholar]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [PubMed]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 2482. [Google Scholar] [CrossRef] [PubMed]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Matsumoto, N.; Yoshida, Y.; Mikawa, R.; Katayama, A.; et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat. Aging 2021, 1, 1117–1126. [Google Scholar]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Gasek, N.S.; Zhou, Y.; Cohn, R.L.; Martin, D.E.; Zuo, W.; Flynn, W.F.; Guo, C.; Jellison, E.R.; et al. Targeting p21(Cip1) highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab 2022, 34, 75–89.e8. [Google Scholar] [CrossRef]
- Wang, B.; Wang, L.; Gasek, N.S.; Zhou, Y.; Kim, T.; Guo, C.; Jellison, E.R.; Haynes, L.; Yadav, S.; Tchkonia, T.; et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 2021, 1, 962–973. [Google Scholar]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e6. [Google Scholar]
- Kim, S.; Kim, C. Transcriptomic Analysis of Cellular Senescence: One Step Closer to Senescence Atlas. Mol. Cells 2021, 44, 136–145. [Google Scholar] [PubMed]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Huby, T.; Witztum, J.L.; Ouzilleau, B.; Miller, E.R.; Saint-Charles, F.; Aucouturier, P.; Chapman, M.J.; Lesnik, P. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation 2009, 119, 1795–1804. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022, 13, 467. [Google Scholar]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [PubMed]
- Zhao, F.; Satyanarayana, G.; Zhang, Z.; Zhao, J.; Ma, X.L.; Wang, Y. Endothelial Autophagy in Coronary Microvascular Dysfunction and Cardiovascular Disease. Cells 2022, 11, 2081. [Google Scholar] [PubMed]
- Choy, J.C.; Granville, D.J.; Hunt, D.W.; McManus, B.M. Endothelial cell apoptosis: Biochemical characteristics and potential implications for atherosclerosis. J. Mol. Cell. Cardiol. 2001, 33, 1673–1690. [Google Scholar]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Michel, J.B. Anoikis in the cardiovascular system: Known and unknown extracellular mediators. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2146–2154. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar]
- Ju, J.; Liu, Y.; Liang, H.; Yang, B. The role of pyroptosis in endothelial dysfunction induced by diseases. Front. Immunol. 2022, 13, 1093985. [Google Scholar] [PubMed]
- Zdanyte, M.; Borst, O.; Münzer, P. NET-(works) in arterial and venous thrombo-occlusive diseases. Front. Cardiovasc. Med. 2023, 10, 1155512. [Google Scholar] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar]
- Zhang, X.; Ren, Z.; Xu, W.; Jiang, Z. Necroptosis in atherosclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2022, 534, 22–28. [Google Scholar]
- Zhang, H.; Zhou, S.; Sun, M.; Hua, M.; Liu, Z.; Mu, G.; Wang, Z.; Xiang, Q.; Cui, Y. Ferroptosis of Endothelial Cells in Vascular Diseases. Nutrients 2022, 14, 4506. [Google Scholar]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar]
- LaRocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316. [Google Scholar]
- LaRocca, T.J.; Gioscia-Ryan, R.A.; Hearon, C.M., Jr.; Seals, D.R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 2013, 134, 314–320. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.N.; Xu, S.B.; Wu, H.; Dai, B.; Jian, D.D.; Yang, M.; Wu, Y.T.; Feng, Q.; Zhu, J.H.; et al. MicroRNA-214-3p: A link between autophagy and endothelial cell dysfunction in atherosclerosis. Acta Physiol. 2018, 222, e12973. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, A.; He, S.; He, S.; Li, C.; Mei, W.; Lu, Q. Autophagy-Related Genes in Atherosclerosis. J. Healthc. Eng. 2021, 2021, 6402206. [Google Scholar]
- Patella, F.; Neilson, L.J.; Athineos, D.; Erami, Z.; Anderson, K.I.; Blyth, K.; Ryan, K.M.; Zanivan, S. In-Depth Proteomics Identifies a Role for Autophagy in Controlling Reactive Oxygen Species Mediated Endothelial Permeability. J. Proteome Res. 2016, 15, 2187–2197. [Google Scholar]
- Zhu, L.; Wu, G.; Yang, X.; Jia, X.; Li, J.; Bai, X.; Li, W.; Zhao, Y.; Li, Y.; Cheng, W.; et al. Low density lipoprotein mimics insulin action on autophagy and glucose uptake in endothelial cells. Sci. Rep. 2019, 9, 3020. [Google Scholar] [PubMed]
