

The Intrinsically Disordered N Terminus in Atg12 from Yeast Is Necessary for the Functional Structure of the Protein


 , , and
, , and 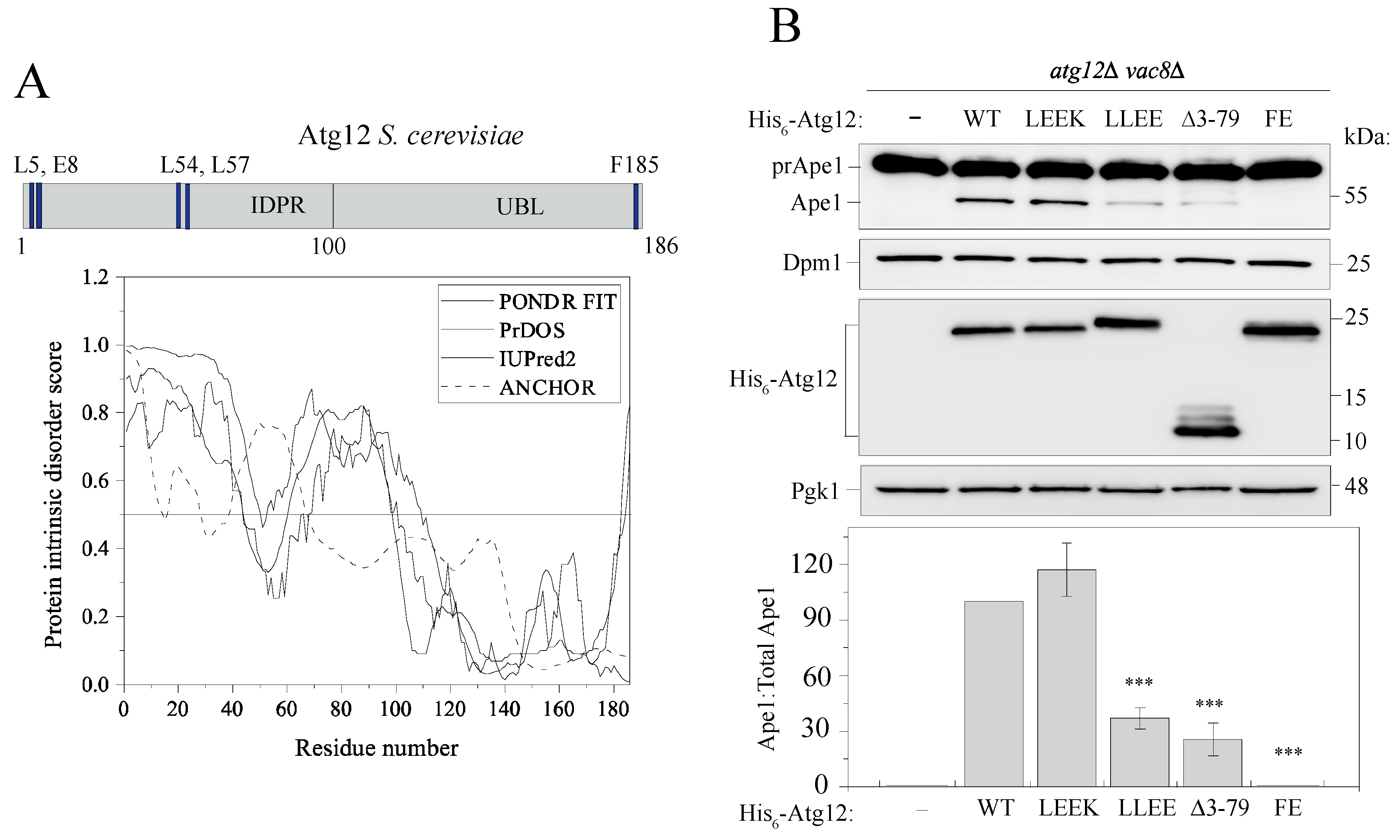{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
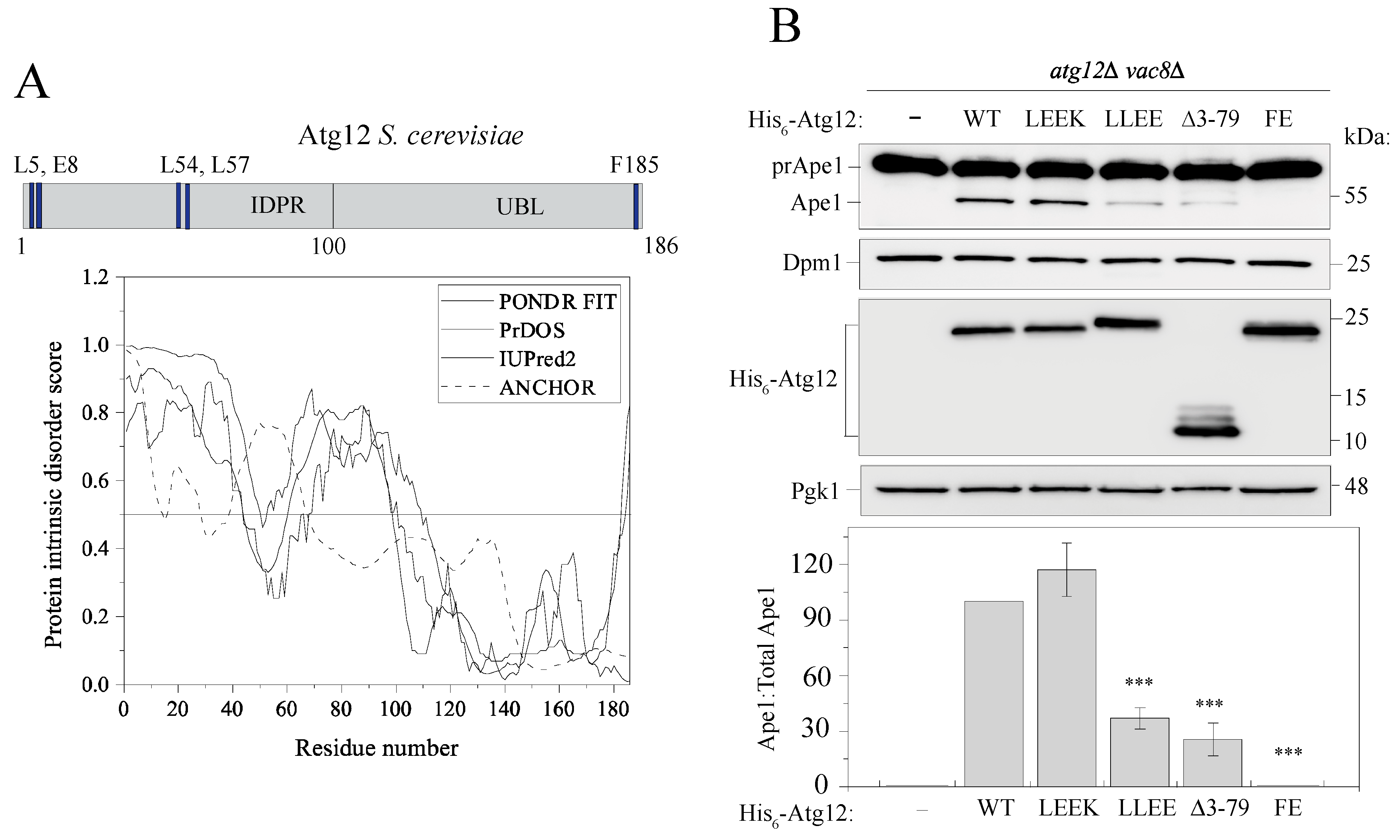
2.1. Atg12 Architecture and Evolutionarily Conserved Elements in the N Terminus of Atg12
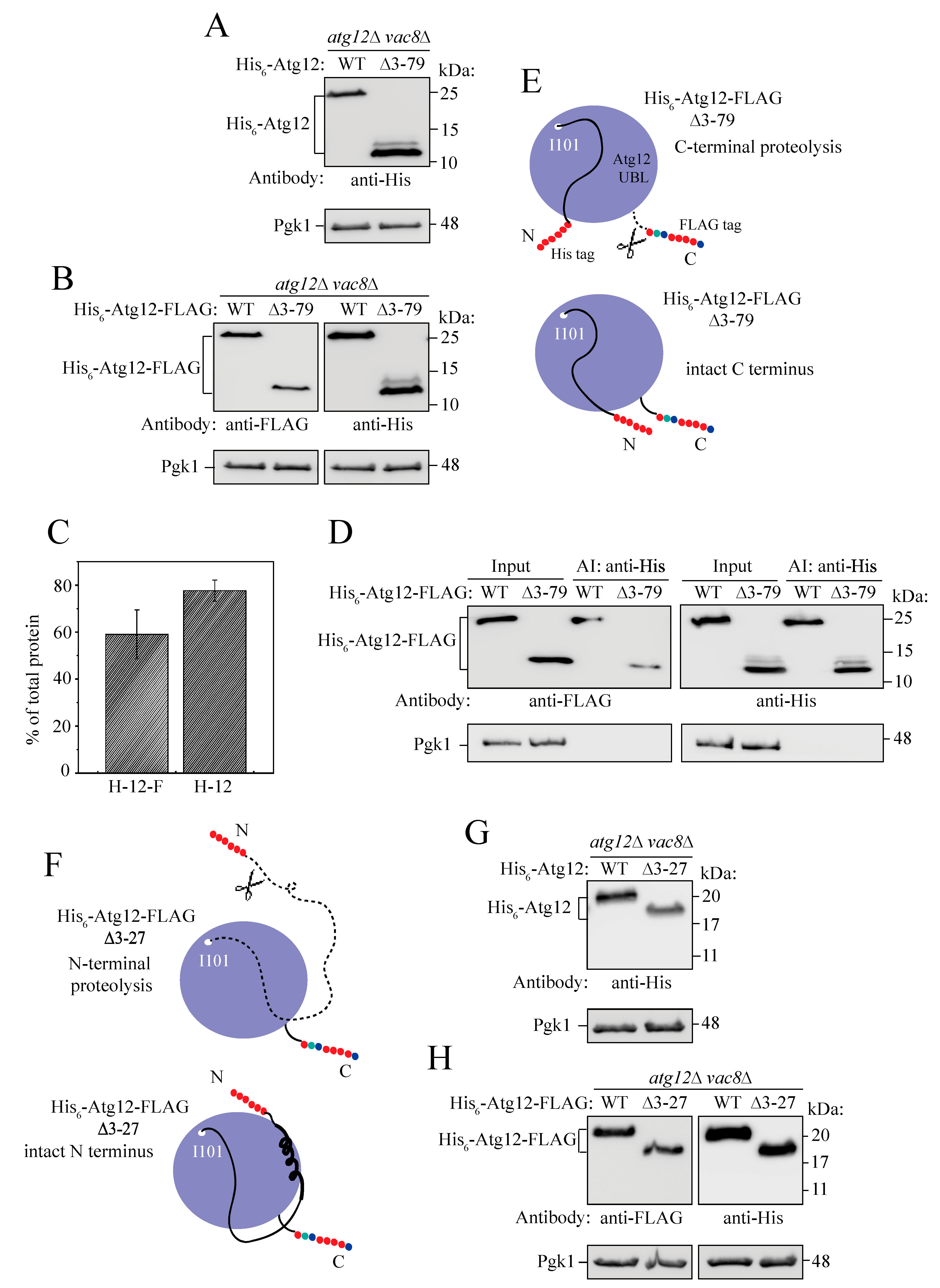
2.2. The IDPR Is Important for Protein Function in Nonselective Autophagy
2.3. The Atg12 IDPR Does Not Associate the Protein with Cellular Membranes
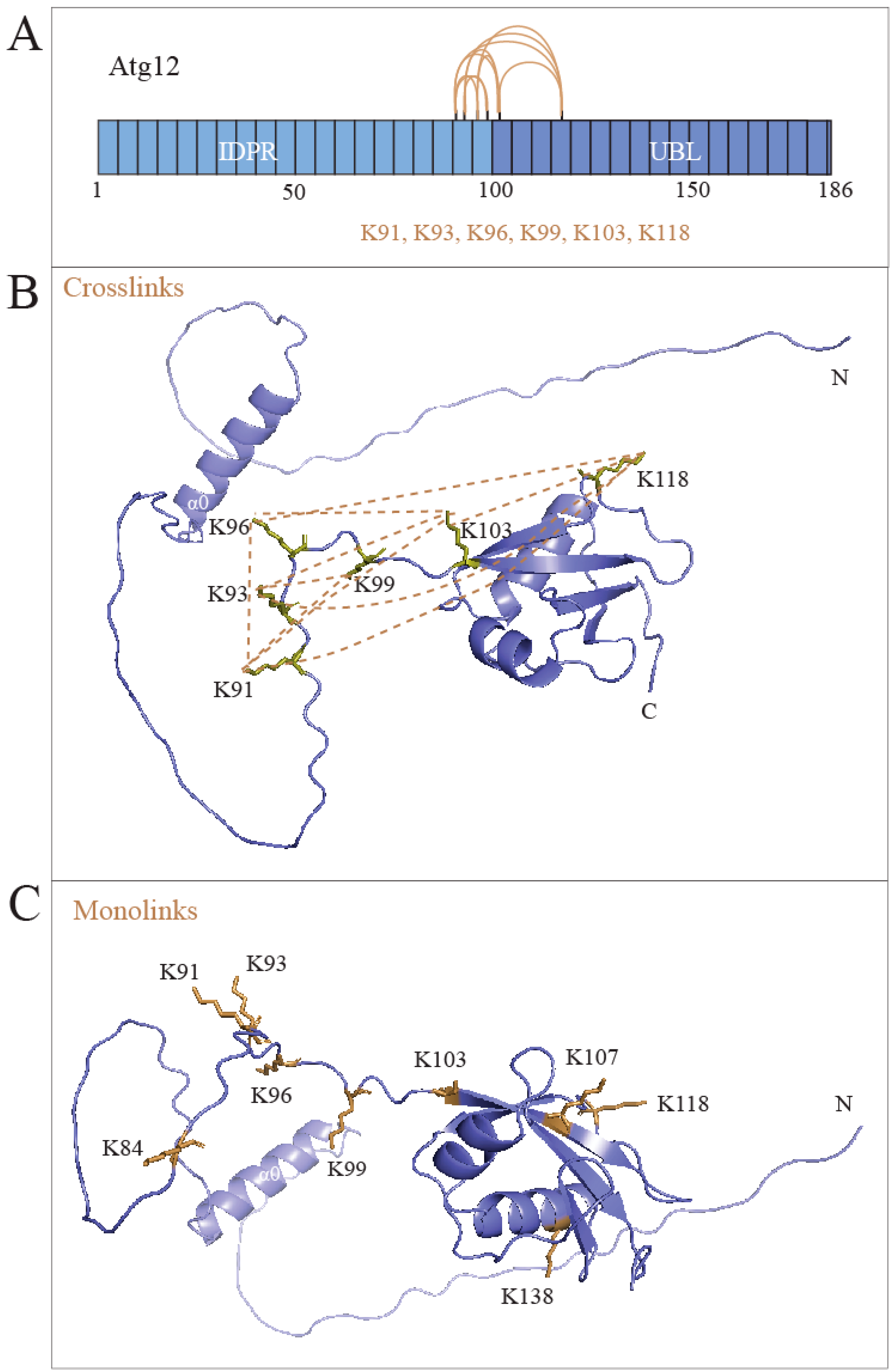
2.4. The IDPR Is Positioned near the UBL Domain of Atg12
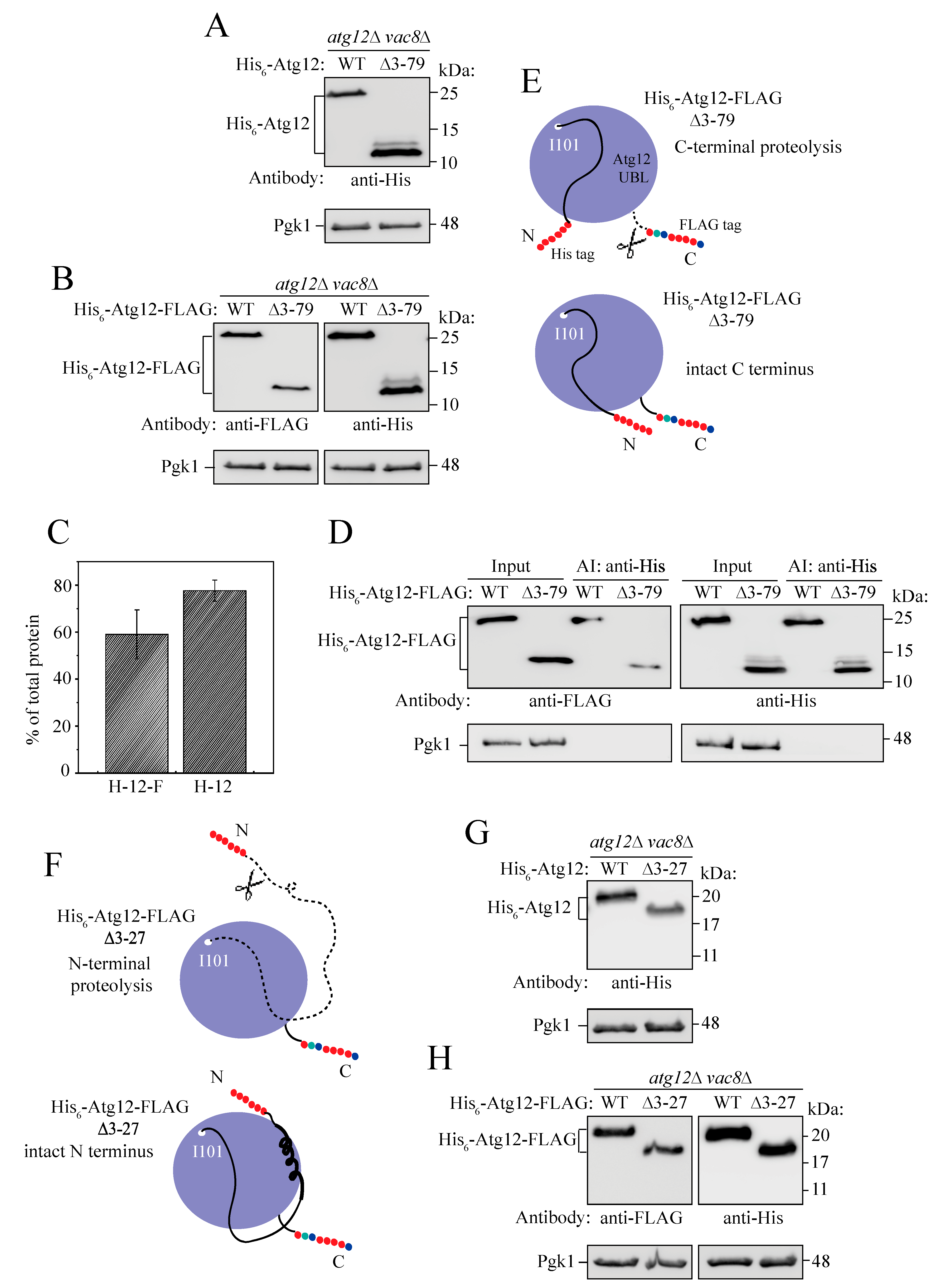
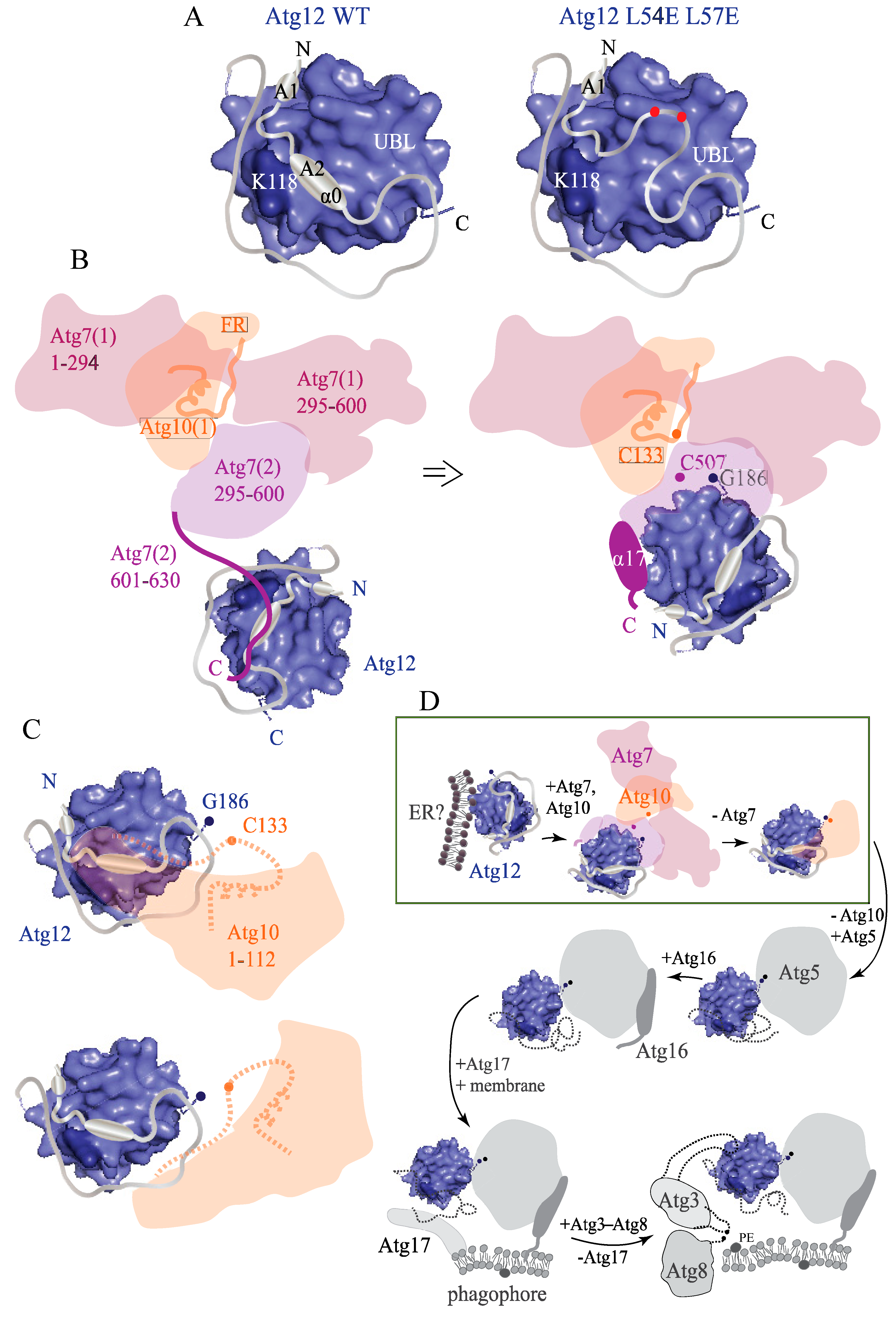
2.5. Intramolecular Interactions between the IDPR and UBL Domain Secure the Intact Structure of Unconjugated Atg12
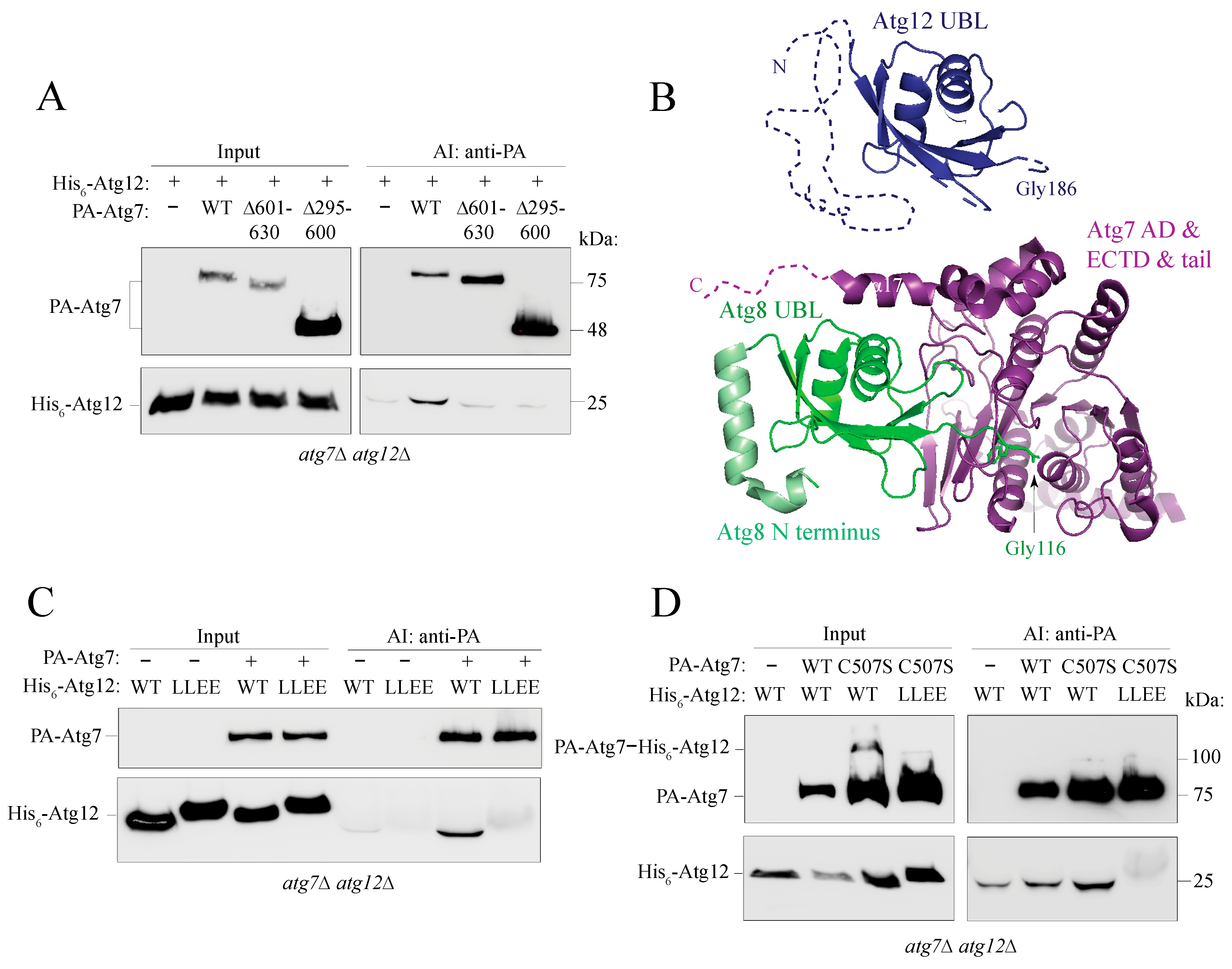
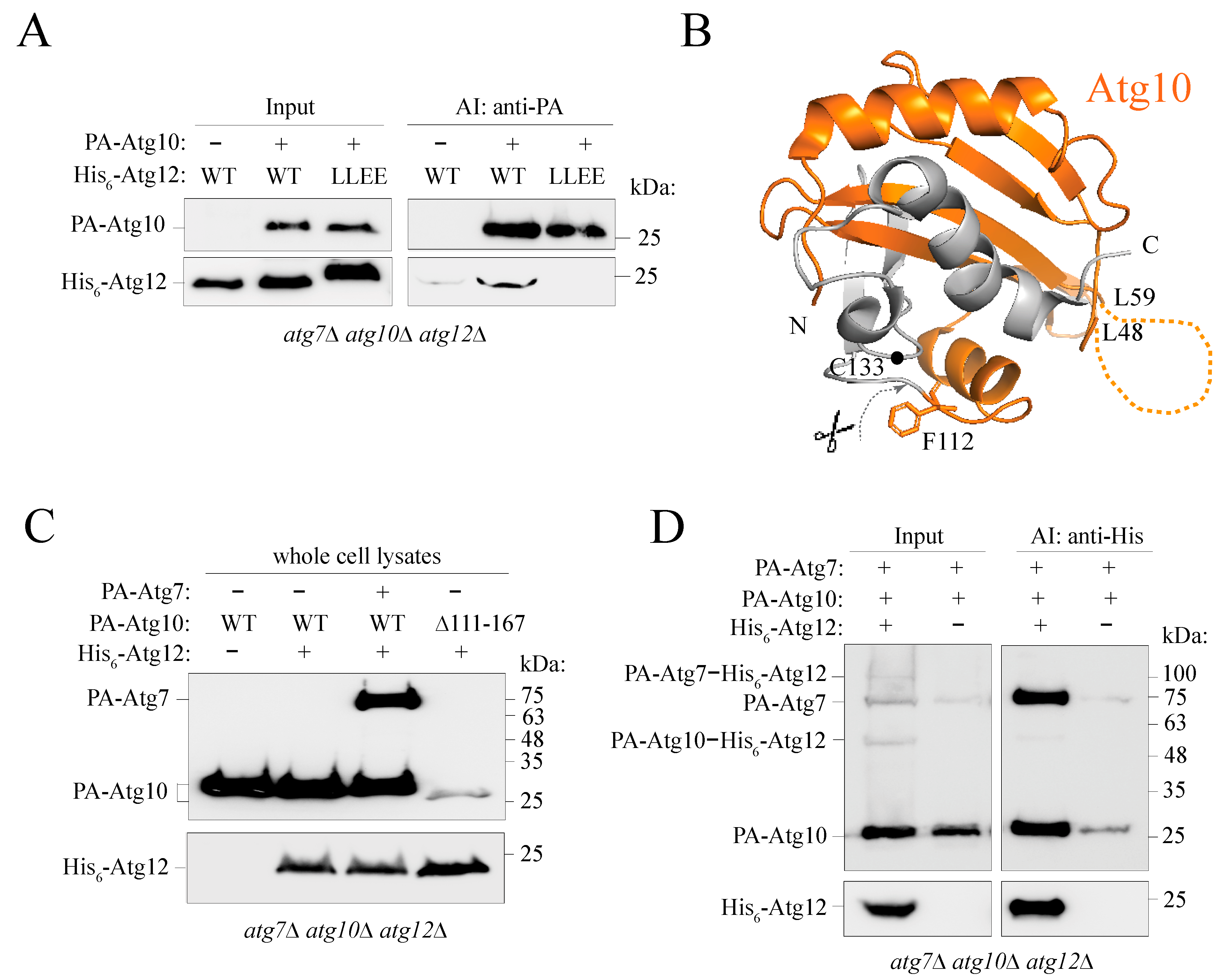
2.6. The Putative α0 Helix in the IDPR Is Needed for Binding of Atg12 to Atg7
2.7. Atg12 Interacts with Unconjugated Partial Atg10 in an IDPR-Dependent Manner
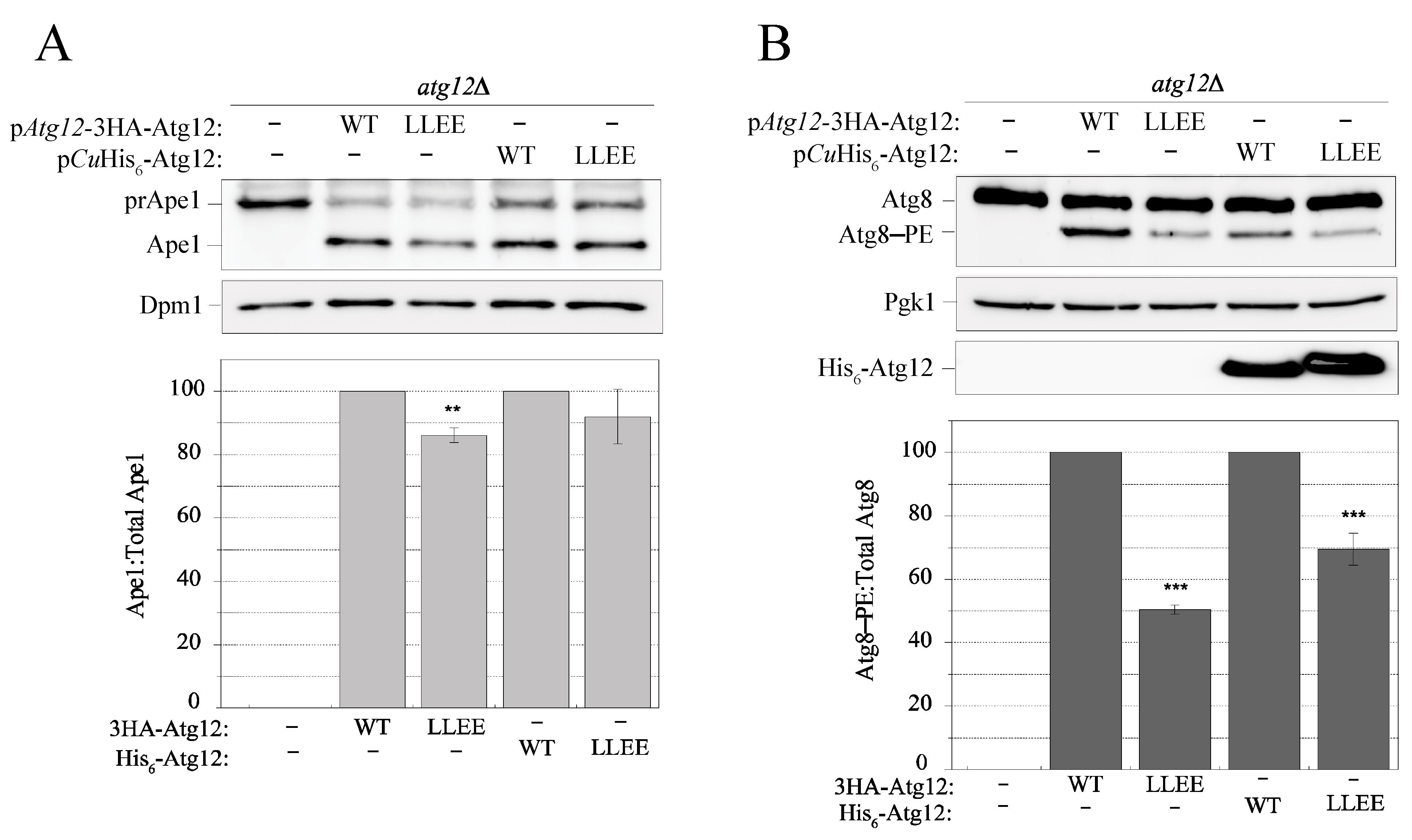
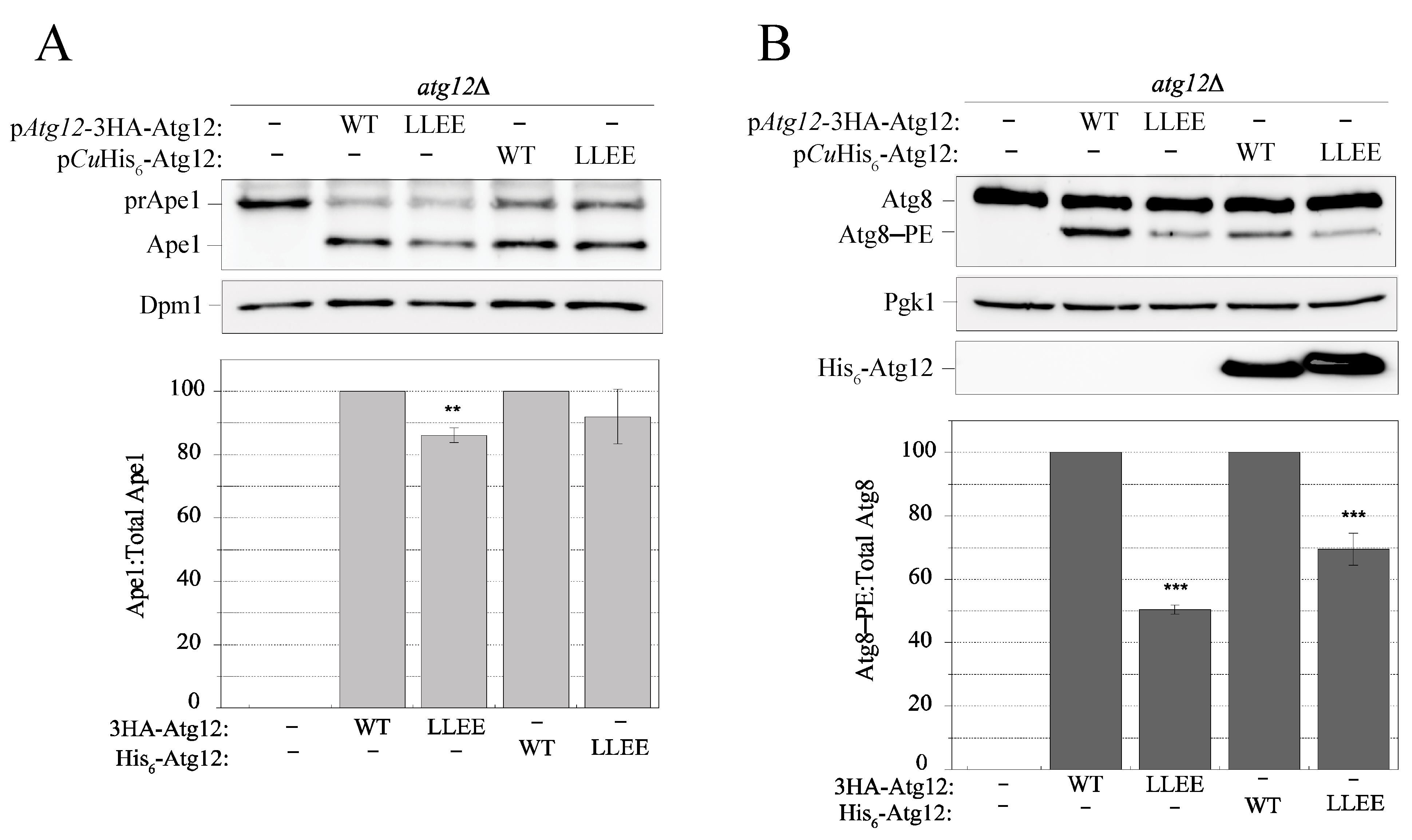
2.8. Disruption of the IDPR by the L54E L57E Mutation Decreases Efficiency of the Cvt Pathway and Atg8 Lipidation
3. Discussion
4. Materials and Methods
