
Exploring TCR-like CAR-Engineered Lymphocyte Cytotoxicity against MAGE-A4
 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Results
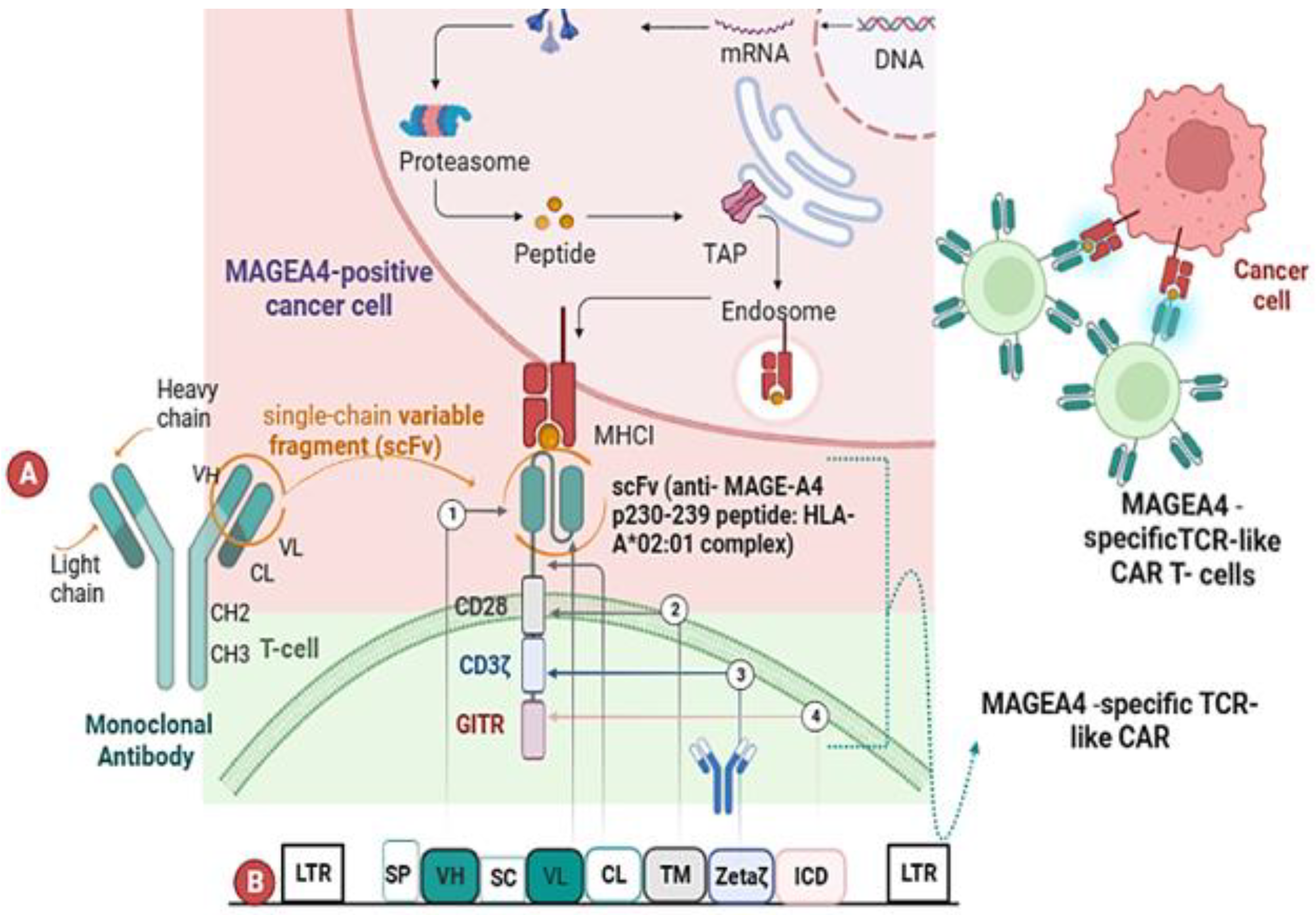
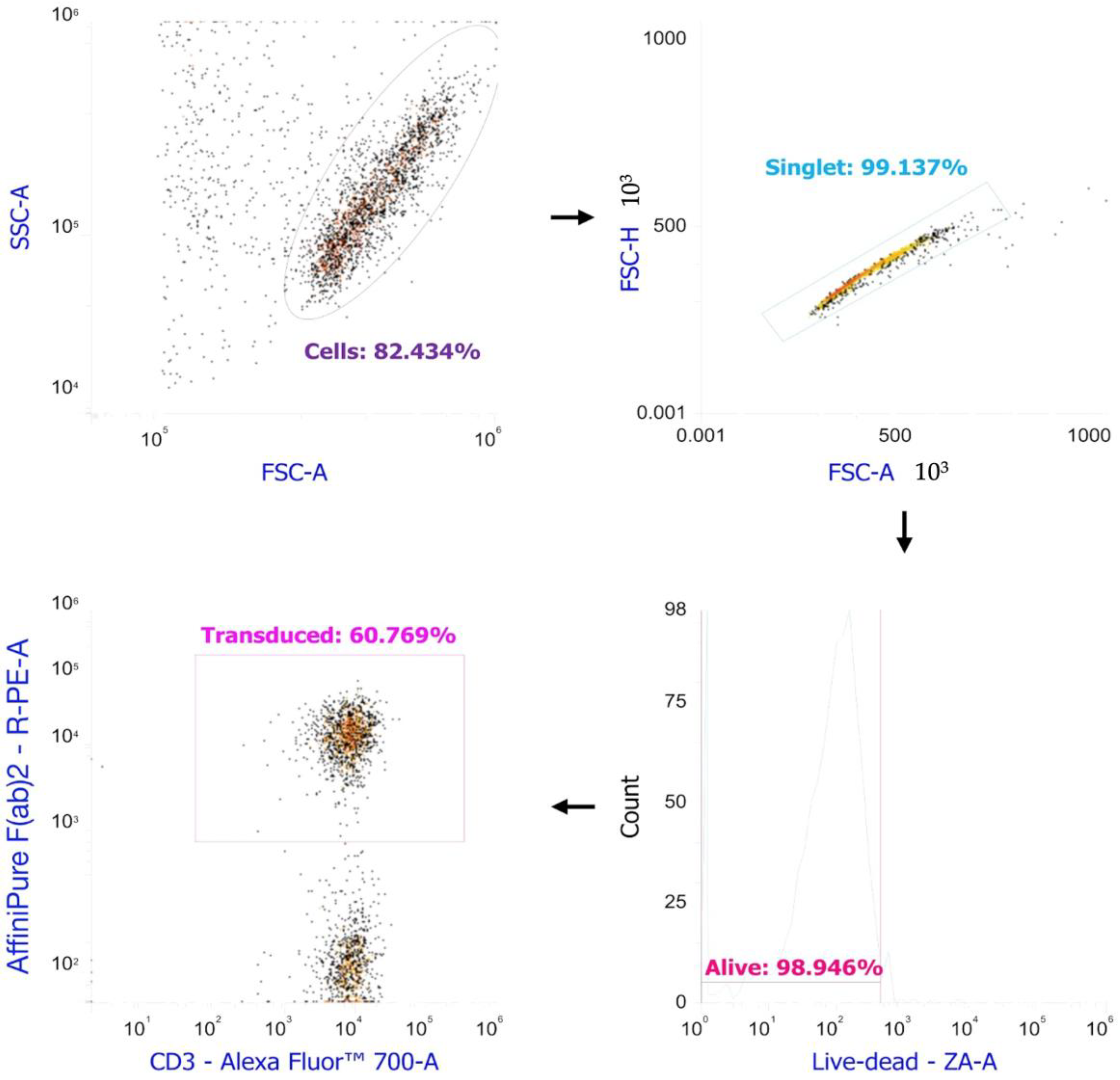
2.1. Anti-MAGE-A4 TCR-like CAR-T Cells Manifest Cytotoxicity Markers
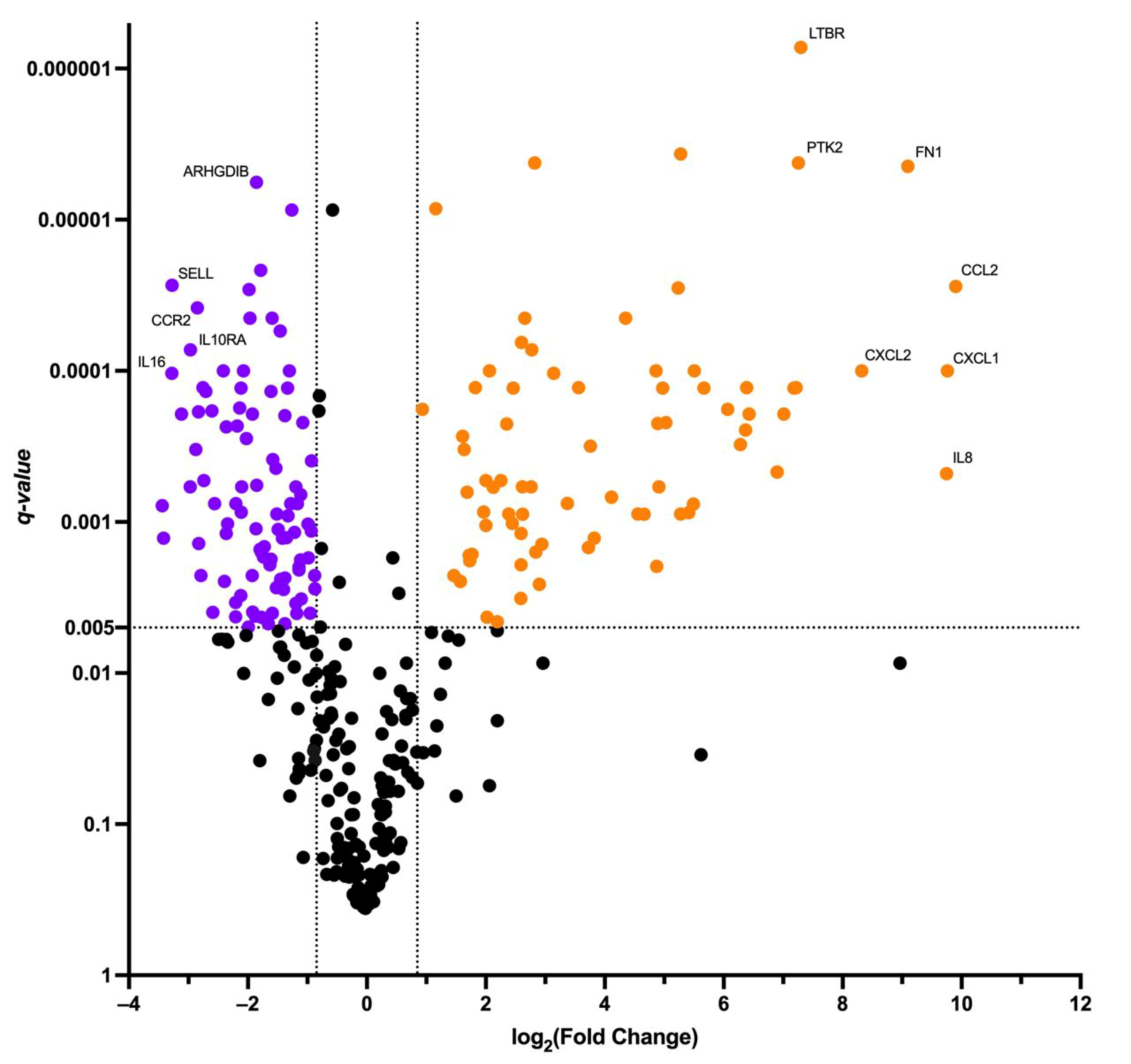
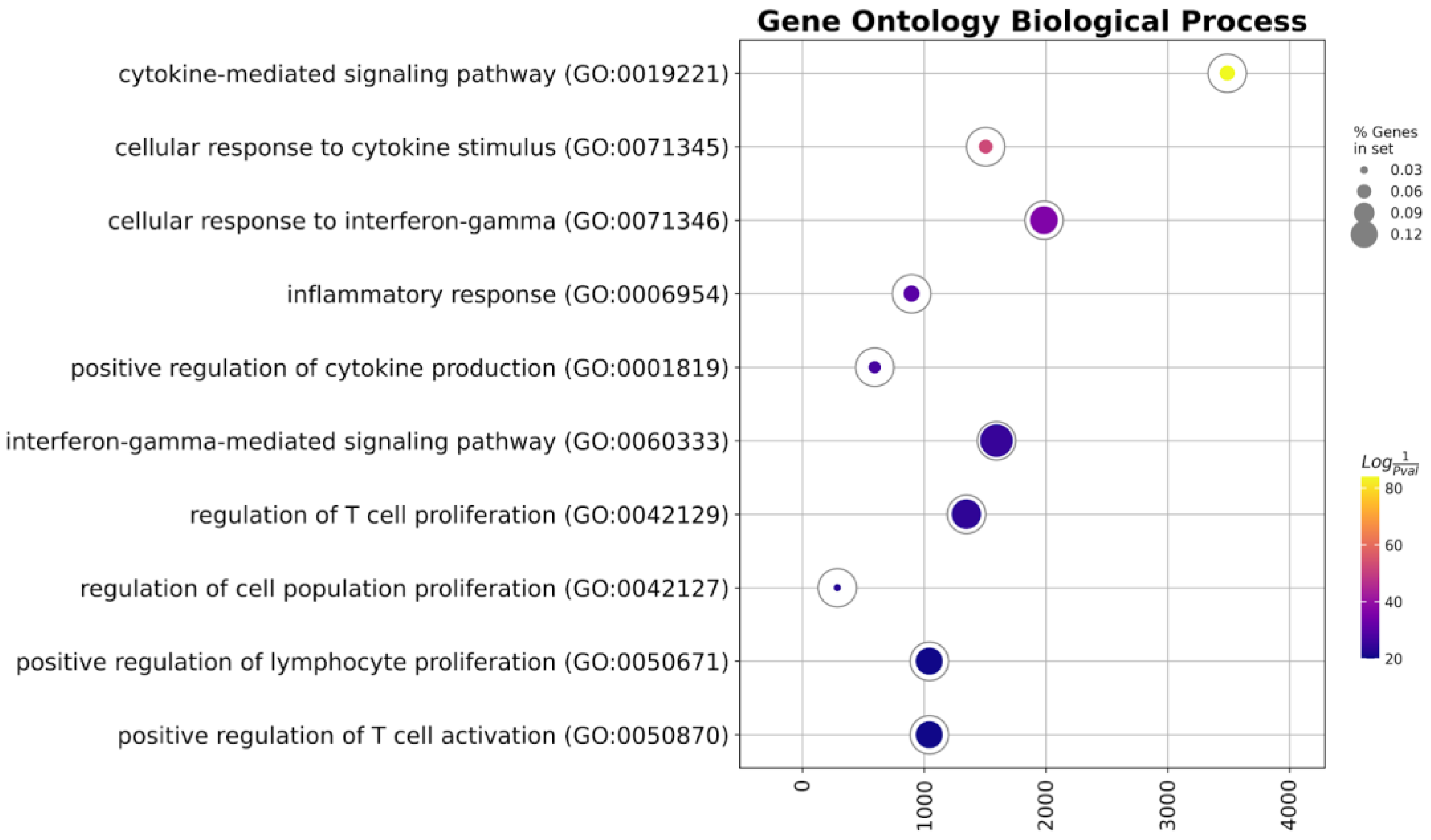
2.2. Transcriptional Profiling Reveals Immunological Features of Generated TCR-like CAR-T Cells
2.3. TCR-like CAR-T Cells Exhibit Antigen-Dependent Activation and Cytotoxicity In Vitro
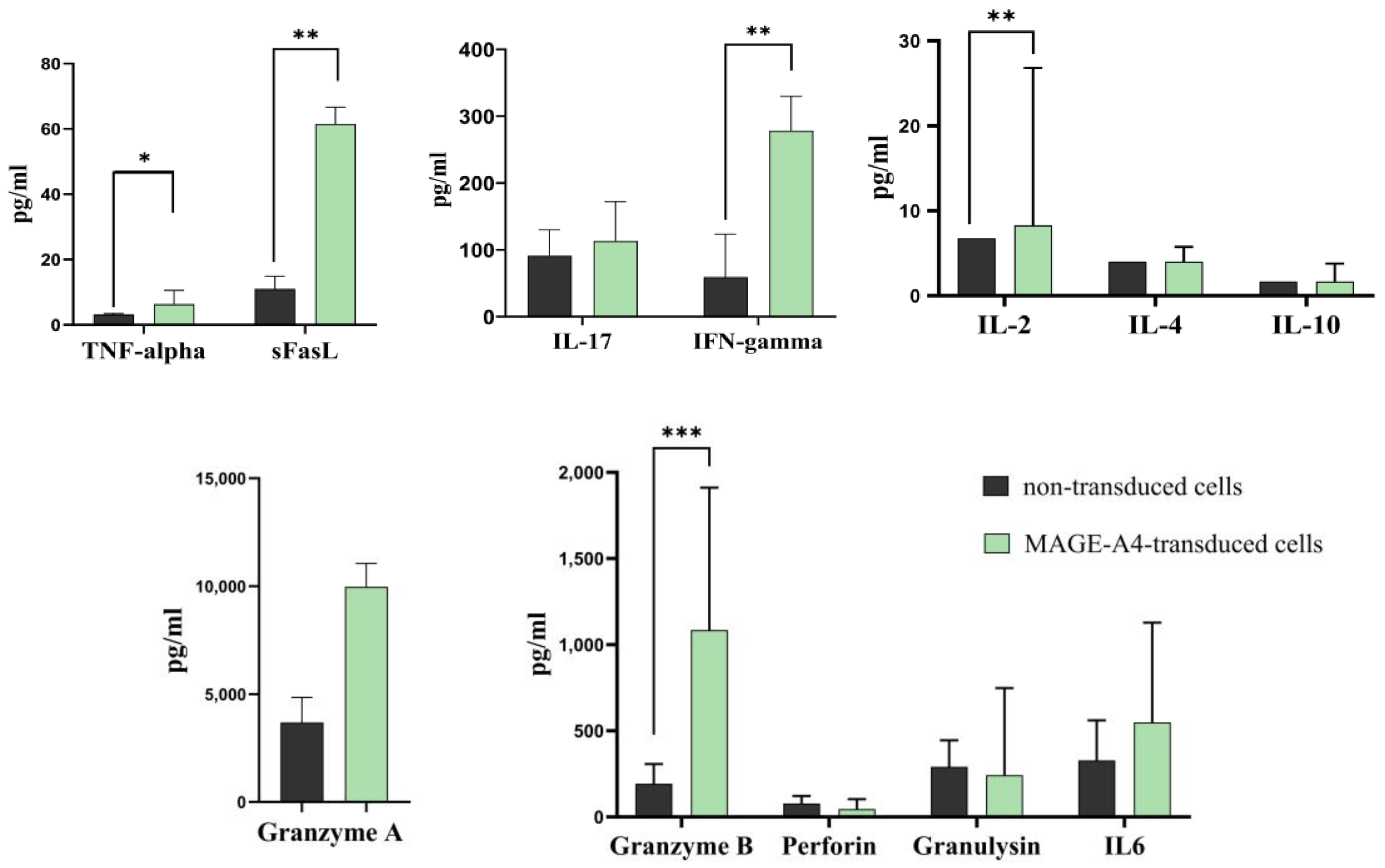
2.4. TCR-like CAR-T Cells Elicit the Secretion of Key Cytokines in Response to Tumor Cells
2.5. Anti-MAGEA4 TCR-like CAR Slows Tumor Growth in Melanoma Model In Vivo
3. Discussion
4. Materials and Methods
4.1. Study Population and Interventions
4.2. PBMC Isolation
4.3. Retroviral Particles
4.4. Induction of T-Cell Proliferation
4.5. Retroviral Transduction of the Anti-CD3 Primed PBMCs
4.6. Evaluation of the Efficiency of Transduction
4.7. Cell Lines
4.8. Phenotyping for Markers of Activation and Cytotoxicity by Flow Cytometry
4.9. HSNE Dimensionality Reduction and Clustering
4.10. Magnetic Separation of Transduced T-Cells after Co-Culturing with Tumor Cells
4.11. Total RNA Extraction
4.12. Gene Expression Profiling by Nanostring
4.13. Differential Gene Expression Testing
4.14. LDH Cytotoxicity Assay
4.15. Quantification of Cytokine Production
4.16. In Vivo Efficacy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rath, J.A.; Arber, C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells 2020, 9, 1485. [Google Scholar] [CrossRef]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Gao, L.; Wen, Q.; Zhong, J.F.; Zhang, C.; et al. Recent Advances in CAR-T Cell Engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef]
- Chodon, T.; Comin-Anduix, B.; Chmielowski, B.; Koya, R.C.; Wu, Z.; Auerbach, M.; Ng, C.; Avramis, E.; Seja, E.; Villanueva, A.; et al. Adoptive Transfer of MART-1 T-Cell Receptor Transgenic Lymphocytes and Dendritic Cell Vaccination in Patients with Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2457–2465. [Google Scholar] [CrossRef]