- Yuan, P.; Hu, Q.; He, X.; Long, Y.; Song, X.; Wu, F.; He, Y.; Zhou, X. Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death Dis. 2020, 11, 141. [Google Scholar] [PubMed]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell 2016, 15, 187–191. [Google Scholar]
- Xie, Y.; You, S.J.; Zhang, Y.L.; Han, Q.; Cao, Y.J.; Xu, X.S.; Yang, Y.P.; Li, J.; Liu, C.F. Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells. Mol. Med. Rep. 2011, 4, 459–464. [Google Scholar] [PubMed]
- Zhang, X.; Yu, L.; Xu, H. Lysosome calcium in ROS regulation of autophagy. Autophagy 2016, 12, 1954–1955. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Choi, S.H. ASK1 Mediates Apoptosis and Autophagy during oxLDL-CD36 Signaling in Senescent Endothelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2840437. [Google Scholar]
- Meng, Q.; Pu, L.; Lu, Q.; Wang, B.; Li, S.; Liu, B.; Li, F. Morin hydrate inhibits atherosclerosis and LPS-induced endothelial cells inflammatory responses by modulating the NFκB signaling-mediated autophagy. Int. Immunopharmacol. 2021, 100, 108096. [Google Scholar] [PubMed]
- Reglero-Real, N.; Pérez-Gutiérrez, L.; Yoshimura, A.; Rolas, L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; Saleeb, R.S.; Gonzalez-Nuñez, M.; Austin-Williams, S.N.; et al. Autophagy modulates endothelial junctions to restrain neutrophil diapedesis during inflammation. Immunity 2021, 54, 1989–2004.e9. [Google Scholar] [PubMed]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fan, Y.; Qiao, C.; Liang, W.; Hu, W.; Zhu, T.; Zhang, J.; Chen, Y.E. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci. Signal. 2017, 10, eaah4214. [Google Scholar]
- Kim, J.Y.; Mondaca-Ruff, D.; Singh, S.; Wang, Y. SIRT1 and Autophagy: Implications in Endocrine Disorders. Front. Endocrinol. 2022, 13, 930919. [Google Scholar]
- Xu, Y.; Wan, W. Acetylation in the regulation of autophagy. Autophagy 2023, 19, 379–387. [Google Scholar]
- Gu, S.; Zhou, C.; Pei, J.; Wu, Y.; Wan, S.; Zhao, X.; Hu, J.; Hua, X. Esomeprazole inhibits hypoxia/endothelial dysfunction-induced autophagy in preeclampsia. Cell Tissue Res. 2022, 388, 181–194. [Google Scholar]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Hao, R.; Wang, W.; Gao, H.; Wang, C. SIRT1/Atg5/autophagy are involved in the antiatherosclerosis effects of ursolic acid. Mol. Cell. Biochem. 2016, 420, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Duan, W.; Wu, G.; Zhang, D.; Wang, L.; Chen, D.; Chen, Z.; Yang, B. Protective effect of hydrogen sulfide on endothelial cells through Sirt1-FoxO1-mediated autophagy. Ann. Transl. Med. 2020, 8, 1586. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, X.; Zhang, Z.; Liu, J.; Wu, C.; Chen, Y.; Zhou, J.; Zhang, J.; Zhang, X. Autophagy promotes directed migration of HUVEC in response to electric fields through the ROS/SIRT1/FOXO1 pathway. Free Radic. Biol. Med. 2022, 192, 213–223. [Google Scholar] [CrossRef]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Hua, Y.; Zhang, J.; Liu, Q.; Su, J.; Zhao, Y.; Zheng, G.; Yang, Z.; Zhuo, D.; Ma, C.; Fan, G. The Induction of Endothelial Autophagy and Its Role in the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 831847. [Google Scholar] [CrossRef]
- De Meyer, G.R.; Grootaert, M.O.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in vascular disease. Circ. Res. 2015, 116, 468–479. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef]
- Chau, Y.P.; Lin, S.Y.; Chen, J.H.; Tai, M.H. Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol. Histopathol. 2003, 18, 715–726. [Google Scholar] [PubMed]
