
Exploring Hydrophilic PD-L1 Radiotracers Utilizing Phosphonic Acids: Insights into Unforeseen Pharmacokinetics



Abstract
:1. Introduction
2. Results
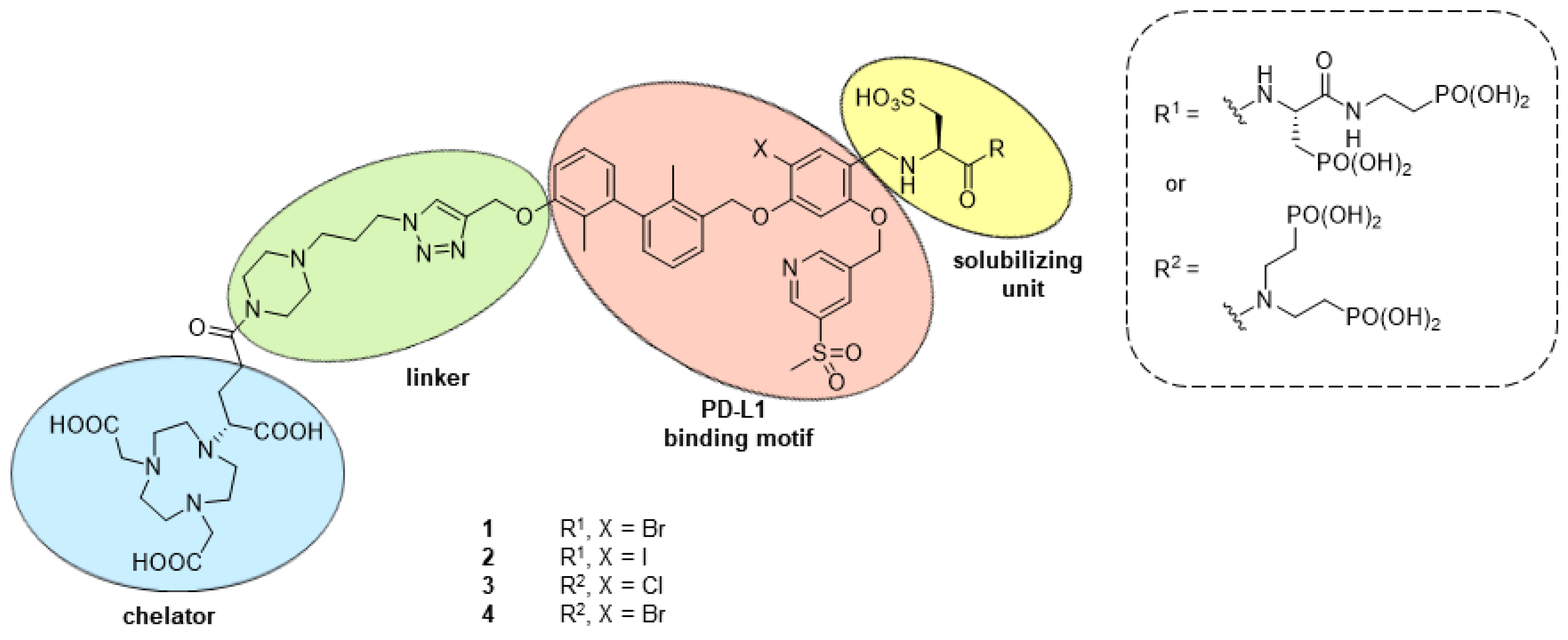
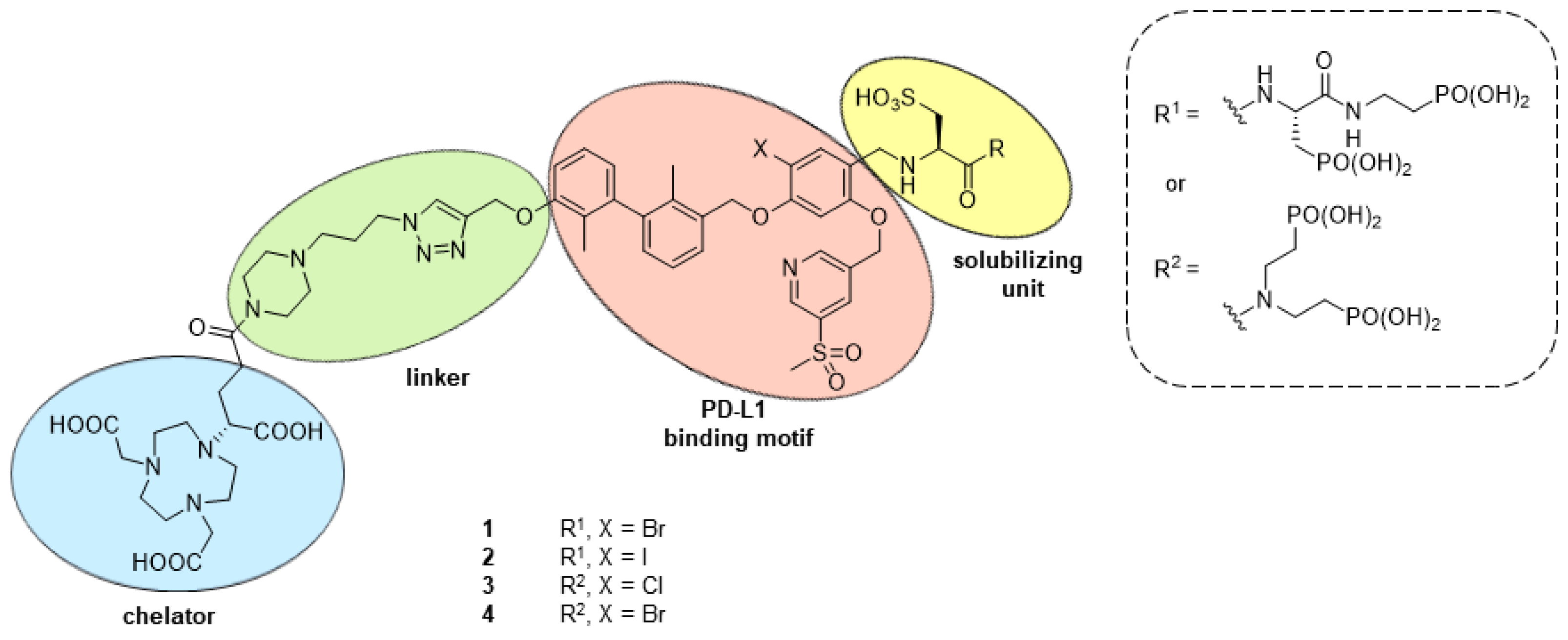
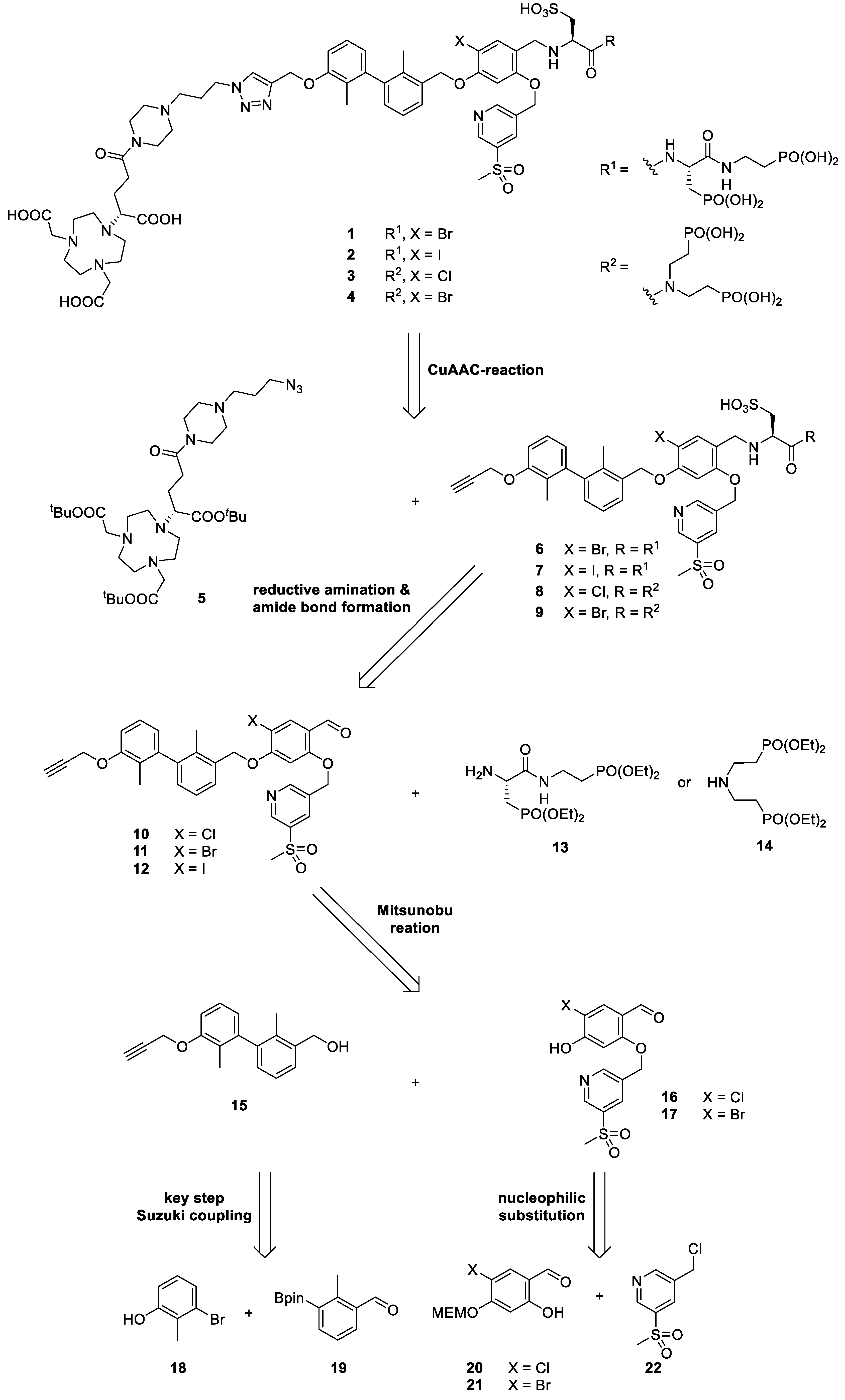
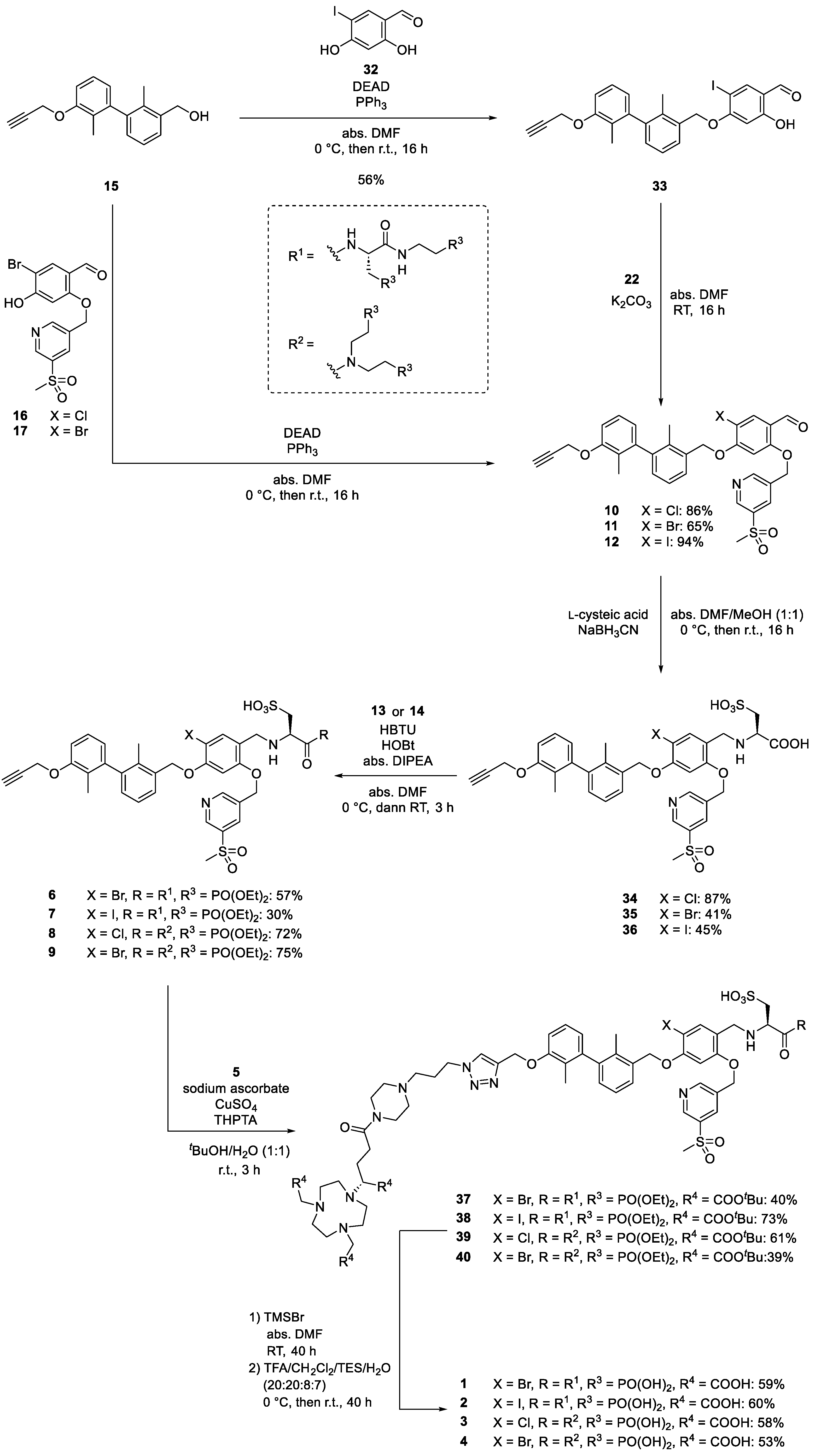
2.1. Design and Synthethic Strategy
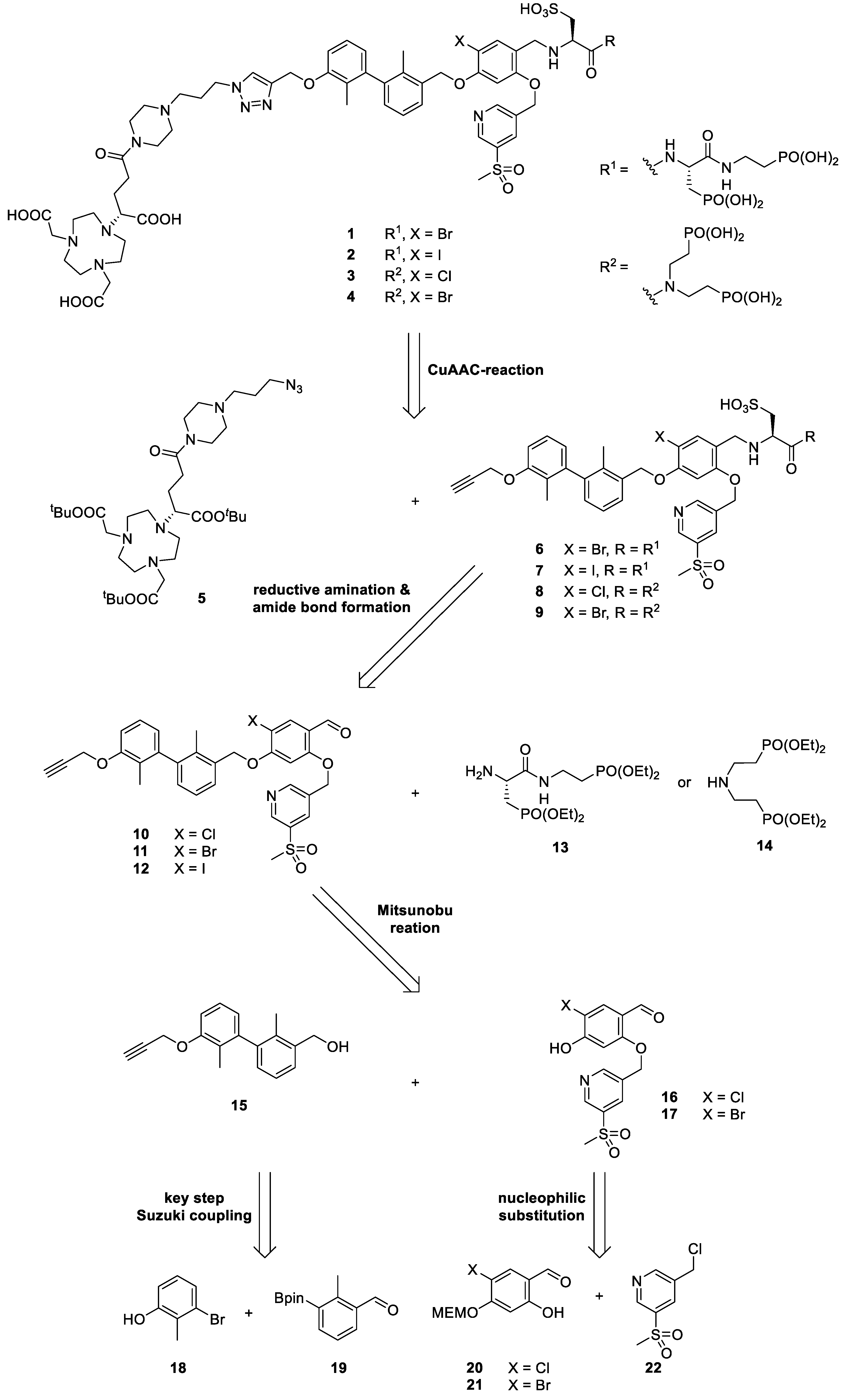
2.2. Retrosynthetical Analysis and Forward Synthesis
2.3. Radiochemistry
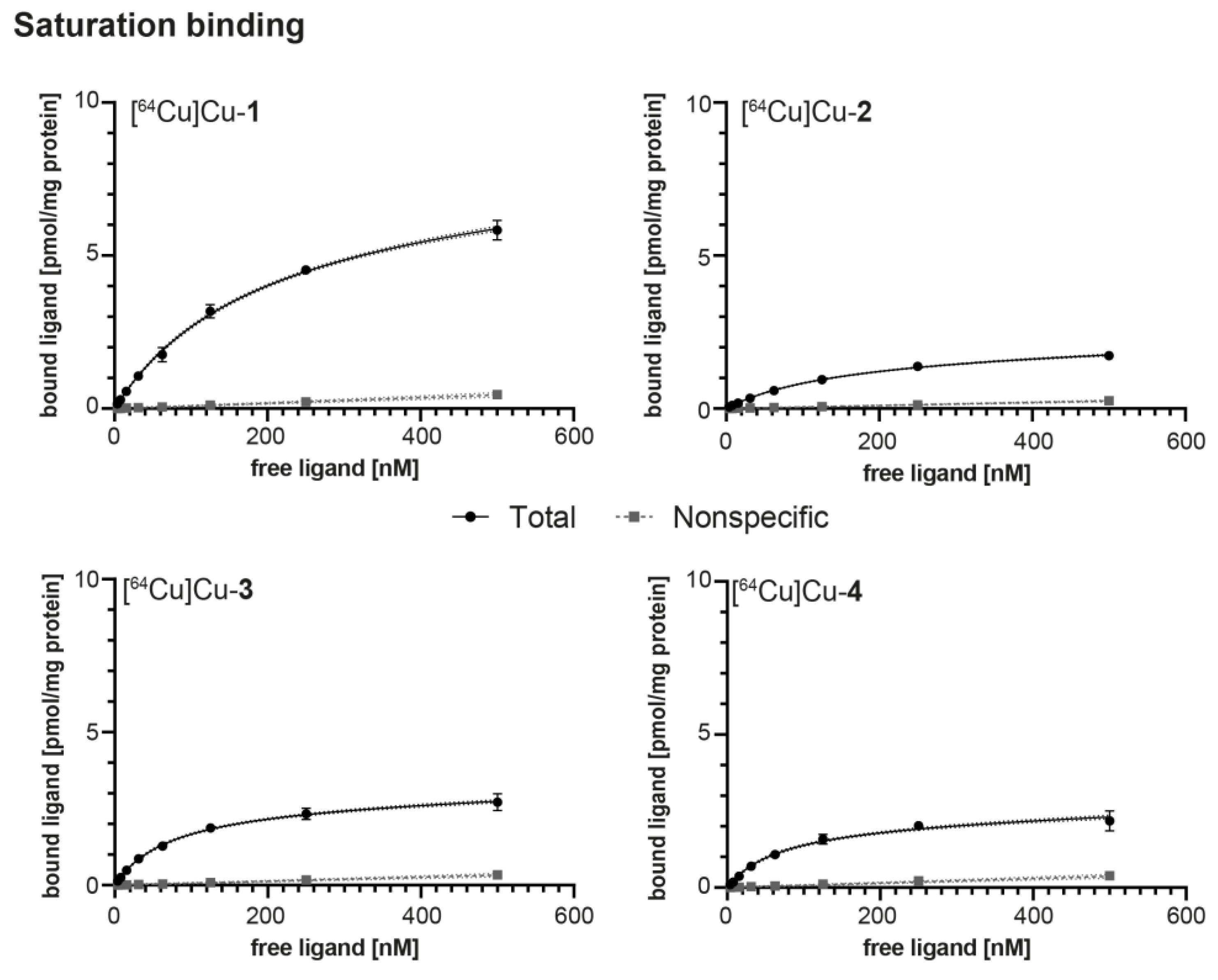
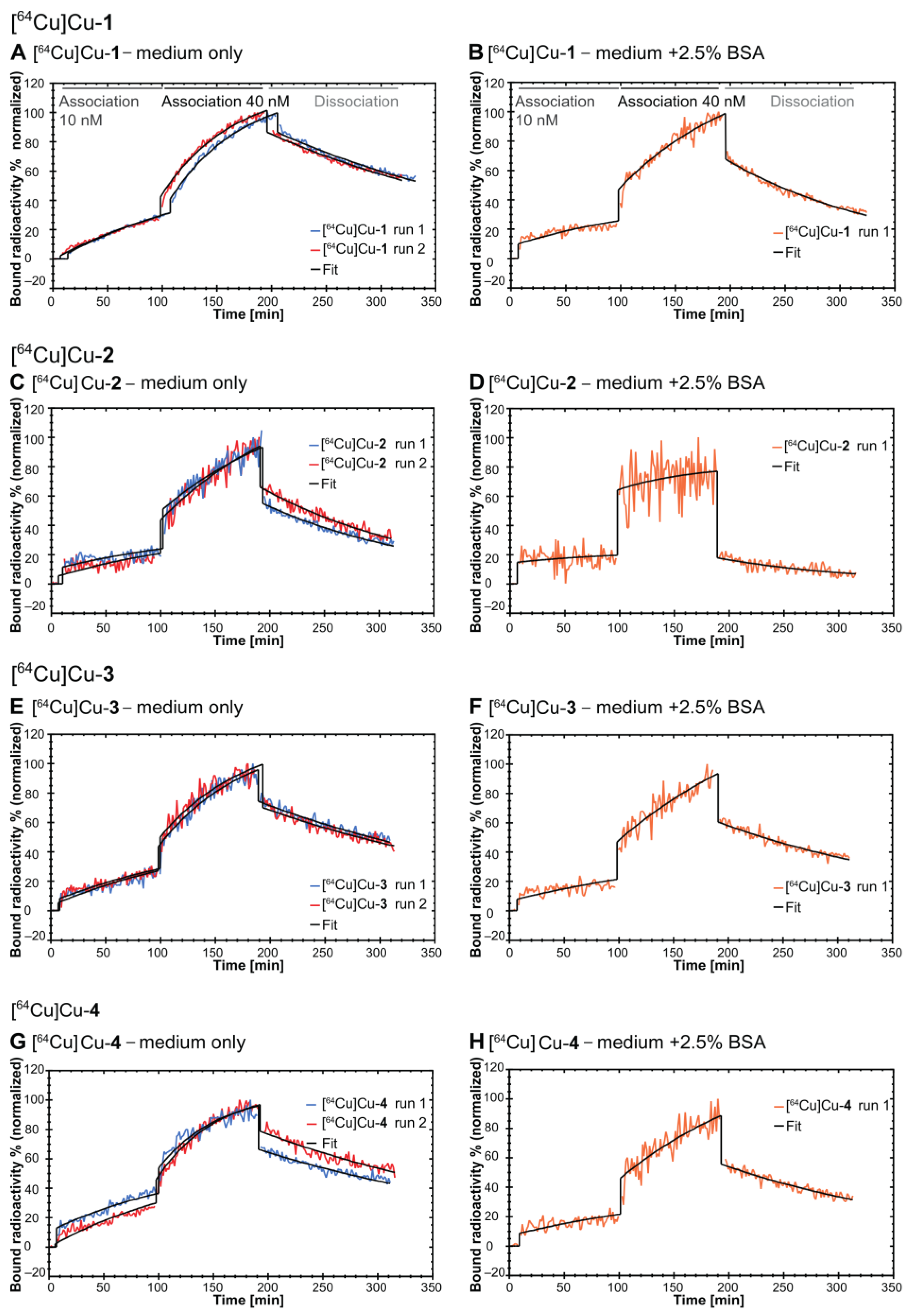
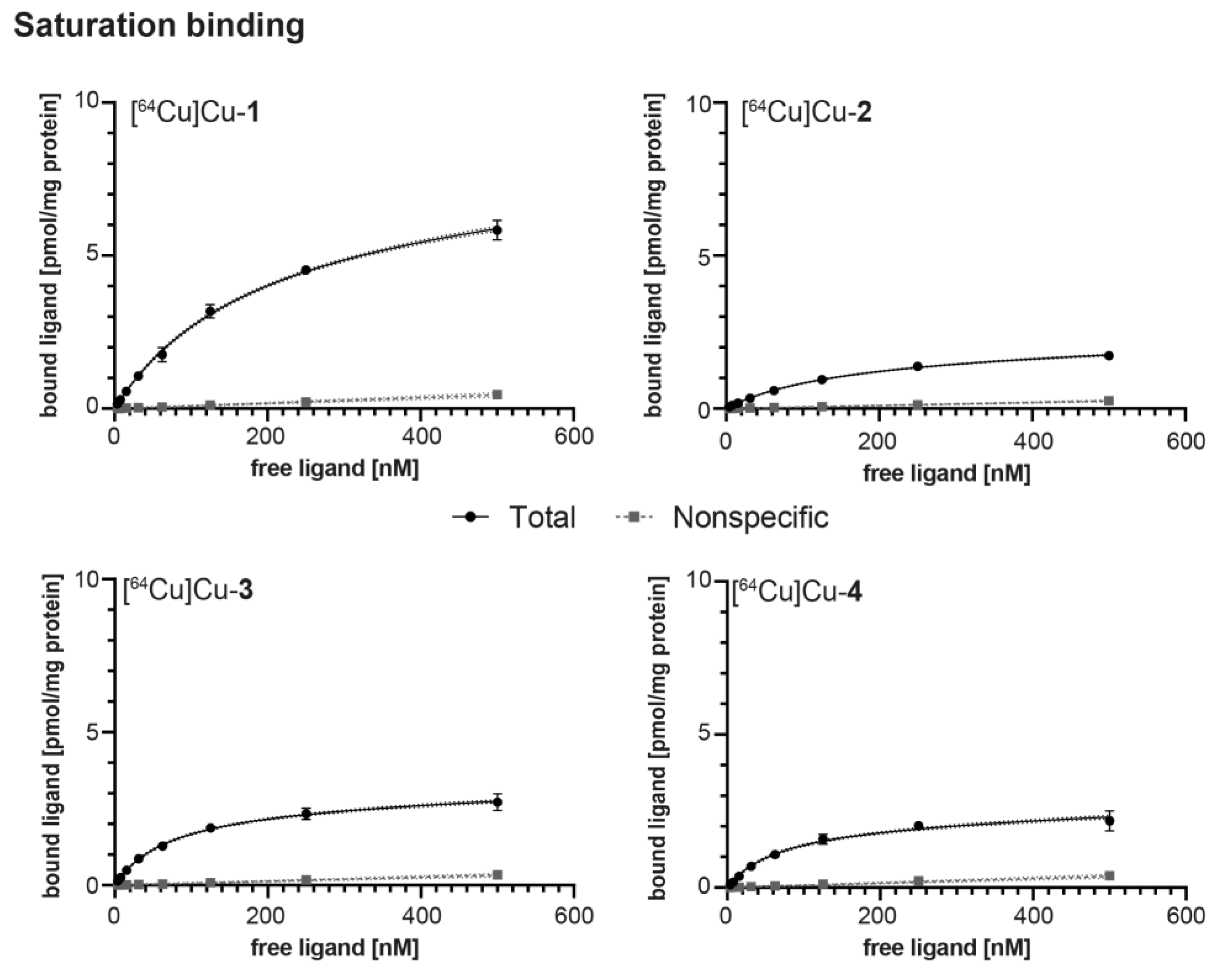
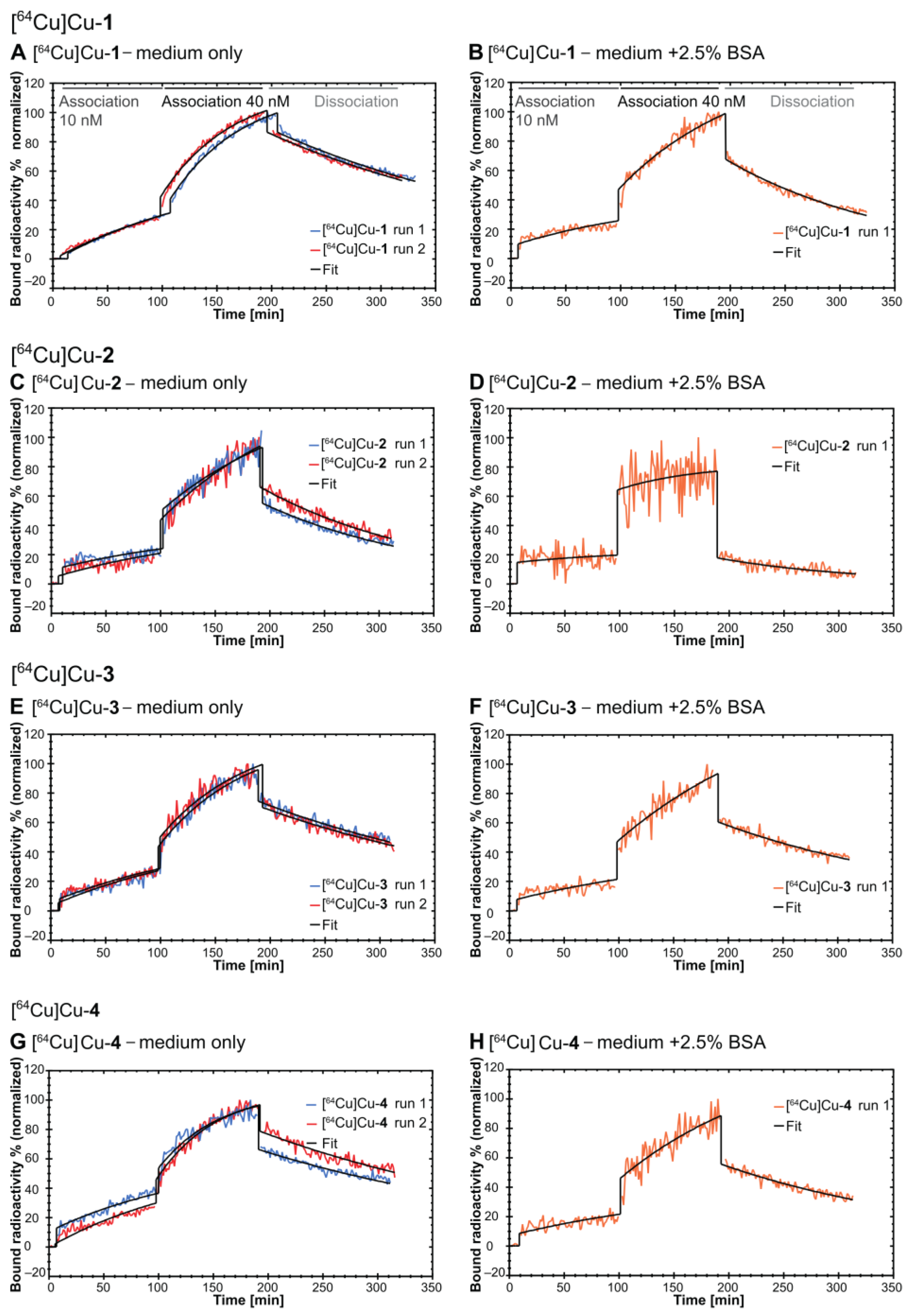
2.4. In Vitro Evaluation
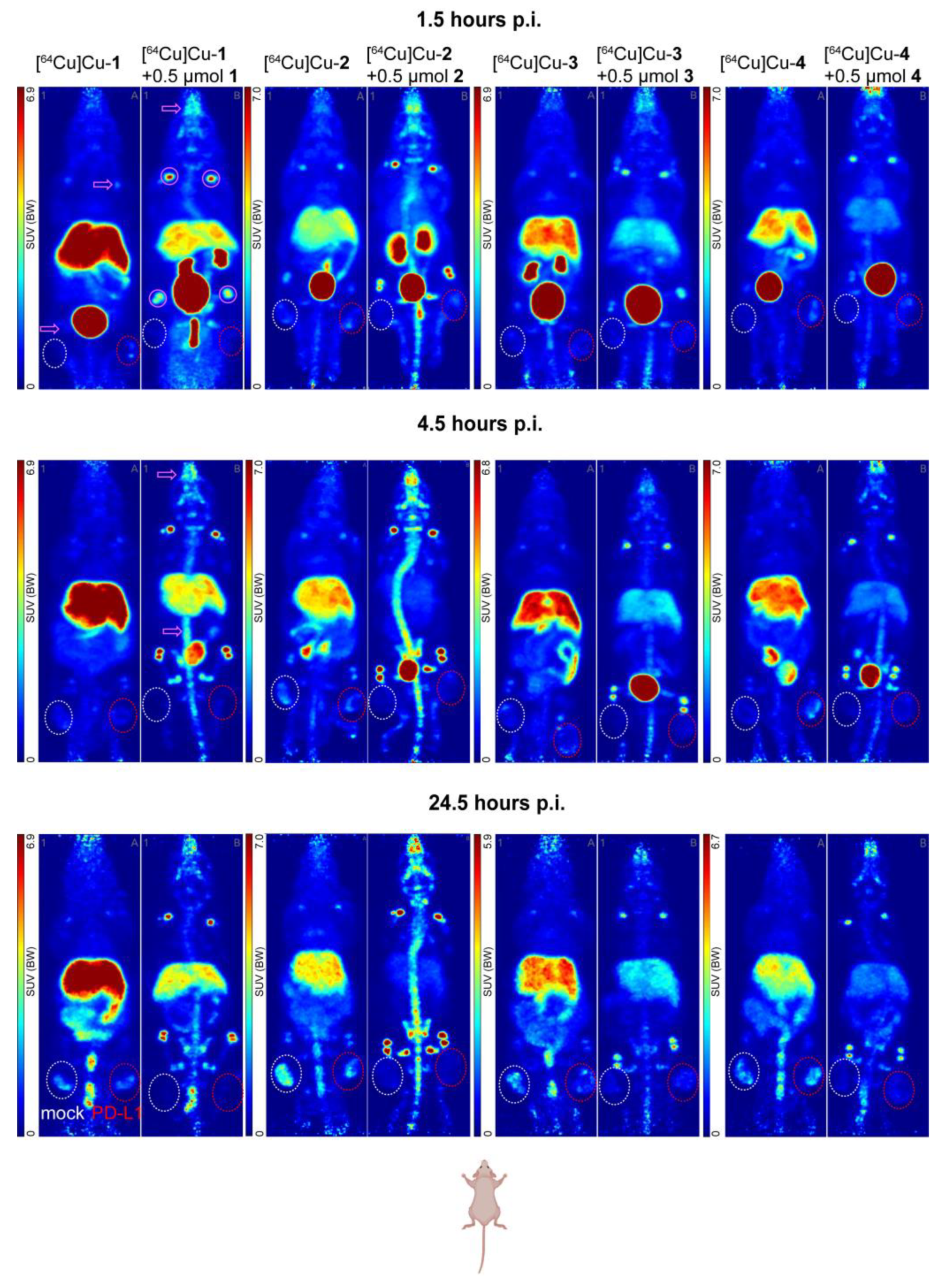
2.5. In Vivo Evaluation
3. Discussion
4. Materials and Methods
4.1. Radiochemistry
4.2. Log D7.4 Determination
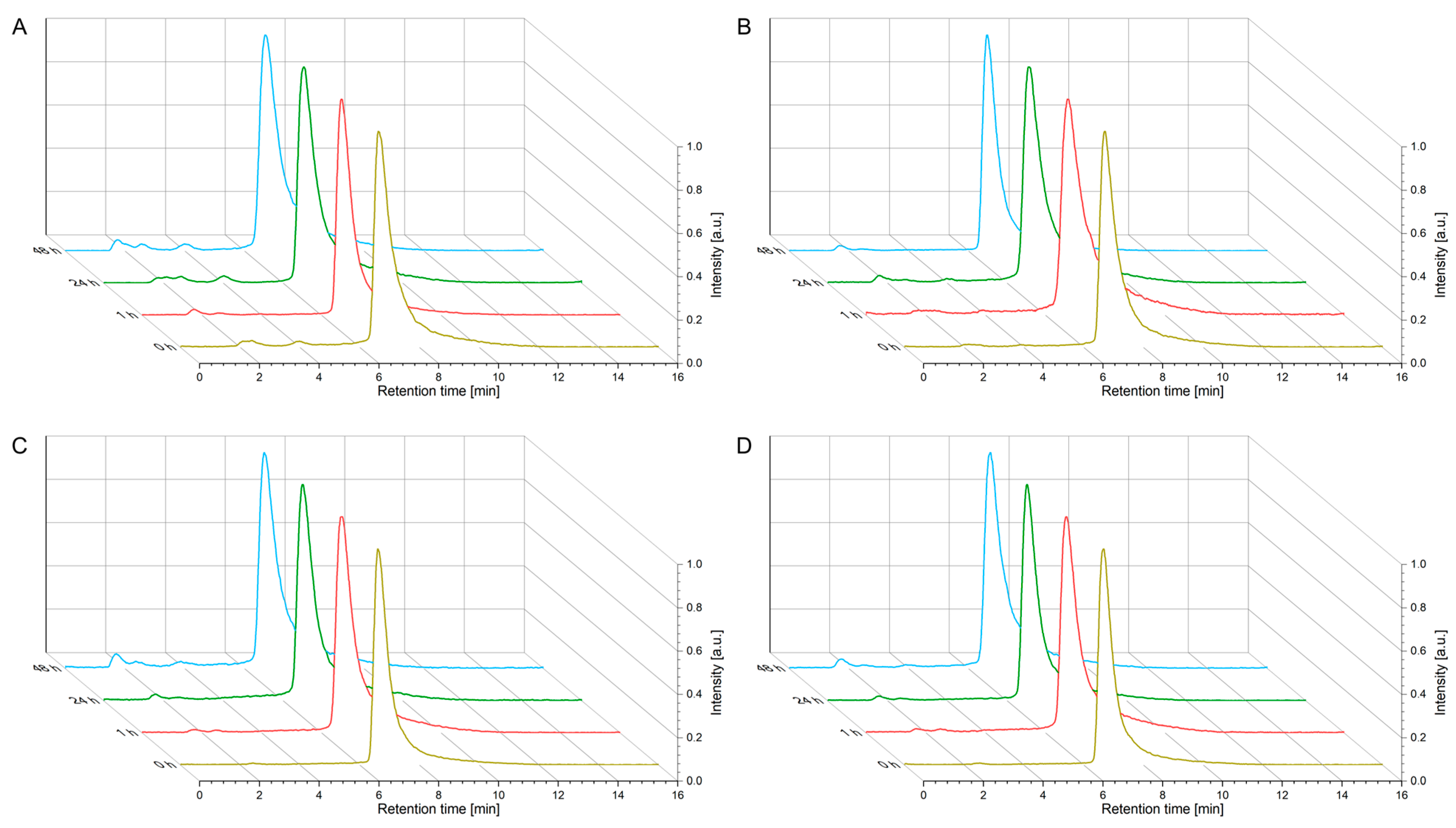
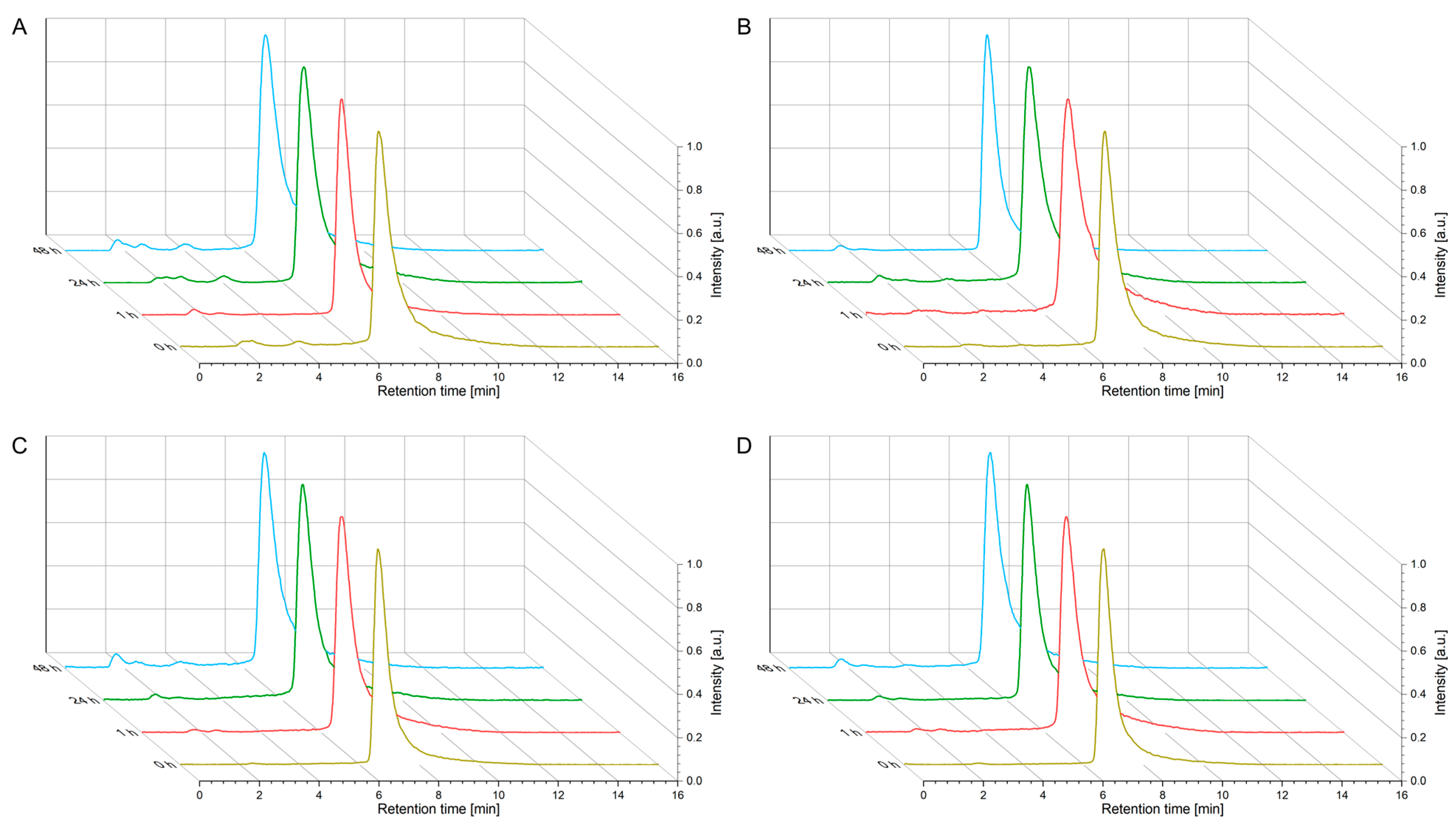
4.3. Kinetic Stability Studies (Human Serum)
4.4. Cell Lines and Cell Culture
4.5. Saturation Binding Studies
4.6. Real-Time Radioligand Binding Studies
4.7. Animals and PET Imaging
4.8. Data and Statistical Analysis
4.9. Synthesis & General Remarks
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef]
- Lotfinejad, P.; Kazemi, T.; Mokhtarzadeh, A.; Shanehbandi, D.; Niaragh, F.J.; Safaei, S.; Asadi, M.; Baradaran, B. PD-1/PD-L1 Axis Importance and Tumor Microenvironment Immune Cells. Life Sci. 2020, 259, 118297. [Google Scholar] [CrossRef] [PubMed]
- Hakozaki, T.; Hosomi, Y.; Kitadai, R.; Kitagawa, S.; Okuma, Y. Efficacy of Immune Checkpoint Inhibitor Monotherapy for Patients with Massive Non-small-Cell Lung Cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2957–2966. [Google Scholar] [CrossRef]
- Guardascione, M.; Toffoli, G. Immune Checkpoint Inhibitors as Monotherapy or Within a Combinatorial Strategy in Advanced Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 6302. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Su, W.; Lu, T.; Wang, Y.; Dong, Y.; Qin, Y.; Liu, D.; Sun, L.; Jiao, W. First-Line Immune-Checkpoint Inhibitors in non-Small Cell Lung Cancer: Current Landscape and Future Progress. Front. Pharmacol. 2020, 11, 1591. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Sternheim, N.; Agarwal, P.; Suchomel, J.; Vadhavkar, S.; Bruno, R.; Ballinger, M.; Bernaards, C.A.; Chan, P.; Ruppel, J. Evaluation of Aatezolizumab Immunogenicity: Clinical Pharmacology (Part 1). J. Clin. Transl. Sci. 2022, 15, 130–140. [Google Scholar] [CrossRef]