- Abramson, J.S. Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus. Med. Rev. 2020, 34, 29–33. [Google Scholar] [CrossRef]
- Fagerberg, L.; Jonasson, K.; von Heijne, G.; Uhlén, M.; Berglund, L. Prediction of the Human Membrane Proteome. Proteomics 2010, 10, 1141–1149. [Google Scholar] [CrossRef]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-Specific Chimeric Antigen Receptor-Modified T Cells for the Treatment of Refractory and/or Recurrent Neuroblastoma in Pediatric Patients. J. Cancer Res. Clin. Oncol. 2022, 148, 2643–2652. [Google Scholar] [CrossRef]
- Liu, H.; Lei, W.; Zhang, C.; Yang, C.; Wei, J.; Guo, Q.; Guo, X.; Chen, Z.; Lu, Y.; Young, K.H.; et al. CD19-Specific CAR T Cells That Express a PD-1/CD28 Chimeric Switch-Receptor Are Effective in Patients with PD-L1–Positive B-Cell Lymphoma. Clin. Cancer Res. 2021, 27, 473–484. [Google Scholar] [CrossRef]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I.; et al. Antigen-Independent Activation Enhances the Efficacy of 4-1BB-Costimulated CD22 CAR T Cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef]
- Baulu, E.; Gardet, C.; Chuvin, N.; Depil, S. TCR-Engineered T Cell Therapy in Solid Tumors: State of the Art and Perspectives. Sci. Adv. 2023, 9, eadf3700. [Google Scholar] [CrossRef]
- Ishihara, M.; Nishida, Y.; Kitano, S.; Kawai, A.; Muraoka, D.; Momose, F.; Harada, N.; Miyahara, Y.; Seo, N.; Hattori, H.; et al. A Phase 1 Trial of NY-ESO-1-Specific TCR-Engineered T-Cell Therapy Combined with a Lymph Node-Targeting Nanoparticulate Peptide Vaccine for the Treatment of Advanced Soft Tissue Sarcoma. Int. J. Cancer 2023, 152, 2554–2566. [Google Scholar] [CrossRef]
- Raskin, S.; Van Pelt, S.; Toner, K.; Balakrishnan, P.B.; Dave, H.; Bollard, C.M.; Yvon, E. Novel TCR-like CAR-T Cells Targeting an HLA*0201-Restricted SSX2 Epitope Display Strong Activity against Acute Myeloid Leukemia. Mol. Ther. Methods Clin. Dev. 2021, 23, 296–306. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Xiong, W.; Yin, B.; Huang, Y.; Chu, J.; Xing, C.; Qian, C.; Du, Y.; Duan, T.; et al. Development of a TCR-like Antibody and Chimeric Antigen Receptor against NY-ESO-1/HLA-A2 for Cancer Immunotherapy. J. Immunother. Cancer 2022, 10, e004035. [Google Scholar] [CrossRef]