4.1. Yeast Plasmids, Strains and Media
4.2. Yeast In Vivo Assays
4.3. Subcellular Fractionation
4.4. Affinity-Isolation Experiments
4.5. Overexpression and Ni-NTA Purification of His6-Atg12 and BS3 Crosslinking
4.6. Anion Exchange and Size-Exclusion Chromatography
4.7. Crosslinking Mass Spectrometry
4.8. Bioinformatics Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Misushima, N. The ATG conjugation system in autophagy. Curr. Opin. Cell. Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Akioka, M.; Kondo-Kakuta, C.; Yamamoto, H.; Ohsumi, Y. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci. 2013, 126, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Noda, N.N.; Nakatogawa, H.; Ohsumi, Y.; Inagaki, F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J. Biol. Chem. 2010, 285, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the ∼350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [PubMed]
- Lystad, A.; Carlsson, S.R.; de la Ballina, L.R.; Kauffman, K.J.; Nag, S.; Yoshimori, T.; Melia, T.J.; Simonsen, A. Distinct functions of ATG16L1 isoforms in membrane bindings and LC3B lipidation in autophagy-related process. Nat. Cell Biol. 2019, 21, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Kotani, T.; Kirisako, H.; Sakoh-Nakatogawa, M.; Oikawa, Y.; Kimura, Y.; Hirano, H.; Yamamoto, H.; Ohsumi, Y.; Nakatogawa, H. Two distinct mechanisms target the autophagy-related E3 complex to the pre-autophagosomal structure. eLife 2019, 8, e43088. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef]