- Chi, H.; Hansen, B.E.; Tang, W.Y.; Schouten, J.N.; Sprengers, D.; Taimr, P.; Janssen, H.L.; de Knegt, R.J. Multiple biopsy passes and the risk of complications of percutaneous liver biopsy. Eur. J. Gastroenterol. Hepatol. 2017, 29, 36–41. [Google Scholar] [CrossRef]
- Wahie, S.; Lawrence, C.M. Wound complications following diagnostic skin biopsies in dermatology inpatients. Arch. Dermatol. 2007, 143, 1267–1271. [Google Scholar] [CrossRef]
- Krutzek, F.; Kopka, K.; Stadlbauer, S. Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis. Pharmaceuticals 2022, 15, 747. [Google Scholar] [CrossRef]
- Kasban, H.; El-Bendary, M.; Salama, D. A Comparative Study of Medical Imaging Techniques. Int. J. Intell. Syst. 2015, 4, 37–58. [Google Scholar]
- Vento, J.; Mulgaonkar, A.; Woolford, L.; Nham, K.; Christie, A.; Bagrodia, A.; de Leon, A.D.; Hannan, R.; Bowman, I.; McKay, R.M.; et al. PD-L1 Detection Using 89Zr-Atezolizumab Immuno-PET in Renal Cell Carcinoma Tumorgrafts from a Patient with Favorable Nivolumab Response. J. Immunother. Cancer 2019, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Jagoda, E.M.; Vasalatiy, O.; Basuli, F.; Opina, A.C.L.; Williams, M.R.; Wong, K.; Lane, K.C.; Adler, S.; Ton, A.T.; Szajek, L.P.; et al. Immuno-PET Imaging of the Programmed Cell Death-1 Ligand (PD-L1) Using a Zirconium-89 Labeled Therapeutic Antibody, Avelumab. Mol. Imaging 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, M.; Clump, D.A.; Srivastava, R.M.; Sun, L.; Zeng, D.; Diaz-Perez, J.A.; Anderson, C.J.; Edwards, W.B.; Ferris, R.L. Preclinical ImmunoPET/CT Imaging Using Zr-89-Labeled Anti-PD-L1 Monoclonal Antibody for Assessing Radiation-Induced PD-L1 Upregulation in Head and Neck Cancer and Melanoma. OncoImmunology 2017, 6, e1329071. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zou, S.; Cheng, S.; Song, S.; Wang, P.; Zhu, X. Monitoring the Response of PD-L1 Expression to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Nonsmall-Cell Lung Cancer Xenografts by Immuno-PET Imaging. Mol. Pharm. 2019, 16, 3469–3476. [Google Scholar] [CrossRef]
- Xing, Y.; Chand, G.; Liu, C.; Cook, G.J.R.; O’Doherty, J.; Zhao, L.; Wong, N.C.L.; Meszaros, L.K.; Ting, H.H.; Zhao, J. Early Phase I Study of a 99mTc-Labeled Anti–Programmed Death Ligand-1 (PD-L1) Single-Domain Antibody in SPECT/CT Assessment of PD-L1 Expression in Non–Small Cell Lung Cancer. J. Nucl. Med. 2019, 60, 1213–1220. [Google Scholar] [CrossRef]
- Bridoux, J.; Broos, K.; Lecocq, Q.; Debie, P.; Martin, C.; Ballet, S.; Raes, G.; Neyt, S.; Vanhove, C.; Breckpot, K.; et al. Anti-Human PD-L1 Nanobody for Immuno-PET Imaging: Validation of a Conjugation Strategy for Clinical Translation. Biomolecules 2020, 10, 1388. [Google Scholar] [CrossRef]