- Watanabe, K.; Nishikawa, H. Engineering Strategies for Broad Application of TCR-T- and CAR-T-Cell Therapies. Int. Immunol. 2021, 33, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Zajac, P.; Schultz-Thater, E.; Tornillo, L.; Sadowski, C.; Trella, E.; Mengus, C.; Iezzi, G.; Spagnoli, G.C. MAGE-A Antigens and Cancer Immunotherapy. Front. Med. 2017, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xu, Z.; Liu, Y.; Wang, X.; Liu, X.; Gao, Y.; Jin, Y. The Expression of Cancer-Testis Antigen in Ovarian Cancer and the Development of Immunotherapy. Am. J. Cancer Res. 2022, 12, 681–694. [Google Scholar]
- Zhang, Y.; Zhang, Y.; Zhang, L. Expression of Cancer-Testis Antigens in Esophageal Cancer and Their Progress in Immunotherapy. J. Cancer Res. Clin. Oncol. 2019, 145, 281–291. [Google Scholar] [CrossRef]
- Alsalloum, A.; Shevchenko, J.A.; Sennikov, S. The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy? Cancers 2023, 15, 1779. [Google Scholar]
- Golubovskaya, V.M.; Berahovich, R.; Xu, Q.; Zhou, H.; Xu, S.; Guan, J.; Harto, H.; Li, L.; Wu, L. GITR Domain inside CAR Co-Stimulates Activity of CAR-T Cells against Cancer. Front. Biosci. (Landmark Ed.) 2018, 23, 2245–2254. [Google Scholar] [CrossRef]
- He, Y.; Vlaming, M.; van Meerten, T.; Bremer, E. The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers 2022, 14, 299. [Google Scholar] [CrossRef]
- Hauer, J.; Püschner, S.; Ramakrishnan, P.; Simon, U.; Bongers, M.; Federle, C.; Engelmann, H. TNF Receptor (TNFR)-Associated Factor (TRAF) 3 Serves as an Inhibitor of TRAF2/5-Mediated Activation of the Noncanonical NF-KappaB Pathway by TRAF-Binding TNFRs. Proc. Natl. Acad. Sci. USA 2005, 102, 2874–2879. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Wei, W.; Li, Y. TCR Engineered T Cells for Solid Tumor Immunotherapy. Exp. Hematol. Oncol. 