- Kato, Y.; Furusyo, N.; Tanaka, Y.; Ueyama, T.; Yamasaki, S.; Murata, M.; Hayashi, J. The Relation between Serum Endostatin Level and Carotid Atherosclerosis in Healthy Residents of Japan: Results from the Kyushu and Okinawa Population Study (KOPS). J. Atheroscler. Thromb. 2017, 24, 1023–1030. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ärnlöv, J.; Ruge, T.; Ingelsson, E.; Larsson, A.; Sundström, J.; Lind, L. Serum endostatin and risk of mortality in the elderly: Findings from 2 community-based cohorts. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Qian, S.; Zhang, R.; Guo, D.; Wang, A.; Peng, Y.; Peng, H.; Li, Q.; Ju, Z.; Geng, D.; et al. Endostatin as a novel prognostic biomarker in acute ischemic stroke. Atherosclerosis 2020, 293, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef]
- Zeng, X.; Chen, J.; Miller, Y.I.; Javaherian, K.; Moulton, K.S. Endostatin binds biglycan and LDL and interferes with LDL retention to the subendothelial matrix during atherosclerosis. J. Lipid Res. 2005, 46, 1849–1859. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Zheng, Y.; Li, B.; Meng, X.; Fu, X.; Fei, Y. Simulated ischemia/reperfusion-induced p65-Beclin 1-dependent autophagic cell death in human umbilical vein endothelial cells. Sci. Rep. 2016, 6, 37448. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Z.; Han, W.; Li, H. Zoledronate induces autophagic cell death in human umbilical vein endothelial cells via Beclin-1 dependent pathway activation. Mol. Med. Rep. 2016, 14, 4747–4754. [Google Scholar] [CrossRef]
- Csordas, A.; Kreutmayer, S.; Ploner, C.; Braun, P.R.; Karlas, A.; Backovic, A.; Wick, G.; Bernhard, D. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc. Res. 2011, 92, 141–148. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Li, B.; Fei, Y.; He, Y.; Chen, J.; Wang, P.; Liu, X. Reactive oxygen species contribute to simulated ischemia/reperfusion-induced autophagic cell death in human umbilical vein endothelial cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2014, 20, 1017–1023. [Google Scholar]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. CMLS 2019, 76, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Zhang, Q.; Liu, J.; Li, R.; Wang, D.; Peng, W.; Wu, C. Suppression of apoptosis in vascular endothelial cell, the promising way for natural medicines to treat atherosclerosis. Pharmacol. Res. 2021, 168, 105599. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die. J. Mol. Biol. 2022, 434, 167378. [Google Scholar] [CrossRef]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Gerrity, R.G.; Richardson, M.; Somer, J.B.; Bell, F.P.; Schwartz, C.J. Endothelial cell morphology in areas of in vivo Evans blue uptake in the aorta of young pigs. II. Ultrastructure of the intima in areas of differing permeability to proteins. Am. J. Pathol. 1977, 89, 313–334. [Google Scholar]
- Tricot, O.; Mallat, Z.; Heymes, C.; Belmin, J.; Lesèche, G.; Tedgui, A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation 2000, 101, 2450–2453. [Google Scholar] [CrossRef]
- Dimmeler, S.; Haendeler, J.; Zeiher, A.M. Regulation of endothelial cell apoptosis in atherothrombosis. Curr. Opin. Lipidol. 2002, 13, 531–536. [Google Scholar] [CrossRef]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Qu, K.; Yan, F.; Qin, X.; Zhang, K.; He, W.; Dong, M.; Wu, G. Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis. Front. Physiol. 2022, 13, 1084604. [Google Scholar] [CrossRef]
- Su, Q.; Wang, Y.; Yang, X.; Li, X.D.; Qi, Y.F.; He, X.J.; Wang, Y.J. Inhibition of Endoplasmic Reticulum Stress Apoptosis by Estrogen Protects Human Umbilical Vein Endothelial Cells Through the PI3 Kinase-Akt Signaling Pathway. J. Cell. Biochem. 2017, 118, 4568–4574. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Momi, S.; Guglielmini, G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem. Pharmacol. 2019, 166, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Haendeler, J.; Galle, J.; Zeiher, A.M. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases. A mechanistic clue to the ‘response to injury’ hypothesis. Circulation 1997, 95, 1760–1763. [Google Scholar] [CrossRef]
- Harada-Shiba, M.; Kinoshita, M.; Kamido, H.; Shimokado, K. Oxidized low density lipoprotein induces apoptosis in cultured human umbilical vein endothelial cells by common and unique mechanisms. J. Biol. Chem. 1998, 273, 9681–9687. [Google Scholar] [CrossRef]
- Sata, M.; Walsh, K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J. Clin. Investig. 1998, 102, 1682–1689. [Google Scholar] [CrossRef]