- Lv, G.; Sun, X.; Qiu, L.; Sun, Y.; Li, K.; Liu, Q.; Zhao, Q.; Qin, S.; Lin, J. PET Imaging of Tumor PD-L1 Expression with a Highly Specific Nonblocking Single-Domain Antibody. J. Nucl. Med. 2020, 61, 117–122. [Google Scholar] [CrossRef]
- González Trotter, D.E.; Meng, X.; McQuade, P.; Rubins, D.; Klimas, M.; Zeng, Z.; Connolly, B.M.; Miller, P.J.; O’Malley, S.S.; Lin, S.-A.; et al. In Vivo Imaging of the Programmed Death Ligand 1 by 18F PET. J. Nucl. Med. 2017, 58, 1852–1857. [Google Scholar] [CrossRef]
- Rubins, D.J.; Meng, X.; McQuade, P.; Klimas, M.; Getty, K.; Lin, S.-A.; Connolly, B.M.; O’Malley, S.S.; Haley, H.; Purcell, M.; et al. In Vivo Evaluation and Dosimetry Estimate for a High Affinity Affibody PET Tracer Targeting PD-L1. Mol. Imaging Biol. 2020, 23, 241–249. [Google Scholar] [CrossRef]
- Sharma, G.; Braga, M.C.; Da Pieve, C.; Szopa, W.; Starzetz, T.; Plate, K.H.; Kaspera, W.; Kramer-Marek, G. Immuno-PET Imaging of Tumour PD-L1 Expression in Glioblastoma. Cancers 2023, 15, 3131. [Google Scholar] [CrossRef]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; Van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole Body PD-1 and PD-L1 Positron Emission Tomography in Patients with Non-Small-Cell Lung Cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef]
- Stutvoet, T.S.; Van Der Veen, E.L.; Kol, A.; Antunes, I.F.; De Vries, E.F.J.; Hospers, G.A.P.; De Vries, E.G.E.; De Jong, S.; Lub-De Hooge, M.N. Molecular Imaging of PD-L1 Expression and Dynamics with the Adnectin-Based PET Tracer 18F-BMS-986192. J. Nucl. Med. 2020, 12, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Huisman, M.C.; Niemeijer, A.-L.N.; Windhorst, A.D.; Schuit, R.C.; Leung, D.; Hayes, W.; Poot, A.; Bahce, I.; Radonic, T.; Oprea-Lager, D.E.; et al. Quantification of PD-L1 Expression with 18F-BMS-986192 PET/CT in Patients with Advanced-Stage Non–Small Cell Lung Cancer. J. Nucl. Med. 2020, 61, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Verhoeff, S.R.; van den Heuvel, M.M.; van Herpen, C.M.; Piet, B.; Aarntzen, E.H.; Heskamp, S. Programmed Cell Death-1/Ligand-1 PET Imaging: A Novel Tool to Optimize Immunotherapy? PET Clin. 2020, 15, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Xenaki, K.T.; Oliveira, S.; van Bergen En Henegouwen, P.M. Antibody or antibody fragments: Implications for molecular imaging and targeted therapy of solid tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Farid, S.S. Process Economics of Industrial Monoclonal Antibody Manufacture. J. Chromatogr. B Biomed. Appl. 2007, 848, 8–18. [Google Scholar] [CrossRef]