2022, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q. Novel Chimeric Antigen Receptor T Cells Based on T-Cell Receptor-like Antibodies. Blood Sci. 2019, 1, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric Antigen Receptor T Cells Persist and Induce Sustained Remissions in Relapsed Refractory Chronic Lymphocytic Leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and Viable Identification of Antigen-Specific CD8+ T Cells by a Flow Cytometric Assay for Degranulation. J. Immunol. Methods 2003, 281, 65–78. [Google Scholar] [CrossRef]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The Role of CD95 and CD95 Ligand in Cancer. Cell Death Differ. 2015, 22, 549–559. [Google Scholar] [CrossRef]
- Hombach, A.A.; Abken, H. Most Do, but Some Do Not: CD4+CD25− T Cells, but Not CD4+CD25+ Treg Cells, Are Cytolytic When Redirected by a Chimeric Antigen Receptor (CAR). Cancers 2017, 9, 112. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From Activation Marker to Metabolic Gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef]
- Holgado, M.P.; Sananez, I.; Raiden, S.; Geffner, J.R.; Arruvito, L. CD32 Ligation Promotes the Activation of CD4(+) T Cells. Front. Immunol. 2018, 9, 2814. [Google Scholar] [CrossRef]
- Hedrick, J.A.; Zlotnik, A. Chemokines and Lymphocyte Biology. Curr. Opin. Immunol. 1996, 8, 343–347. [Google Scholar] [CrossRef]
- Castellino, F.; Huang, A.Y.; Altan-Bonnet, G.; Stoll, S.; Scheinecker, C.; Germain, R.N. Chemokines Enhance Immunity by Guiding Naive CD8+ T Cells to Sites of CD4+ T Cell–Dendritic Cell Interaction. Nature 2006, 440, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.H.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 411–433. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.M.; Juedes, A.; Szabo, S.J.; von Herrath, M.; Glimcher, L.H. Antigen-Driven Effector CD8 T Cell Function Regulated by T-Bet. Proc. Natl. Acad. Sci. USA 2003, 100, 15818–15823. [Google Scholar] [CrossRef]
- Townsend, M.J.; Weinmann, A.S.; Matsuda, J.L.; Salomon, R.; Farnham, P.J.; Biron, C.A.; Gapin, L.; Glimcher, L.H. T-Bet Regulates the Terminal Maturation and Homeostasis of NK and Valpha14i NKT Cells. Immunity 2004, 20, 477–494. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Huang, A.C.; Weinmann, A.S. The Lineage-Defining Factors T-Bet and Bcl-6 Collaborate to Regulate Th1 Gene Expression Patterns. J. Exp. Med. 2011, 208, 1001–1013. [Google Scholar] [CrossRef]