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Nègre-Salvayre, A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2158–2166. [Google Scholar] [CrossRef]
- Li, D.; Mehta, J.L. Upregulation of endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and implications in apoptosis of human coronary artery endothelial cells: Evidence from use of antisense LOX-1 mRNA and chemical inhibitors. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1116–1122. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Grechko, A.V.; Shakhpazyan, N.K.; Orekhov, A.N. The Role of KLF2 in the Regulation of Atherosclerosis Development and Potential Use of KLF2-Targeted Therapy. Biomedicines 2022, 10, 254. [Google Scholar] [CrossRef]
- Dekker, R.J.; van Soest, S.; Fontijn, R.D.; Salamanca, S.; de Groot, P.G.; VanBavel, E.; Pannekoek, H.; Horrevoets, A.J. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002, 100, 1689–1698. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, Q.; Wang, Z. PTP1B inhibition ameliorates inflammatory injury and dysfunction in ox-LDL-induced HUVECs by activating the AMPK/SIRT1 signaling pathway via negative regulation of KLF2. Exp. Ther. Med. 2022, 24, 467. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xuan, W.; Jia, Z.; Li, H.; Li, M.; Liang, X.; Su, D. HRD1 prevents atherosclerosis-mediated endothelial cell apoptosis by promoting LOX-1 degradation. Cell Cycle 2020, 19, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Hsu, P.P.; Chen, B.P.; Yuan, S.; Usami, S.; Shyy, J.Y.; Li, Y.S.; Chien, S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. USA 2000, 97, 9385–9389. [Google Scholar] [CrossRef]
- Malek, A.M.; Jiang, L.; Lee, I.; Sessa, W.C.; Izumo, S.; Alper, S.L. Induction of nitric oxide synthase mRNA by shear stress requires intracellular calcium and G-protein signals and is modulated by PI 3 kinase. Biochem. Biophys. Res. Commun. 1999, 254, 231–242. [Google Scholar] [CrossRef]
- Tardy, Y.; Resnick, N.; Nagel, T.; Gimbrone, M.A., Jr.; Dewey, C.F., Jr. Shear stress gradients remodel endothelial monolayers in vitro via a cell proliferation-migration-loss cycle. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3102–3106. [Google Scholar] [CrossRef] [PubMed]
- Chien, S. Effects of disturbed flow on endothelial cells. Ann. Biomed. Eng. 2008, 36, 554–562. [Google Scholar] [CrossRef]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.S.; Lee, H.; Nigro, P.; Thomas, T.; Le, N.T.; Chang, E.; McClain, C.; Reinhart-King, C.A.; King, M.R.; Berk, B.C.; et al. PKCζ mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J. Cell Biol. 2011, 193, 867–884. [Google Scholar] [CrossRef]
- Birukov, K.G.; Jacobson, J.R.; Flores, A.A.; Ye, S.Q.; Birukova, A.A.; Verin, A.D.; Garcia, J.G. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L785–L797. [Google Scholar] [CrossRef]
- Wang, J.; Fan, B.; Wei, Y.; Suo, X.; Ding, Y. A simple multi-well stretching device to induce inflammatory responses of vascular endothelial cells. Lab A Chip 2016, 16, 360–367. [Google Scholar] [CrossRef]
- Dong, G.; Huang, X.; Jiang, S.; Ni, L.; Chen, S. Simvastatin Mitigates Apoptosis and Transforming Growth Factor-Beta Upregulation in Stretch-Induced Endothelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6026051. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chiou, K.R.; Bugayenko, A.; Irani, K.; Kass, D.A. Reduced wall compliance suppresses Akt-dependent apoptosis protection stimulated by pulse perfusion. Circ. Res. 2005, 97, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Ensenat, D.; Wang, H.; Schafer, A.I.; Durante, W. Physiologic cyclic stretch inhibits apoptosis in vascular endothelium. FEBS Lett. 2003, 541, 52–56. [Google Scholar] [CrossRef]
- Raaz, U.; Kuhn, H.; Wirtz, H.; Hammerschmidt, S. Rapamycin reduces high-amplitude, mechanical stretch-induced apoptosis in pulmonary microvascular endothelial cells. Microvasc. Res. 2009, 77, 297–303. [Google Scholar] [CrossRef]