- Chatterjee, S.; Lesniak, W.G.; Miller, M.S.; Lisok, A.; Sikorska, E.; Wharram, B.; Kumar, D.; Gabrielson, M.; Pomper, M.G.; Gabelli, S.B.; et al. Rapid PD-L1 Detection in Tumors with PET Using a Highly Specific Peptide. Biochem. Biophys. Res. Commun. 2017, 483, 258–263. [Google Scholar] [CrossRef]
- Jiang, J.; Li, D.; Liu, T.; Xia, L.; Guo, X.; Meng, X.; Liu, F.; Wang, F.; Yang, Z.; Zhu, H. Noninvasive Evaluation of PD-L1 Expression Using Copper-64 labeled Peptide WL12 by Micro-PET Imaging in Chinese Hamster Ovary Cell Tumor Model. Bioorganic Med. Chem. Lett. 2021, 40, 127901. [Google Scholar] [CrossRef]
- De Silva, R.A.; Kumar, D.; Lisok, A.; Chatterjee, S.; Wharram, B.; Venkateswara Rao, K.; Mease, R.; Dannals, R.F.; Pomper, M.G.; Nimmagadda, S. Peptide-Based 68Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol. Pharm. 2018, 15, 3946–3952. [Google Scholar] [CrossRef]
- Xiang, Q.; Li, D.; Cheng, C.; Xu, K.; Zuo, C. 68Ga-HBED-CC-WL-12 PET in Diagnosing and Differentiating Pancreatic Cancers in Murine Models. Pharmaceuticals 2023, 16, 80. [Google Scholar] [CrossRef]
- Lesniak, W.G.; Mease, R.C.; Chatterjee, S.; Kumar, D.; Lisok, A.; Wharram, B.; Kalagadda, V.R.; Emens, L.A.; Pomper, M.G.; Nimmagadda, S. Development of [18F]FPy-WL12 as a PD-L1 Specific PET Imaging Peptide. Mol. Imaging 2019, 18, 1536012119852189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiang, J.; Yang, X.; Liu, T.; Ding, J.; Nimmagadda, S.; Pomper, M.G.; Zhu, H.; Zhao, J.; Yang, Z. First-In-Human Evaluation of a PD-L1-Binding Peptide Radiotracer in Non-Small Cell Lung Cancer Patients with PET. J. Nucl. Med. 2021, 63, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, L.; Liang, B.; Zhu, S.; Lv, G.; Qiu, L.; Lin, J. Design, Synthesis, and Biological Evaluation of a Small-Molecule PET Agent for Imaging PD-L1 Expression. Pharmaceuticals 2023, 16, 213. [Google Scholar] [CrossRef]
- Miao, Y.; Lv, G.; Chen, Y.; Qiu, L.; Xie, M.; Lin, J. One-Step Radiosynthesis and Initial Evaluation of a Small Molecule PET Tracer for PD-L1 Imaging. Bioorg. Med. Chem. Lett. 2020, 30, 127572. [Google Scholar] [CrossRef]