- Ichii, H.; Sakamoto, A.; Kuroda, Y.; Tokuhisa, T. Bcl6 Acts as an Amplifier for the Generation and Proliferative Capacity of Central Memory CD8+ T Cells. J. Immunol. 2004, 173, 883–891. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, Y.; Tang, S.; Zhou, L.; Huang, C.; Cao, Y.; Huang, H.; Wu, X.; Meng, D.; Ye, L.; et al. Cutting Edge: Transcription Factor BCL6 Is Required for the Generation, but Not Maintenance, of Memory CD8+ T Cells in Acute Viral Infection. J. Immunol. 2019, 203, 323–327. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Levings, M.K.; Bacchetta, R.; Schulz, U.; Roncarolo, M.G. The Role of IL-10 and TGF-Beta in the Differentiation and Effector Function of T Regulatory Cells. Int. Arch. Allergy Immunol. 2002, 129, 263–276. [Google Scholar] [CrossRef]
- Sad, S.; Mosmann, T.R. Interleukin (IL) 4, in the Absence of Antigen Stimulation, Induces an Anergy-like State in Differentiated CD8+ TC1 Cells: Loss of IL-2 Synthesis and Autonomous Proliferation but Retention of Cytotoxicity and Synthesis of Other Cytokines. J. Exp. Med. 1995, 182, 1505–1515. [Google Scholar] [CrossRef]
- de Waal Malefyt, R.; Haanen, J.; Spits, H.; Roncarolo, M.G.; te Velde, A.; Figdor, C.; Johnson, K.; Kastelein, R.; Yssel, H.; de Vries, J.E. Interleukin 10 (IL-10) and Viral IL-10 Strongly Reduce Antigen-Specific Human T Cell Proliferation by Diminishing the Antigen-Presenting Capacity of Monocytes via Downregulation of Class II Major Histocompatibility Complex Expression. J. Exp. Med. 1991, 174, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, Z.; Kloepper, J.; Ren, J.; Tay, R.E.; Kazer, S.W.; Kiner, E.; Krishnan, S.; Posada, J.M.; Ghosh, M.; Mamessier, E.; et al. Targeting Treg Cells with GITR Activation Alleviates Resistance to Immunotherapy in Murine Glioblastomas. Nat. Commun. 2021, 12, 2582. [Google Scholar] [CrossRef] [PubMed]