- Hong, S.B.; Kim, B.-W.; Kim, J.H.; Song, H.K. Structure of the autophagic E2 enzyme Atg10. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.E.; Mao, K.; Taherbhoy, A.M.; Yu, S.; Olszewski, J.L.; Duda, D.M.; Kurinov, I.; Deng, A.; Fenn, T.D.; Klionsky, D.J.; et al. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7-Atg3 and Atg7-Atg10 structures. Nat. Struct. Mol. Biol. 2012, 19, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hong, S.B.; Lee, J.K.; Han, S.; Roh, K.-H.; Lee, K.-E.; Kim, Y.K.; Choi, E.-J.; Song, H.K. Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy 2015, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Suzuki, N.N.; Obara, K.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of Atg5·Atg16, a complex essential for autophagy. J. Biol. Chem. 2007, 282, 6763–6772. [Google Scholar] [CrossRef] [PubMed]
- Metlagel, Z.; Otomo, C.; Takaesu, G.; Otomo, T. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc. Natl. Acad. Sci. USA 2013, 110, 18844–18849. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Fujioka, Y.; Hanada, T.; Ohsumi, Y.; Inagaki, F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013, 14, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Satoo, K.; Fujioka, Y.; Kumeta, H.; Ogura, K.; Nakatogawa, H.; Ohsumi, Y.; Inagaki, F. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol. Cell 2011, 44, 462–475. [Google Scholar] [CrossRef]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef]
- Yamada, Y.; Suzuki, N.N.; Hanada, T.; Ichimura, Y.; Kumeta, H.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J. Biol. Chem. 2007, 282, 8036–8043. [Google Scholar] [CrossRef]
- Sugawara, K.; Suzuki, N.N.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J. Biol. Chem. 2005, 280, 40058–40065. [Google Scholar] [CrossRef]
- Hanada, T.; Ohsumi, Y. Structure-function relationship of Atg12, a ubiquitin-like modifier essential for autophagy. Autophagy 2005, 1, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Csizmok, V.; Follis, A.V.; Kriwacki, R.W.; Forman-Kay, J.D. Dynamic Protein Interaction Networks and New Structural Paradigms in Signaling. Chem. Rev. 2016, 116, 6424–6462. [Google Scholar] [CrossRef]
- Gsponer, J.; Babu, M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009, 99, 94–103. [Google Scholar] [CrossRef]