- Lv, G.; Miao, Y.; Chen, Y.; Lu, C.; Wang, X.; Xie, M.; Qiu, L.; Lin, J. Promising Potential of a 18F-Labelled Small-Molecular Radiotracer to Evaluate PD-L1 Expression in Tumors by PET Imaging. Bioorg. Chem. 2021, 115, 105294. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Yang, J.; Xu, B.; Magiera-Mularz, K.; Skalniak, L.; Musielak, B.; Kholodovych, V.; Holak, T.A.; Hu, L. Design, Synthesis, Evaluation, and Structural Studies of C2-Symmetric Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein–Protein Interaction. J. Med. Chem. 2019, 62, 7250–7263. [Google Scholar] [CrossRef]
- Wang, T.; Cai, S.; Wang, M.; Zhang, W.; Zhang, K.; Chen, D.; Li, Z.; Jiang, S. Novel Biphenyl Pyridines as Potent Small-Molecule Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2021, 64, 7390–7403. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Luo, L.; Wang, Z.; Hu, N.; Wang, W.; Xie, F.; Liang, E.; Yan, X.; Xiao, J.; Li, S. Design, Synthesis, and Biological Evaluation of Linear Aliphatic Amine-Linked Triaryl Derivatives as Potent Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction with Promising Antitumor Effects In Vivo. J. Med. Chem. 2020, 63, 13825–13850. [Google Scholar] [CrossRef]
- Qin, M.; Meng, Y.; Yang, H.; Liu, L.; Zhang, H.; Wang, S.; Liu, C.; Wu, X.; Wu, D.; Tian, Y.; et al. Discovery of 4-Arylindolines Containing a Thiazole Moiety as Potential Antitumor Agents Inhibiting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2021, 64, 5519–5534. [Google Scholar] [CrossRef]
- Song, Z.; Liu, B.; Peng, X.; Gu, W.; Sun, Y.; Xing, L.; Xu, Y.; Geng, M.; Ai, J.; Zhang, A. Design, Synthesis, and Pharmacological Evaluation of Biaryl-Containing PD-1/PD-L1 Interaction Inhibitors Bearing a Unique Difluoromethyleneoxy Linkage. J. Med. Chem. 2021, 64, 16687–16702. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, F.; Yan, Z.; Shen, L.; Zhang, X.; He, F.; Wang, H.; Lu, X.; Yu, K.; Zhao, Y.; et al. Discovery of a Novel, Potent, and Selective Small-Molecule Inhibitor of PD-1/PD-L1 Interaction with Robust in Vivo Anti-Tumor Efficacy. Br. J. Pharmacol. 2021, 178, 2651–2670. [Google Scholar] [CrossRef] [PubMed]
- Koblish, H.K.; Wu, L.; Wang, L.; Liu, P.C.; Wynn, R.; Rios-Doria, J.; Spitz, S.; Liu, H.; Volgina, A.; Zolotarjova, N. Characterization of INCB086550, a Potent and Novel Small-Molecule PD-L1 Inhibitor. Cancer Discov. 2022, 12, 1482–1499. [Google Scholar] [CrossRef]
- Surmiak, E.; Magiera-Mularz, K.; Musielak, B.; Muszak, D.; Kocik-Krol, J.; Kitel, R.; Plewka, J.; Holak, T.A.; Skalniak, L. PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 11797. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, S.; Cheng, Y.; Zhang, W.; Wang, M.; Sun, H.; Guo, B.; Li, Z.; Xiao, Y.; Jiang, S. Discovery of Small-Molecule Inhibitors of the PD-1/PD-L1 Axis That Promote PD-L1 Internalization and Degradation. J. Med. Chem. 2022, 65, 3879–3893. [Google Scholar] [CrossRef] [PubMed]