- Meiraz, A.; Garber, O.G.; Harari, S.; Hassin, D.; Berke, G. Switch from Perforin-Expressing to Perforin-Deficient CD8(+) T Cells Accounts for Two Distinct Types of Effector Cytotoxic T Lymphocytes In Vivo. Immunology 2009, 128, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Hassin, D.; Garber, O.G.; Meiraz, A.; Schiffenbauer, Y.S.; Berke, G. Cytotoxic T Lymphocyte Perforin and Fas Ligand Working in Concert Even When Fas Ligand Lytic Action Is Still Not Detectable. Immunology 2011, 133, 190–196. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Mechanisms of Granule-Dependent Killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef]
- Mareeva, T.; Martinez-Hackert, E.; Sykulev, Y. How a T Cell Receptor-like Antibody Recognizes Major Histocompatibility Complex-Bound Peptide. J. Biol. Chem. 2008, 283, 29053–29059. [Google Scholar] [CrossRef]
- Maruta, M.; Ochi, T.; Tanimoto, K.; Asai, H.; Saitou, T.; Fujiwara, H.; Imamura, T.; Takenaka, K.; Yasukawa, M. Direct Comparison of Target-Reactivity and Cross-Reactivity Induced by CAR- and BiTE-Redirected T Cells for the Development of Antibody-Based T-Cell Therapy. Sci. Rep. 2019, 9, 13293. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Abila, B.; Mostafa Kamel, Y. CAR-T: What Is Next? Cancers 2023, 15, 663. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-Dose Decitabine Priming Endows CAR T Cells with Enhanced and Persistent Antitumour Potential via Epigenetic Reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef]
- Rotte, A.; Frigault, M.J.; Ansari, A.; Gliner, B.; Heery, C.; Shah, B. Dose-Response Correlation for CAR-T Cells: A Systematic Review of Clinical Studies. J. Immunother. Cancer 2022, 10, e005678. [Google Scholar] [CrossRef] [PubMed]
- Melsen, J.E.; van Ostaijen-Ten Dam, M.M.; Lankester, A.C.; Schilham, M.W.; van den Akker, E.B. A Comprehensive Workflow for Applying Single-Cell Clustering and Pseudotime Analysis to Flow Cytometry Data. J. Immunol. 2020, 205, 864–871. [Google Scholar] [CrossRef]
- Höllt, T.; Pezzotti, N.; van Unen, V.; Koning, F.; Eisemann, E.; Lelieveldt, B.; Vilanova, A. Cytosplore: Interactive Immune Cell Phenotyping for Large Single-Cell Datasets. Comput. Graph. Forum 2016, 35, 171–180. [Google Scholar] [CrossRef]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A Comprehensive Package for Performing Gene Set Enrichment Analysis in Python. Bioinformatics 2023, 39, btac757. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Overlap | q-Value | Combined Score | Genes |
