

Altered Mitochondrial Morphology and Bioenergetics in a New Yeast Model Expressing Aβ42

 and
and
Abstract
1. Introduction
2. Results
2.1. Development and Primary Characterization of Y. lipolytica Cells Expressing Aβ42
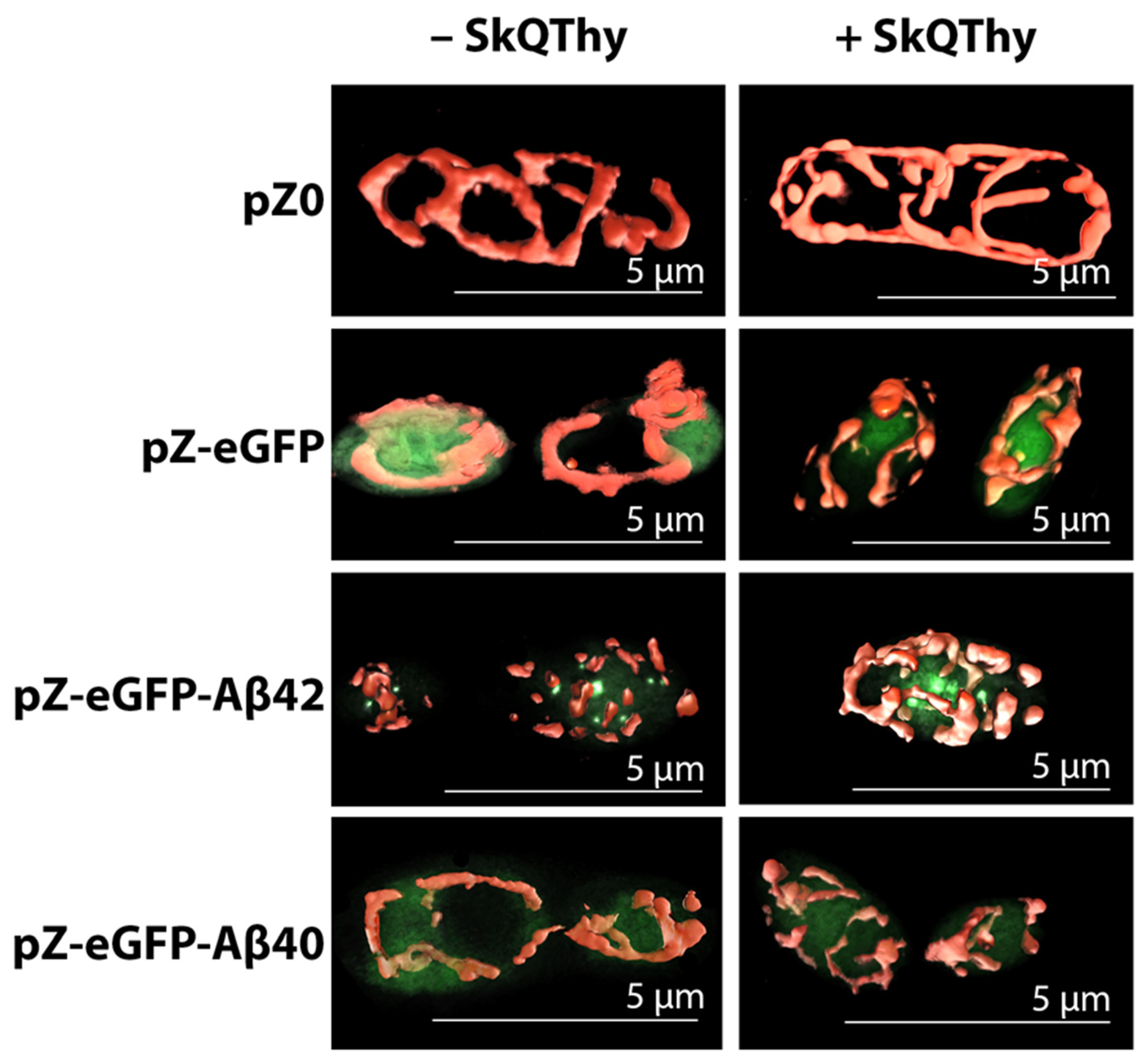
2.2. Morphology of Mitochondria in Yeast Cells
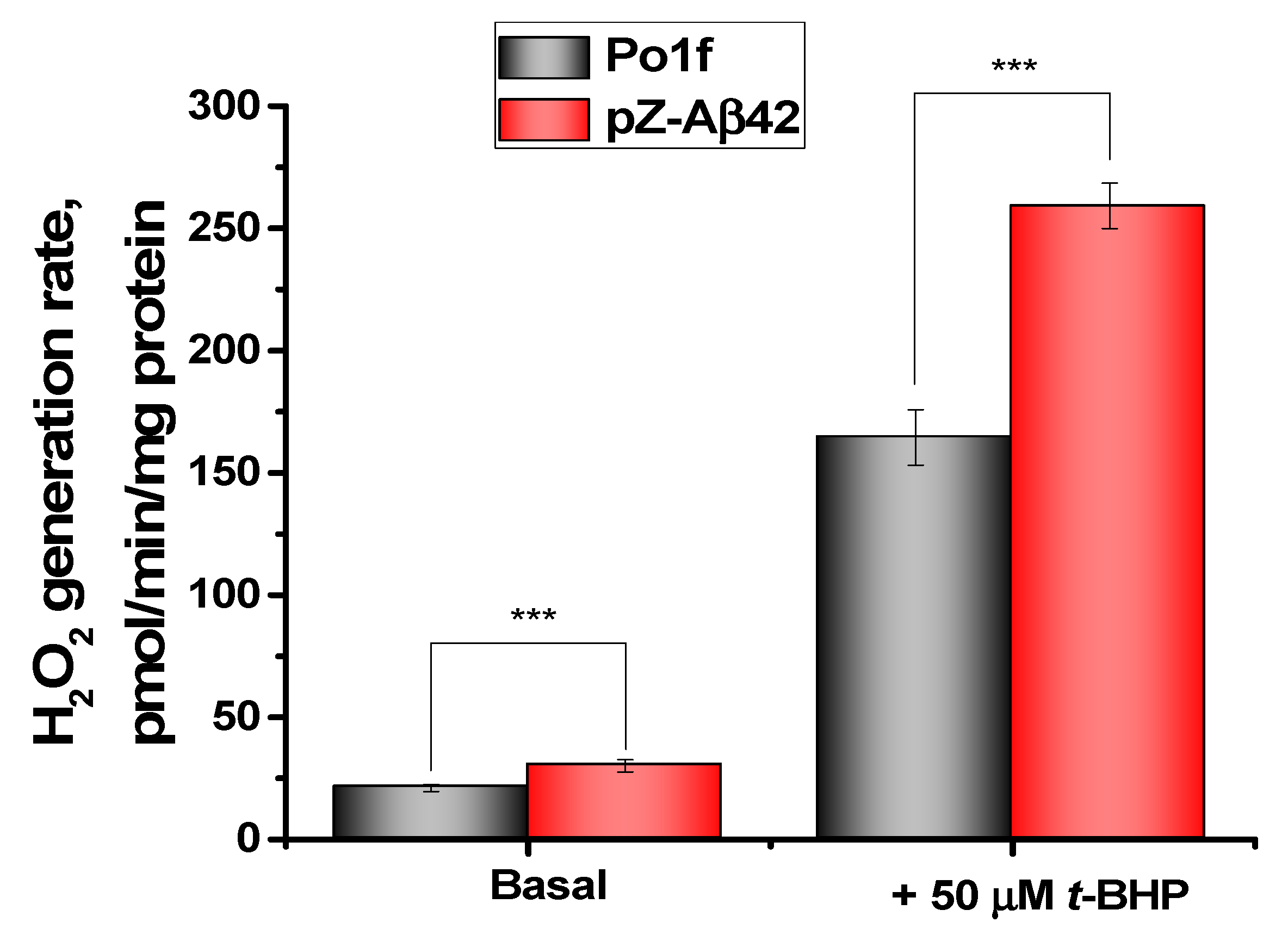
2.3. Assessment of Oxidative Status and Viability of Y. lipolytica Cells
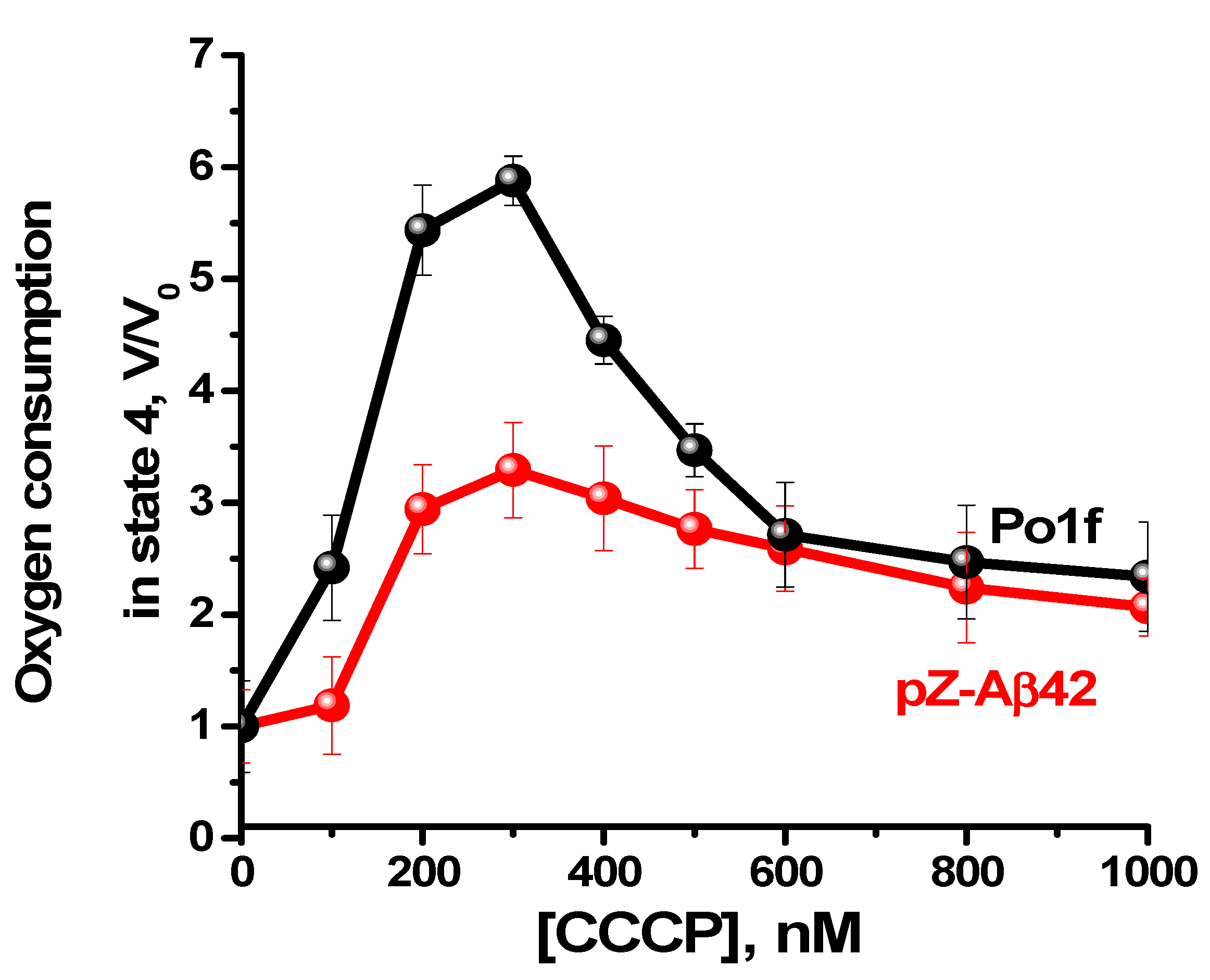
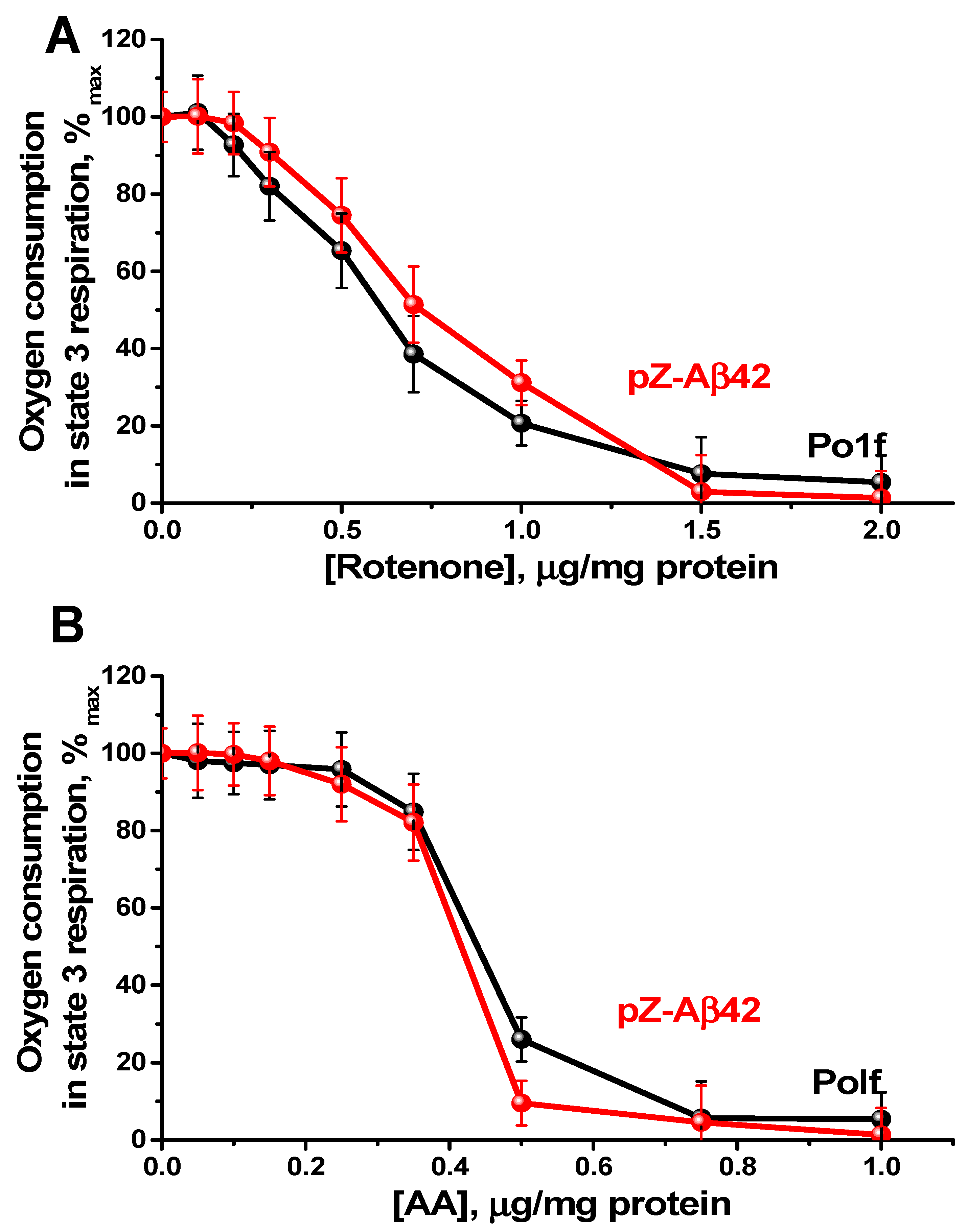
2.4. Characterization of Mitochondria Isolated from Yeast Cells