- Zhuang, F.; Bao, H.; Shi, Q.; Li, J.; Jiang, Z.L.; Wang, Y.; Qi, Y.X. Endothelial microparticles induced by cyclic stretch activate Src and modulate cell apoptosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 13586–13596. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Y.; Fard, J.K.; Ghafoor, D.; Eid, A.H.; Sahebkar, A. Paradoxical effects of statins on endothelial and cancer cells: The impact of concentrations. Cancer Cell Int. 2023, 23, 43. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nuki, T.; Gomita, K.; Weyand, C.M.; Hagiwara, N. Statins reduce endothelial cell apoptosis via inhibition of TRAIL expression on activated CD4 T cells in acute coronary syndrome. Atherosclerosis 2010, 213, 33–39. [Google Scholar] [CrossRef]
- Safaeian, L.; Mirian, M.; Bahrizadeh, S. Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H(2)O(2)-induced oxidative stress. Arch. Physiol. Biochem. 2022, 128, 1681–1686. [Google Scholar] [CrossRef]
- Zeng, J.; Tao, J.; Xi, L.; Wang, Z.; Liu, L. PCSK9 mediates the oxidative low-density lipoprotein-induced pyroptosis of vascular endothelial cells via the UQCRC1/ROS pathway. Int. J. Mol. Med. 2021, 47, 53. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, Q.; Li, H.; Meng, Z.; Hao, M.; Ma, X.; Lin, W.; Kuang, H. Dapagliflozin Reduces Apoptosis of Diabetic Retina and Human Retinal Microvascular Endothelial Cells Through ERK1/2/cPLA2/AA/ROS Pathway Independent of Hypoglycemic. Front. Pharmacol. 2022, 13, 827896. [Google Scholar] [CrossRef]
- Faridvand, Y.; Kazemzadeh, H.; Vahedian, V.; Mirzajanzadeh, P.; Nejabati, H.R.; Safaie, N.; Maroufi, N.F.; Pezeshkian, M.; Nouri, M.; Jodati, A. Dapagliflozin attenuates high glucose-induced endothelial cell apoptosis and inflammation through AMPK/SIRT1 activation. Clin. Exp. Pharmacol. Physiol. 2022, 49, 643–651. [Google Scholar] [CrossRef]
- Yang, J.; Sato, K.; Aprahamian, T.; Brown, N.J.; Hutcheson, J.; Bialik, A.; Perlman, H.; Walsh, K. Endothelial overexpression of Fas ligand decreases atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Carino, K.; Johnston, L.; Servinsky, L.; Machamer, C.E.; Kolb, T.M.; Lam, H.; Dudek, S.M.; An, S.S.; Rane, M.J.; et al. A nonapoptotic endothelial barrier-protective role for caspase-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1118–L1126. [Google Scholar] [CrossRef] [PubMed]
- Avrutsky, M.I.; Ortiz, C.C.; Johnson, K.V.; Potenski, A.M.; Chen, C.W.; Lawson, J.M.; White, A.J.; Yuen, S.K.; Morales, F.N.; Canepa, E.; et al. Endothelial activation of caspase-9 promotes neurovascular injury in retinal vein occlusion. Nat. Commun. 2020, 11, 3173. [Google Scholar] [CrossRef]
- Bader, S.M.; Preston, S.P.; Saliba, K.; Lipszyc, A.; Grant, Z.L.; Mackiewicz, L.; Baldi, A.; Hempel, A.; Clark, M.P.; Peiris, T.; et al. Endothelial Caspase-8 prevents fatal necroptotic hemorrhage caused by commensal bacteria. Cell Death Differ. 2023, 30, 27–36. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.J.; Czabotar, P.E.; Murphy, J.M. The Structural Basis of Necroptotic Cell Death Signaling. Trends Biochem. Sci. 2019, 44, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Robichaud, S.; Nguyen, M.A.; Geoffrion, M.; Wyatt, H.; Cottee, M.L.; Dennison, T.; Pietrangelo, A.; Lee, R.; Lagace, T.A.; et al. Loss of MLKL (Mixed Lineage Kinase Domain-Like Protein) Decreases Necrotic Core but Increases Macrophage Lipid Accumulation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1155–1167. [Google Scholar] [CrossRef]
- Coornaert, I.; Puylaert, P.; Marcasolli, G.; Grootaert, M.O.J.; Vandenabeele, P.; De Meyer, G.R.Y.; Martinet, W. Impact of myeloid RIPK1 gene deletion on atherogenesis in Apoe-deficient mice. Atherosclerosis 2021, 322, 51–60. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef]
- Colijn, S.; Muthukumar, V.; Xie, J.; Gao, S.; Griffin, C.T. Cell-specific and athero-protective roles for RIPK3 in a murine model of atherosclerosis. Dis. Models Mech. 2020, 13, dmm041962. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Nguyen, M.A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates with Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce NF-κB Activation and Atherogenesis in Mice. Circulation 2021, 143, 163–177. [Google Scholar] [CrossRef]