|---|---|---|---|---|
| Cytokine-mediated signaling pathway | 40/621 | 0.0 | 3489.629 | IL22, CIITA, CSF2, CSF1, CXCL1, IL1RAP, CXCL2, ICAM1, SOCS3, PSMB7, PSMB5, IRAK2, CCL4, CCL3, CCL2, NCAM1, HLA-DPA1, IL13RA1, EGR1, CCL20, TNFRSF9, IL13, PRKCD, LIF, FN1, IL2, IL3, IL1A, CXCL10, CXCL11, IL6, BCL6, IFNG, IL1B, HLA-DPB1, HLA-DRA, LTBR, IL6ST, HLA-DRB3, HLA-DRB1 |
| Cellular response to cytokine stimulus | 28/482 | 0.0 | 1506.004 | IL22, CSF2, CSF1, CXCL1, IL1RAP, CXCL2, ICAM1, SOCS3, IRAK2, CCL4, CCL3, CCL2, HLA-DPA1, IL13RA1, EGR1, CCL20, IL13, LIF, FN1, IL2, IL3, IL1A, CXCL10, IL6, BCL6, IL1B, IL6ST, TLR2 |
| Cellular response to interferon-gamma | 15/121 | 0.0 | 1984.336 | CIITA, CCL20, PRKCD, ICAM1, IFNG, CCL4, HLA-DPB1, CCL3, HLA-DRA, CCL2, NCAM1, HLA-DRB3, HLA-DRB1, TLR2, HLA-DPA1 |
| Inflammatory response | 16/230 | 0.0 | 896.4251 | IL22, CIITA, CEBPB, CCL20, CXCL1, CXCL2, IL1A, CXCL10, CXCL11, IL6, IFNG, IL1B, CCL4, CCL3, CCL2, TLR2 |
| Positive regulation of cytokine production | 17/335 | 0.0 | 594.1125 | APP, EGR1, CEBPB, CSF2, IL13, IL2, C3, IL1A, IL6, IFNG, IL1B, HLA-DPB1, CTNNB1, IL6ST, TLR2, CD276, HLA-DPA1 |
| Interferon-gamma-mediated signaling pathway | 10/68 | 0.0 | 1594.699 | CIITA, IFNG, PRKCD, HLA-DPB1, HLA-DRA, NCAM1, HLA-DRB3, HLA-DRB1, ICAM1, HLA-DPA1 |
| Regulation of T-cell proliferation | 10/76 | 0.0 | 1346.906 | IL6, CEBPB, HLA-DMB, IL1B, HLA-DPB1, IL6ST, HLA-DRB1, IDO1, CD276, HLA-DPA1 |
| Regulation of cell population proliferation | 21/764 | 0.0 | 287.0463 | APP, CSF2, CSF1, TNFRSF9, LIF, FN1, CXCL1, IL2, PTK2, IL3, IL1A, CXCL10, BST1, CXCL11, IL6, BCL6, IFNG, IL1B, CTNNB1, IL6ST, IL13RA1 |
| Positive regulation of lymphocyte proliferation | 9/75 | 0.0 | 1042.675 | BST1, IL6, HLA-DMB, IL1B, HLA-DPB1, IL6ST, IL2, HLA-DPA1, CD276 |
| Positive regulation of T-cell activation | 9/75 | 0.0 | 1042.675 | IL6, HLA-DMB, IL1B, HLA-DPB1, CCL2, THY1, IL6ST, HLA-DPA1, CD276 |
| Parameters Median [Q1–Q3] | Control Group Cell (n = 6) | MAGEA4-Transduced T-Cell (n = 6) |
|---|---|---|
| Granzyme A | 3681 [2200–4842] | 9973 [4056–11,047] |