- Brust, P.; van den Hoff, J.; Steinbach, J. Development of 18 F-Labeled Radiotracers for Neuroreceptor Imaging with Positron Emission Tomography. Neurosci. Bull. 2014, 30, 777–811. [Google Scholar] [CrossRef]
- Jennings, M.; Marcu, L.G.; Bezak, E. PET-Specific Parameters and Radiotracers in Theoretical Tumour Modelling. Comput. Math. Methods Med. 2015, 2015, 415923. [Google Scholar] [CrossRef]
- Fowler, J.S.; Volkow, N.D.; Wang, G.-J.; Ding, Y.-S.; Dewey, S.L. PET and Drug Research and Development. J. Nucl. Med. 1999, 40, 1154–1163. [Google Scholar]
- Krutzek, F.; Donat, C.K.; Ullrich, M.; Zarschler, K.; Ludik, M.-C.; Feldmann, A.; Loureiro, L.R.; Kopka, K.; Stadlbauer, S. Design and Biological Evaluation of Small-Molecule PET-Tracers for Imaging of Programmed Death Ligand 1. Cancers 2023, 15, 2638. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Torner, R.; Skalniak, L.; Domling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Yeung, K.-S.; Connolly, T.P.; Frennesson, D.B.; Grant-Young, K.A.; Hewawasam, P.; Langley, D.R.; Meng, Z.; Mull, E.; Parcella, K.E.; Saulnier, M.G.; et al. Compounds Useful as Immunomodulators. Patent WO2015160641, 22 October 2015. [Google Scholar]
- Konieczny, M.; Musielak, B.; Kocik, J.; Skalniak, L.; Sala, D.; Czub, M.; Magiera-Mularz, K.; Rodriguez, I.; Myrcha, M.; Stec, M.; et al. Di-Bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J. Med. Chem. 2020, 63, 11271–11285. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E. Calcium Phosphates as Delivery Systems for Bisphosphonates. J. Funct. Biomater. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H.A. Bisphosphonates: Preclinical Aspects and Use in Osteoporosis. Ann. Med. 1997, 29, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Song, X.P.; Bouillon, C.; Lescrinier, E.; Herdewijn, P. Iminodipropionic Acid as the Leaving Group for DNA Polymerization by HIV-1 Reverse Transcriptase. ChemBioChem 2011, 12, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.E.; Shi, Z.; Chen, S.; Schmidt, J.F.; Reader, J.C.; Hone, N.D.; Civarri, J.P. 2-(Amino-Substituted)-4-Aryl Pyrimidines and Related Compounds Useful for Treating Inflammatory Diseases. International Patent WO 2005/066139, 21 July 2005. [Google Scholar]
- Weekes, D.M.; Ramogida, C.F.; Jaraquemada-Pelaez, M.d.G.; Patrick, B.O.; Apte, C.; Kostelnik, T.I.; Cawthray, J.F.; Murphy, L.; Orvig, C. Dipicolinate Complexes of Gallium (III) and Lanthanum (III). Inorg. Chem. 2016, 55, 12544–12558. [Google Scholar] [CrossRef]