- Theillet, F.-X.; Binolfi, A.; Frembgen-Kesner, T.; Hingorani, K.; Sarkar, M.; Kyne, C.; Li, C.; Crowley, P.B.; Gierasch, L.; Pielak, G.J.; et al. Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs). Chem. Rev. 2014, 114, 6661–6714. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 2019, 74, 909–921.e6. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Tyndall, E.R.; Bui, V.; Tang, Z.; Shen, Y.; Jiang, X.; Flanagan, J.M.; Wang, H.G.; Tian, F. An N terminal conserved region in human ATG3 couples membrane curvature sensitivity to conjugase activity during autophagy. Nat. Commun. 2021, 12, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Fujioka, Y.; Suzuki, S.W.; Noshiro, D.; Suzuki, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ando, T.; Noda, N.N.; et al. The Intrinsically Disordered Protein Atg13 Mediates Supramolecular Assembly of Autophagy Initiation Complexes. Dev. Cell 2016, 38, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Su, M.; Soni, G.; Salem, S.; Colbert, C.L.; Sinha, S.C. Intrinsically disordered regions in autophagy proteins. Proteins 2014, 82, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef]
- Biesaga, M.; Frigolé-Vivas, M.; Salvatella, X. Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr. Opin. Chem. Biol. 2021, 62, 90–100. [Google Scholar] [CrossRef]