| Granzyme B | 140.2 [131.2–233.6] | 743.1 *** [699.2–1011] |
| Granulysin | 336.9 [126.6–383.6] | 242.7 [134.6–747.9] |
| Perforin | 67.74 [38.59–113.5] | 46.23 [38.71–103.8] |
| IL-17 | 99.70 [51.88–116.2] | 113.4 [81.46–172.3] |
| IFN-gamma | 17.65 [13.36–20.49] | 278.3 ** [108.2–329.8] |
| TNF-alpha | 3.05 [3.06–3.21] | 6.360 * [3.05–10.57] |
| FasL | 8.70 [6.70–11.2] | 61.50 ** [15.77–66.72] |
| Fas | Not Detected | Not Detected |
| IL-6 | 248.4 [179.4–645.6] | 549.3 [389.3–1128] |
| IL-2 | 6.81 [6.78–6.89] | 8.28 ** [7.08–26.82] |
| IL-4 | 4.03 [4.01–4.05] | 4.03 [4.02–5.74] |
| IL-10 | 1.70 [1.68–1.72] | 1.73 [1.70–3.77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsalloum, A.; Shevchenko, J.; Fisher, M.; Philippova, J.; Perik-Zavodskii, R.; Perik-Zavodskaia, O.; Alrhmoun, S.; Lopatnikova, J.; Vasily, K.; Volynets, M.; et al. Exploring TCR-like CAR-Engineered Lymphocyte Cytotoxicity against MAGE-A4. Int. J. Mol. Sci. 2023, 24, 15134. https://doi.org/10.3390/ijms242015134
Alsalloum A, Shevchenko J, Fisher M, Philippova J, Perik-Zavodskii R, Perik-Zavodskaia O, Alrhmoun S, Lopatnikova J, Vasily K, Volynets M, et al. Exploring TCR-like CAR-Engineered Lymphocyte Cytotoxicity against MAGE-A4. International Journal of Molecular Sciences. 2023; 24(20):15134. https://doi.org/10.3390/ijms242015134
Chicago/Turabian StyleAlsalloum, Alaa, Julia Shevchenko, Marina Fisher, Julia Philippova, Roman Perik-Zavodskii, Olga Perik-Zavodskaia, Saleh Alrhmoun, Julia Lopatnikova, Kurilin Vasily, Marina Volynets, and et al. 2023. "Exploring TCR-like CAR-Engineered Lymphocyte Cytotoxicity against MAGE-A4" International Journal of Molecular Sciences 24, no. 20: 15134. https://doi.org/10.3390/ijms242015134
APA StyleAlsalloum, A., Shevchenko, J., Fisher, M., Philippova, J., Perik-Zavodskii, R., Perik-Zavodskaia, O., Alrhmoun, S., Lopatnikova, J., Vasily, K., Volynets, M., Zavjalov, E., Solovjeva, O., Akahori, Y., Shiku, H., Silkov, A., & Sennikov, S. (2023). Exploring TCR-like CAR-Engineered Lymphocyte Cytotoxicity against MAGE-A4. International Journal of Molecular Sciences, 24(20), 15134. https://doi.org/10.3390/ijms242015134