- Fields, G.B.; Noble, R.L. Solid Phase Peptide Synthesis Utilizing 9-Fluorenylmethoxycarbonyl Amino Acids. Int. J. Pept. Protein Res. 1990, 35, 161–214. [Google Scholar] [CrossRef]
- Ueki, M.; Amemiya, M. Removal of 9-Fluorenylmethyloxycarbonyl (Fmoc) Group with Tetrabutylammonium Fluoride. Tetrahedron Lett. 1987, 28, 6617–6620. [Google Scholar] [CrossRef]
- Chen, C.-C.; Rajagopal, B.; Liu, X.Y.; Chen, K.L.; Tyan, Y.-C.; Lin, F.; Lin, P.-C. A Mild Removal of Fmoc Group Using Sodium Azide. Amino Acids 2014, 46, 367–374. [Google Scholar] [CrossRef]
- Pretze, M.; Mamat, C. Automated Preparation of [18F]AFP and [18F]BFP: Two Novel Bifunctional 18F-Labeling Building Blocks for Huisgen-Click. J. Fluorine Chem. 2013, 150, 25–35. [Google Scholar] [CrossRef]
- Enders, D.; Fronert, J.; Bisschops, T.; Boeck, F. Asymmetric Total Synthesis of Smyrindiol Employing an Organocatalytic Aldol Key Step. Beilstein J. Org. Chem. 2012, 8, 1112–1117. [Google Scholar] [CrossRef]
- Błażewska, K.M. McKenna Reaction: Which Oxygen Attacks Bromotrimethylsilane? J. Org. Chem. 2014, 79, 408–412. [Google Scholar] [CrossRef]
- Justyna, K.; Małolepsza, J.; Kusy, D.; Maniukiewicz, W.; Błażewska, K.M. The McKenna Reaction–Avoiding Side Reactions in Phosphonate Deprotection. Beilstein J. Org. Chem 2020, 16, 1436–1446. [Google Scholar] [CrossRef]
- Zaias, J.; Mineau, M.; Cray, C.; Yoon, D.; Altman, N.H. Reference Values for Serum Proteins of Common laboratory Rodent Strains. J. Am. Assoc. Lab. Anim. Sci. 2009, 48, 387–390. [Google Scholar] [PubMed]
- Park, J.-J.; Thi, E.P.; Carpio, V.H.; Bi, Y.; Cole, A.G.; Dorsey, B.D.; Fan, K.; Harasym, T.; Iott, C.L.; Kadhim, S. Checkpoint Inhibition Through Small Molecule-Induced Internalization of Programmed Death-Ligand 1. Nat. Commun. 2021, 12, 1222. [Google Scholar] [CrossRef] [PubMed]
- Mönkkönen, J.; Urtti, A.; Paronen, P.; Elo, H.A.; Ylitalo, P. The Uptake of Clodronate (Dichloromethylene Bisphosphonate) by Macrophages in vivo and in vitro. Drug Metab. Dispos. 1989, 17, 690–693. [Google Scholar] [PubMed]
- Harris, T.D.; Kalogeropoulos, S.; Nguyen, T.; Dwyer, G.; Edwards, D.S.; Liu, S.; Bartis, J.; Ellars, C.; Onthank, D.; Yalamanchili, P. Structure—Activity Relationships of 111In-and 99mTc-Labeled Quinolin-4-one Peptidomimetics as Ligands for the Vitronectin Receptor: Potential Tumor Imaging Agents. Bioconjugate Chem. 2006, 17, 1294–1313. [Google Scholar] [CrossRef]