- Longhena, F.; Spano, P.; Bellucci, A. Targeting of Disordered Proteins by Small Molecules in Neurodegenerative Diseases. In Targeting Trafficking in Drug Development; Ulloa-Aguirre, A., Tao, Y.X., Eds.; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2018; Volume 245, pp. 85–110. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velázquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell. Mol. Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef]
- Uversky, V.N.; Davé, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef]
- Kulkarni, P.; Getzenberg, R.H. Disorder, Promiscuous Interactions, and Stochasticity Regulate State Switching in the Unstable Prostate. J. Cell. Biochem. 2016, 117, 2235–2240. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Alam, J.M.; Noda, N.N. Membrane-binding domains in autophagy. Chem. Phys. Lipids 2019, 218, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leary, K.A.; Ragusa, M.J. Characterization of protein-membrane interactions in yeast autophagy. Cells 2022, 11, 1876. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Simon, I.; Dosztányi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.B.; Kim, B.-W.; Lee, K.-E.; Kim, S.W.; Jeon, H.; Kim, J.; Song, H.K. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat. Struct. Mol. Biol. 2011, 18, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Schwarten, M.; Stoldt, M.; Mohrlüder, J.; Willbold, D. Solution structure of Atg8 reveals conformational polymorphism of the N-terminal domain. Biochem. Biophys. Res. Commun. 2010, 395, 426–431. [Google Scholar] [CrossRef]