- Gan, I.; Jiang, J.; Lian, D.; Huang, X.; Fuhrmann, B.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2019, 19, 686–698. [Google Scholar] [CrossRef]
- Kwok, C.; Pavlosky, A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Necroptosis Is Involved in CD4+ T Cell-Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017, 101, 2026–2037. [Google Scholar] [CrossRef]
- Zhu, P.; Wang, J.; Du, W.; Ren, J.; Zhang, Y.; Xie, F.; Xu, G. NR4A1 Promotes LPS-Induced Acute Lung Injury through Inhibition of Opa1-Mediated Mitochondrial Fusion and Activation of PGAM5-Related Necroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 6638244. [Google Scholar] [CrossRef] [PubMed]
- Singla, S.; Sysol, J.R.; Dille, B.; Jones, N.; Chen, J.; Machado, R.F. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am. J. Respir. Cell Mol. Biol. 2017, 57, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Paku, S.; Laszlo, V.; Dezso, K.; Nagy, P.; Hoda, M.A.; Klepetko, W.; Renyi-Vamos, F.; Timar, J.; Reynolds, A.R.; Dome, B. The evidence for and against different modes of tumour cell extravasation in the lung: Diapedesis, capillary destruction, necroptosis, and endothelialization. J. Pathol. 2017, 241, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Bolik, J.; Krause, F.; Stevanovic, M.; Gandraß, M.; Thomsen, I.; Schacht, S.S.; Rieser, E.; Müller, M.; Schumacher, N.; Fritsch, J.; et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J. Exp. Med. 2022, 219, e20201039. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Coll, R.C.; Schroder, K.; Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef]
- Jin, X.; Fu, W.; Zhou, J.; Shuai, N.; Yang, Y.; Wang, B. Oxymatrine attenuates oxidized low-density lipoprotein-induced HUVEC injury by inhibiting NLRP3 inflammasome-mediated pyroptosis via the activation of the SIRT1/Nrf2 signaling pathway. Int. J. Mol. Med. 2021, 48, 187. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, L.; Guo, X.; Liu, J.; Xu, L.; Li, Y. Exogenous spermine inhibits high glucose/oxidized LDL-induced oxidative stress and macrophage pyroptosis by activating the Nrf2 pathway. Exp. Ther. Med. 2022, 23, 310. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Das, B.; Sarkar, C.; Rawat, V.S.; Kalita, D.; Deka, S.; Agnihotri, A. Promise of the NLRP3 Inflammasome Inhibitors in In Vivo Disease Models. Molecules 2021, 26, 4996. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Qian, Z.; Zhao, Y.; Wan, C.; Deng, Y.; Zhuang, Y.; Xu, Y.; Zhu, Y.; Lu, S.; Bao, Z. Pyroptosis in the Initiation and Progression of Atherosclerosis. Front. Pharmacol. 2021, 12, 652963. [Google Scholar] [CrossRef]
- Lin, X.; Ouyang, S.; Zhi, C.; Li, P.; Tan, X.; Ma, W.; Yu, J.; Peng, T.; Chen, X.; Li, L.; et al. Focus on ferroptosis, pyroptosis, apoptosis and autophagy of vascular endothelial cells to the strategic targets for the treatment of atherosclerosis. Arch. Biochem. Biophys. 2022, 715, 109098. [Google Scholar] [CrossRef]
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Liu, J.; Chen, X.; Zhang, L.; Pi, J.; Sun, H.; Li, L.; Bauer, R.; Wang, H.; Yu, Z.; et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ. Res. 2019, 125, 590–605. [Google Scholar] [CrossRef]
- Xu, X.; Yang, Y.; Wang, G.; Yin, Y.; Han, S.; Zheng, D.; Zhou, S.; Zhao, Y.; Chen, Y.; Jin, Y. Low shear stress regulates vascular endothelial cell pyroptosis through miR-181b-5p/STAT-3 axis. J. Cell. Physiol. 2021, 236, 318–327. [Google Scholar] [CrossRef]
- Puylaert, P.; Van Praet, M.; Vaes, F.; Neutel, C.H.G.; Roth, L.; Guns, P.J.; De Meyer, G.R.Y.; Martinet, W. Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in Apoe Knock-Out Mice. Biomedicines 2022, 10, 1171. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.L. Iron and the sex difference in heart disease risk. Lancet 1981, 1, 1293–1294. [Google Scholar] [CrossRef] [PubMed]
- Kiechl, S.; Willeit, J.; Egger, G.; Poewe, W.; Oberhollenzer, F. Body iron stores and the risk of carotid atherosclerosis: Prospective results from the Bruneck study. Circulation 1997, 96, 3300–3307. [Google Scholar] [CrossRef]