- Thieme, S.; Walther, M.; Pietzsch, H.-J.; Henniger, J.; Preusche, S.; Mäding, P.; Steinbach, J. Module-Assisted Preparation of 64Cu with High Specific Activity. Appl. Radiat. Isot. 2012, 70, 602–608. [Google Scholar] [CrossRef]
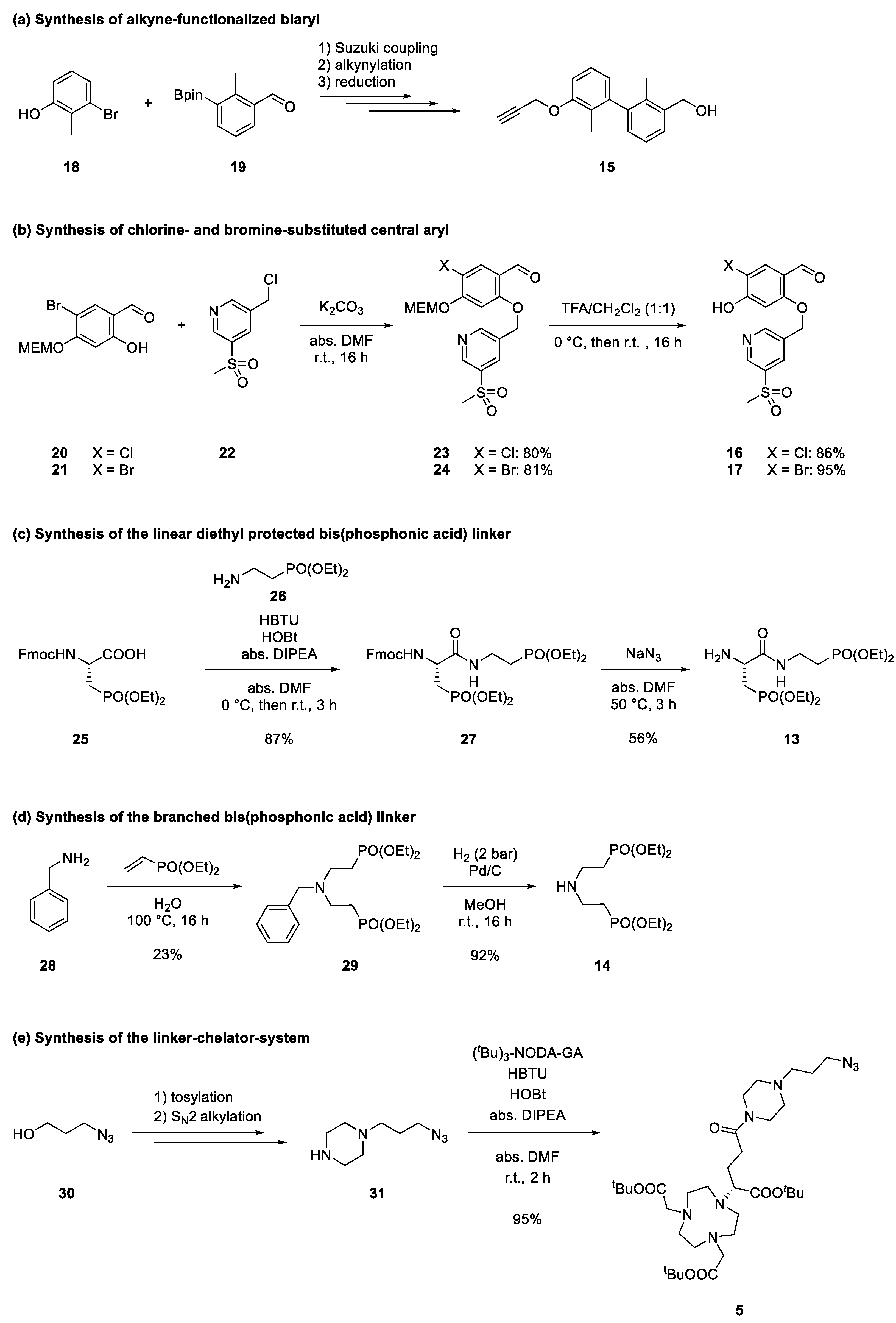



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
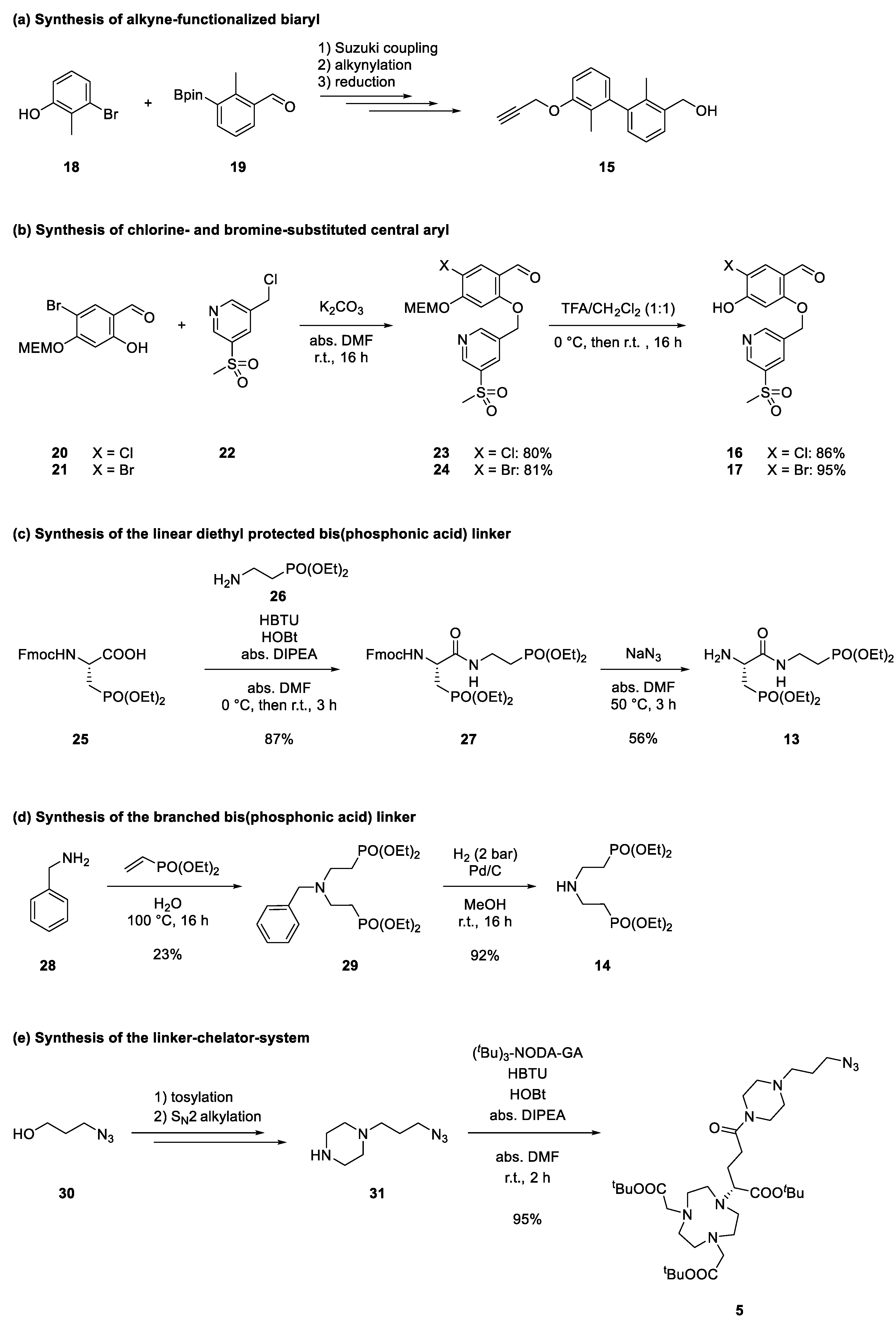{kind=link}
{kind=link}
| Radioligand | KD [nM] 1 | Bmax [pmol × mg−1] 1 | log D7.4 2 | AM [GBq × µmol−1] |
|---|---|---|---|---|
| [64Cu]Cu-1 | 190.7 ± 10.2 | 7.50 ± 0.22 | −4.00 ± 0.14 | 13.5 ± 0.02 |
| [64Cu]Cu-2 | 154.1 ± 26.2 | 1.97 ± 0.23 | −3.03 ± 0.05 | 12.9 ± 0.05 |
| [64Cu]Cu-3 | 76.7 ± 23.5 | 2.81 ± 0.32 | −3.80 ± 0.02 | 13.1 ± 0.02 |
| [64Cu]Cu-4 | 69.6 ± 8.10 | 2.22 ± 0.18 | −3.81 ± 0.08 | 13.0 ± 0.03 |
| Radioligand | ka [(M × s)−1] | kd [s−1] | KD [nM] | Bmax [%] |
|---|---|---|---|---|
| medium only (n = 2; mean ± S.D.) | ||||
| [64Cu]Cu-1 | (4.84 ± 0.38) × 103 | (6.48 ± 0.20) × 10−5 | 13.5 ± 0.64 | 137.8 ± 4.06 |
| [64Cu]Cu-2 | (0.38 ± 0.07) × 103 | (10.5 ± 0.28) × 10−5 | 279 ± 45.3 | 949 ± 140 |
| [64Cu]Cu-3 | (3.74 ± 0.14) × 103 | (6.50 ± 0.17) × 10−5 | 17.4 ± 0.14 | 135.6 ± 8.22 |
| [64Cu]Cu-4 | (4.68 ± 0.73) × 103 | (5.83 ± 0.64)×10−5 | 12.7 ± 3.39 | 118 ± 27.5 |
| medium + 2.5% BSA (n = 1) | ||||
| [64Cu]Cu-1 | 10.2 × 102 | 1.09 × 10−4 | 107 | 382.6 |
| [64Cu]Cu-2 | 6.14 × 102 | 1.47 × 10−4 | 239 | 224 |
| [64Cu]Cu-3 | 5.42 × 102 | 0.77 × 10−4 | 143 | 567.3 |
| [64Cu]Cu-4 | 9.10 × 102 | 0.79 × 10−4 | 86.9 | 328.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krutzek, F.; Donat, C.K.; Stadlbauer, S. Exploring Hydrophilic PD-L1 Radiotracers Utilizing Phosphonic Acids: Insights into Unforeseen Pharmacokinetics. Int. J. Mol. Sci. 2023, 24, 15088. https://doi.org/10.3390/ijms242015088
Krutzek F, Donat CK, Stadlbauer S. Exploring Hydrophilic PD-L1 Radiotracers Utilizing Phosphonic Acids: Insights into Unforeseen Pharmacokinetics. International Journal of Molecular Sciences. 2023; 24(20):15088. https://doi.org/10.3390/ijms242015088
Chicago/Turabian StyleKrutzek, Fabian, Cornelius K. Donat, and Sven Stadlbauer. 2023. "Exploring Hydrophilic PD-L1 Radiotracers Utilizing Phosphonic Acids: Insights into Unforeseen Pharmacokinetics" International Journal of Molecular Sciences 24, no. 20: 15088. https://doi.org/10.3390/ijms242015088
APA StyleKrutzek, F., Donat, C. K., & Stadlbauer, S. (2023). Exploring Hydrophilic PD-L1 Radiotracers Utilizing Phosphonic Acids: Insights into Unforeseen Pharmacokinetics. International Journal of Molecular Sciences, 24(20), 15088. https://doi.org/10.3390/ijms242015088