- Li, C.; Wen, A.; Shen, B.; Lu, J.; Huang, Y.; Chang, Y. FastCloning: A highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 2011, 11, 92. [Google Scholar] [CrossRef]
- Liu, H.T.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 91. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Solon, A.L.; Tan, Z.; Schutt, K.L.; Jepsen, L.; Haynes, S.E.; Nesvizhskii, A.I.; Sept, D.; Stumpff, J.; Ohi, R.; Cianfrocco, M.A. Kinesin-binding protein remodels the kinesin motor to prevent microtubule binding. Sci. Adv. 2021, 7, eabj9812. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-L.; Meng, J.-M.; Cao, Y.; Yin, J.-L.; Fang, R.-Q.; Fan, S.-B.; Liu, C.; Zeng, W.-F.; Ding, Y.-H.; Tan, D.; et al. A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 2019, 10, 3404. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Cao, Y.; Cheong, H.S.; Song, H.; Klionsky, D.J. In vivo reconstitution of autophagy in Saccharomyces cerevisiae. J. Cell Biol. 2008, 182, 703–713. [Google Scholar] [CrossRef]
- Robinson, J.S.; Klionsky, D.J.; Banta, L.M.; Emr, S.D. Protein Sorting in Saccharomyces-Cerevisiae—Isolation of Mutants Defective in the Delivery and Processing of Multiple Vacuolar Hydrolases. Mol. Cell. Biol. 1988, 8, 4936–4948. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popelka, H.; Lahiri, V.; Hawkins, W.D.; da Veiga Leprevost, F.; Nesvizhskii, A.I.; Klionsky, D.J. The Intrinsically Disordered N Terminus in Atg12 from Yeast Is Necessary for the Functional Structure of the Protein. Int. J. Mol. Sci. 2023, 24, 15036. https://doi.org/10.3390/ijms242015036
Popelka H, Lahiri V, Hawkins WD, da Veiga Leprevost F, Nesvizhskii AI, Klionsky DJ. The Intrinsically Disordered N Terminus in Atg12 from Yeast Is Necessary for the Functional Structure of the Protein. International Journal of Molecular Sciences. 2023; 24(20):15036. https://doi.org/10.3390/ijms242015036
Chicago/Turabian StylePopelka, Hana, Vikramjit Lahiri, Wayne D. Hawkins, Felipe da Veiga Leprevost, Alexey I. Nesvizhskii, and Daniel J. Klionsky. 2023. "The Intrinsically Disordered N Terminus in Atg12 from Yeast Is Necessary for the Functional Structure of the Protein" International Journal of Molecular Sciences 24, no. 20: 15036. https://doi.org/10.3390/ijms242015036
APA StylePopelka, H., Lahiri, V., Hawkins, W. D., da Veiga Leprevost, F., Nesvizhskii, A. I., & Klionsky, D. J. (2023). The Intrinsically Disordered N Terminus in Atg12 from Yeast Is Necessary for the Functional Structure of the Protein. International Journal of Molecular Sciences, 24(20), 15036. https://doi.org/10.3390/ijms242015036