- Meyers, D.G.; Jensen, K.C.; Menitove, J.E. A historical cohort study of the effect of lowering body iron through blood donation on incident cardiac events. Transfusion 2002, 42, 1135–1139. [Google Scholar] [CrossRef]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef]
- Lee, T.S.; Shiao, M.S.; Pan, C.C.; Chau, L.Y. Iron-deficient diet reduces atherosclerotic lesions in Apoe-deficient mice. Circulation 1999, 99, 1222–1229. [Google Scholar] [CrossRef]
- Minqin, R.; Rajendran, R.; Pan, N.; Tan, B.K.; Ong, W.Y.; Watt, F.; Halliwell, B. The iron chelator desferrioxamine inhibits atherosclerotic lesion development and decreases lesion iron concentrations in the cholesterol-fed rabbit. Free Radic. Biol. Med. 2005, 38, 1206–1211. [Google Scholar] [CrossRef]
- Zhang, W.J.; Wei, H.; Frei, B. The iron chelator, desferrioxamine, reduces inflammation and atherosclerotic lesion development in experimental mice. Exp. Biol. Med. 2010, 235, 633–641. [Google Scholar] [CrossRef]
- Yang, K.; Song, H.; Yin, D. PDSS2 Inhibits the Ferroptosis of Vascular Endothelial Cells in Atherosclerosis by Activating Nrf2. J. Cardiovasc. Pharmacol. 2021, 77, 767–776. [Google Scholar] [CrossRef]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Zhang, J.; Chen, X.; Zhang, Z.; Li, Q. Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discov. 2021, 7, 235. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yang, N.; Song, Y.; Si, H.; Qin, Q.; Guo, Z. Effect of iron overload on endothelial cell calcification and its mechanism. Ann. Transl. Med. 2021, 9, 1658. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, M.N.; Osama, M. The implications of neutrophil extracellular traps in the pathophysiology of atherosclerosis and atherothrombosis. Exp. Biol. Med. 2020, 245, 1376–1384. [Google Scholar] [CrossRef]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Nija, R.J.; Sanju, S.; Sidharthan, N.; Mony, U. Extracellular Trap by Blood Cells: Clinical Implications. Tissue Eng. Regen. Med. 2020, 17, 141–153. [Google Scholar] [CrossRef]
- de Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar]
- Döring, Y.; Weber, C.; Soehnlein, O. Footprints of neutrophil extracellular traps as predictors of cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1735–1736. [Google Scholar] [CrossRef]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.; Sausen, G.; Salinas, M.; Masson, G.S.; Cole, A.; Beltrami-Moreira, M.; Chatzizisis, Y.; Quillard, T.; Tesmenitsky, Y.; et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ. Res. 2017, 121, 31–42. [Google Scholar] [CrossRef]










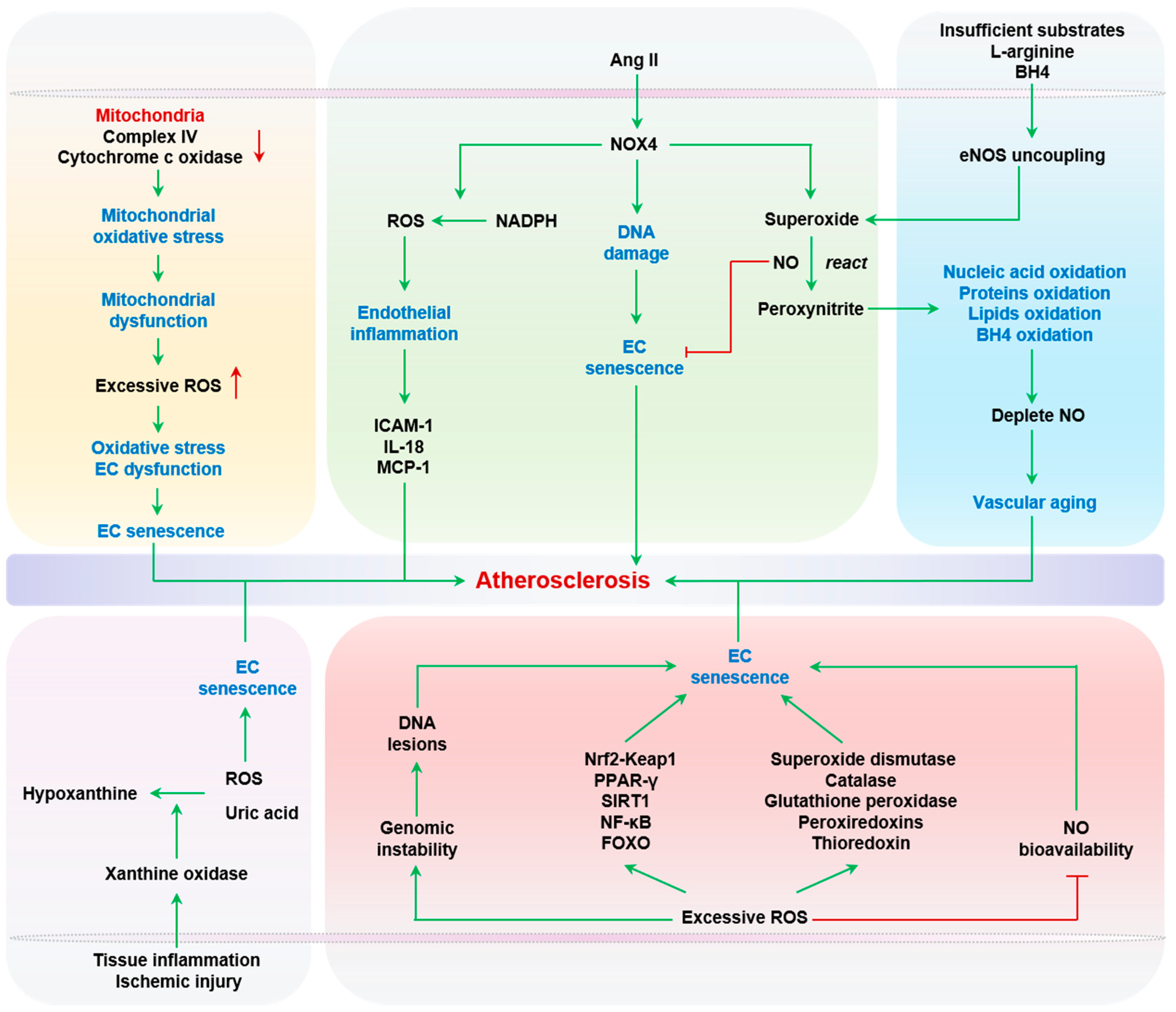{kind=link}
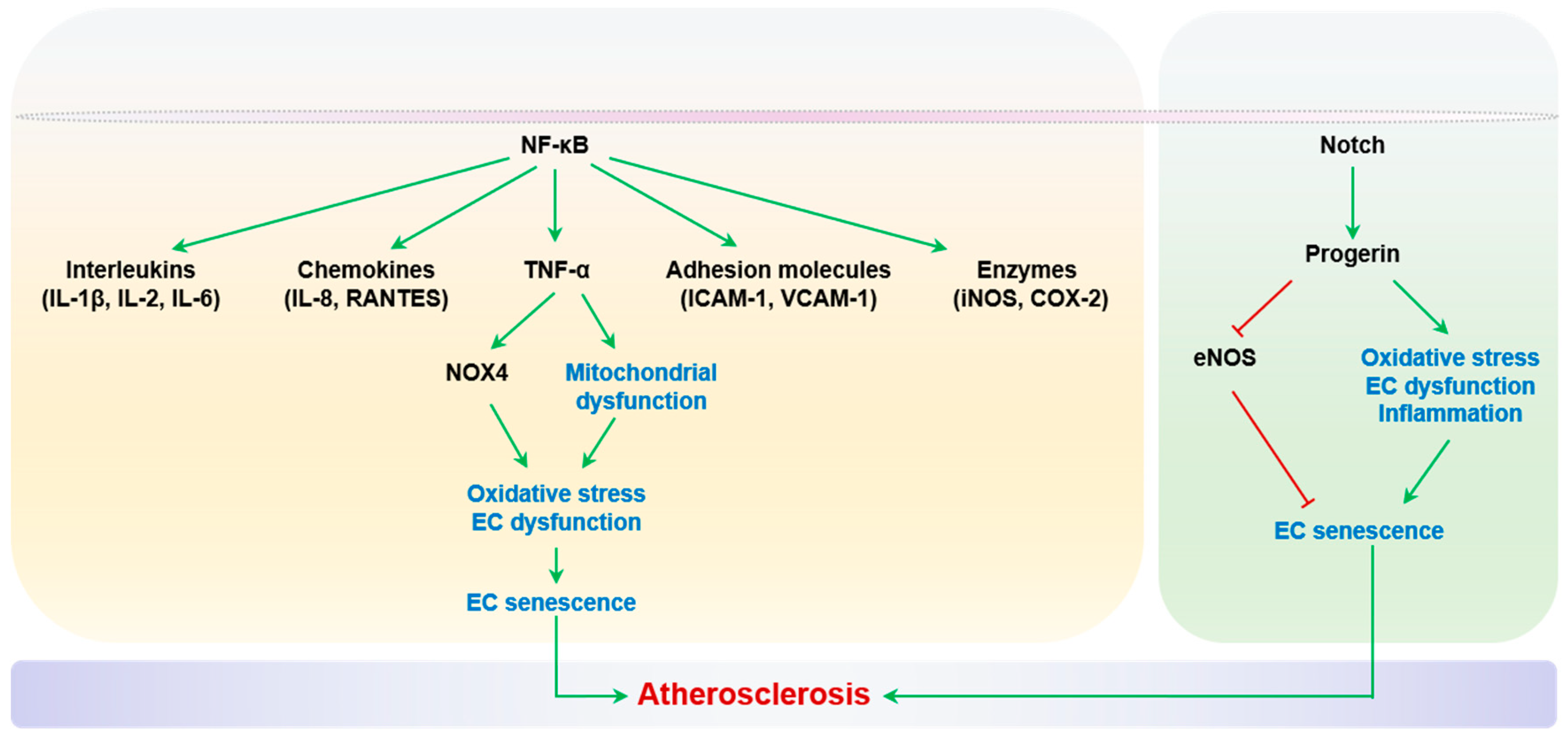{kind=link}
{kind=link}
{kind=link}
{kind=link}
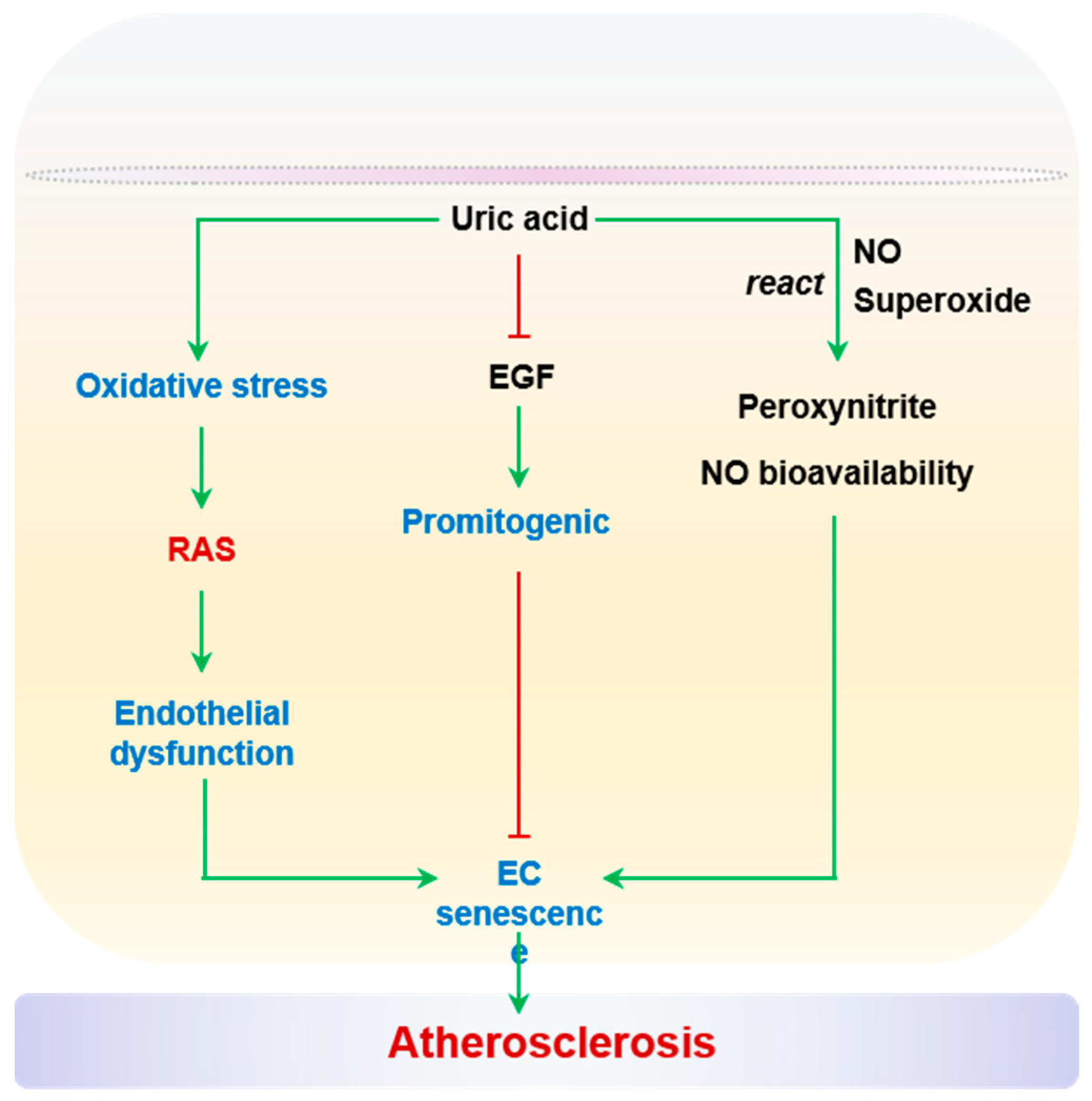{kind=link}
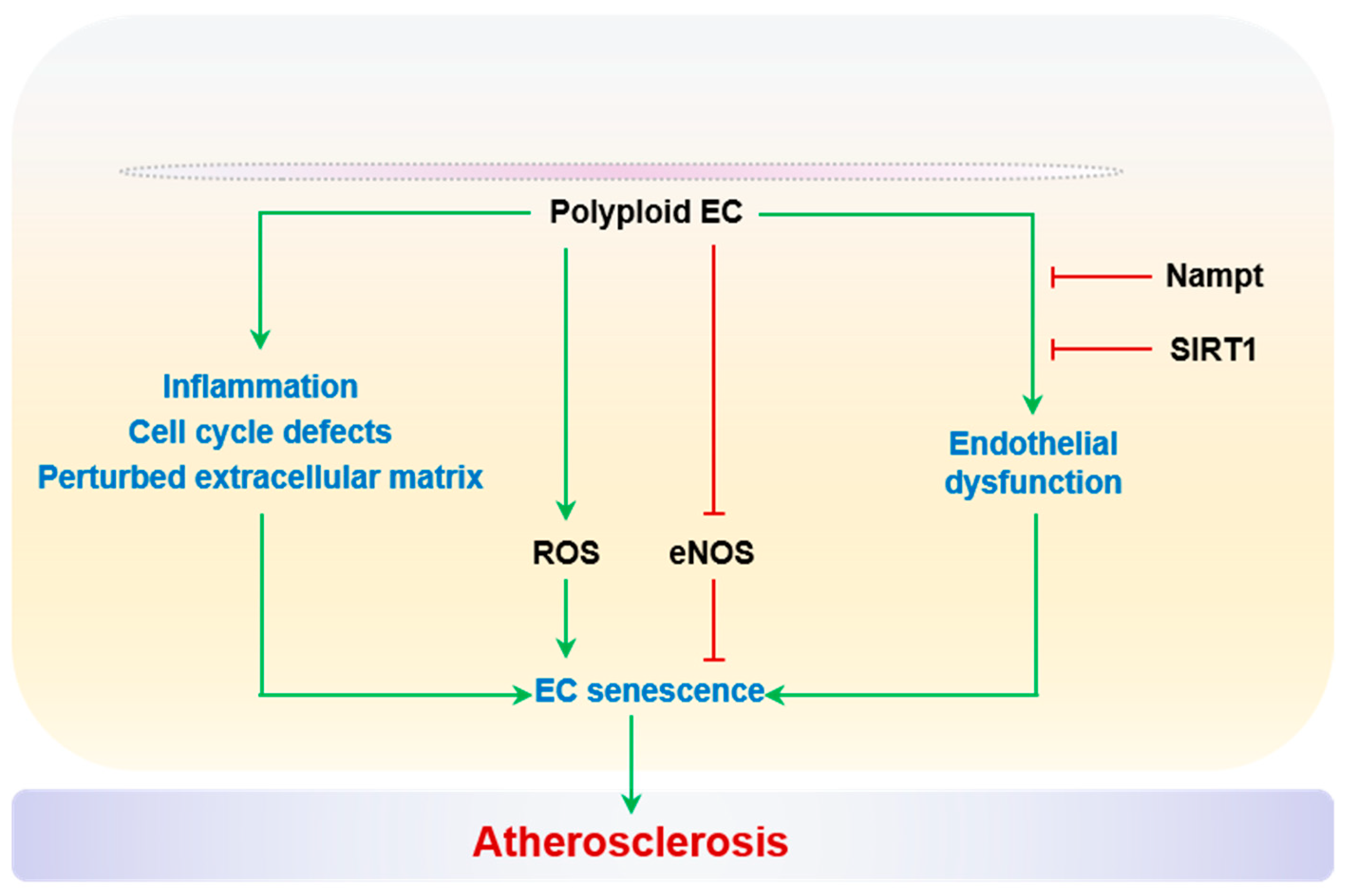{kind=link}
{kind=link}
{kind=link}
| Samples | Indexes | Methods | Advantage | Shortcoming | Ref |
|---|---|---|---|---|---|
| Protein (tissues, cells) | SA-β-gal | SA-β-gal test | Gold standard | Traumatically harvested, time-consuming | [36] |
| Gal-EM | Transmission electron microscopy | Gold standard | Traumatically harvested, expensive instruments | [37] | |
| p16, p21, SAHF | RNAscope Immunohistochemistry | High specificity, quantifiable | Operational complexity, time-consuming | [38] | |
| Lipofuscin | Sudan black B staining | High specificity | Time-consuming | [39] | |
| Serum/Plasma | SASP: IL8/CXCL1/CXCL2/CXCL3 /TNF/IL-1α/IL-1β/ICAM-1/GM-CSF | ELISA Western blot | Mature method, quantifiable | Low specificity | [40] |
| TRX80 | ELISA | Mature method, quantifiable | Low specificity | [41] | |
| TNF-α/IL-1β/IL-6 | ELISA | Mature method, quantifiable | Large sample sizes are required | [42] | |
| Endothelial progenitor cells | Flow cytometry | High specificity, quantifiable | Operational complexity | [43] | |
| DNA | Telomere loss | Southern blot qPCR | High specificity, mature method, quantifiable | Operational complexity, time-consuming | [44] |
| g (POLG) mutation | DNA sequencing | Mature method, quantifiable | Low specificity, expensive | [45] | |
| m.3243A > G | DNA sequencing | Mature method, quantifiable | Low specificity, expensive | [46] | |
| LINE-1 elements | RT-qPCR | Mature method, quantifiable | Low specificity | [47] | |
| γH2AX | Dual isotope SPECT imaging | Quantifiable | Low specificity, expensive | [48] | |
| Non-coding RNA | miR-183/miR-34a | PCR | Mature method, quantifiable | Low specificity | [49] |
| Cell Death | Morphological Features | Biochemical Features | Key Biomarkers | Hallmarks | Related Signaling Molecules and Pathways | |
|---|---|---|---|---|---|---|
| Autophagy | Formation of double-membraned autolysosomes | Increased lysosomal activity | ATG5, ATG7, DRAM3, TFEB | LC3-II/I, autophagic vesicles, autophagic flow | PI3K-AKT-mTOR, MAPK-ERK1/2 pathway | [326,327] |
| Apoptosis | Cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and apoptotic body formation | Activation of caspases, oligonucleosomal DNA fragmentation, modification of nuclear polypeptides | Caspase, p53, Fas, Bcl-2, Bax | Cell morphology, annexin V/PI, mitochondrial membrane potential, cytochrome C | Death receptor, mitochondrial, endoplasmic reticulum pathway, caspase, p53, Bcl-2-mediated pathway | [328,329] |
| Anoikis | Insufficient cell–matrix interactions | Integrins interact with ECM components to form adhesion complexes | Bcl-2, Bcl-XL, Mcl1, Bax, Bak, Bok, Bid, Bik, Bmf, Noxa, Bad, Bim, Puma | Caspases activation, DNA fragmentation | JNK, ERK, PI3K, PKB/AKT pathway, integrin, caveolin-1 | [330,331] |
| Pyroptosis | Apoptosis-like chromatin condensation and DNA fragmentation in the early stage, karyopyknosis, necrosis-like cell membrane pore formation, cellular swelling, membrane rupture | Dependent on caspase-1 and proinflammatory cytokine releases | Caspase-1, IL-1β, IL-18 | Gasdermin D, caspase-4, cell morphology | Caspase-1, NLRP3-mediated pathway | [332,333] |
| NETosis | Enzymes from granules translocate to the nucleus, chromatin de-condensation, internal membranes break down, cytolysis releases NETs | NETs generation | LPS, GM-CSF, IL8, PAD4, HMGB1, TF, MMP9, PMA, TGFβ, MPO, NE, RAGE, Histone, Anti-DsDNA | Promoting the transcription and translation of NE, MPO, and PAD4 | Raf-MEK-ERK, TLR2-fibronectin, caspase-4/5-GSDMD pathway | [334,335] |
| Necroptosis | Rupture of the cellular membrane, progressively translucent cytoplasm and swelling of organelles, moderate chromatin condensation | Drop in ATP levels, Activation of RIP1, RIP3, MLKL | LEF1, RIP1, RIP3 | RIPK1, RIPK3 | TNFα, TNFR1, TLR3, TRAIL, FasL, ROS, PKC-MAPK-AP1-mediated pathway | [336,337] |
| Ferroptosis | Small mitochondria with condensed mitochondrial membrane densities, reduction in or vanishing of mitochondria crista, outer mitochondrial membrane rupture | Inhibition of xCT and reduced GSH, inhibition of GPX4 Iron accumulation, lipid peroxidation | GPX4, Nrf2, LSH, TFR1, xCT | Fe2+, ROS, GSH, lipid peroxidation | MVA, HSF1-HSPB1, p62-Keap1-Nrf2 pathway; LSH pathway | [338,339] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bu, L.-L.; Yuan, H.-H.; Xie, L.-L.; Guo, M.-H.; Liao, D.-F.; Zheng, X.-L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. Int. J. Mol. Sci. 2023, 24, 15160. https://doi.org/10.3390/ijms242015160
Bu L-L, Yuan H-H, Xie L-L, Guo M-H, Liao D-F, Zheng X-L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. International Journal of Molecular Sciences. 2023; 24(20):15160. https://doi.org/10.3390/ijms242015160
Chicago/Turabian StyleBu, Lan-Lan, Huan-Huan Yuan, Ling-Li Xie, Min-Hua Guo, Duan-Fang Liao, and Xi-Long Zheng. 2023. "New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death" International Journal of Molecular Sciences 24, no. 20: 15160. https://doi.org/10.3390/ijms242015160
APA StyleBu, L.-L., Yuan, H.-H., Xie, L.-L., Guo, M.-H., Liao, D.-F., & Zheng, X.-L. (2023). New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. International Journal of Molecular Sciences, 24(20), 15160. https://doi.org/10.3390/ijms242015160