3. Discussion
4. Materials and Methods
4.1. Chemical Reagents
4.2. Cell Cultures
4.3. Plasmid and Yeast Strain Construction
4.4. Mitochondria Visualization in Y. lipolytica Cells by Structural Illumination Microscopy (SIM)
4.5. Mitochondria Visualization in Y. lipolytica Cells by Widefield Fluorescent Microscopy
4.6. Assessment of Oxidative Stress and Cell Viability of Y. lipolytica Cells
4.7. Isolation of Y. lipolytica Mitochondria
4.8. Monitoring of Oxygen Consumption by Yeast Mitochondria
4.9. Assay of ATP Synthesis by Mitochondria
4.10. Assessment of Hydrogen Peroxide Production by Mitochondria
4.11. Mitochondrial Protein Assay
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.D.; Jasem, S.; Licchesi, J.D.F. The Ubiquitin System in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2020, 1233, 195–221. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Chigurupati, S.; Alhowail, A.; Abdeen, A.; Ibrahim, S.F.; Vargas-De-La-Cruz, C.; et al. Decrypting the potential role of α-lipoic acid in Alzheimer’s disease. Life Sci. 2021, 284, 119899. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free. Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef]
- Conway, M.E. Alzheimer’s disease: Targeting the glutamatergic system. Biogerontology 2020, 21, 257–274. [Google Scholar] [CrossRef]
- James, B.D.; Bennett, D.A. Causes and Patterns of Dementia: An Update in the Era of Redefining Alzheimer’s Disease. Annu. Rev. Public Health 2019, 40, 65–84. [Google Scholar] [CrossRef]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef]
- Walia, V.; Kaushik, D.; Mittal, V.; Kumar, K.; Verma, R.; Parashar, J.; Akter, R.; Rahman, M.H.; Bhatia, S.; Al-Harrasi, A.; et al. Delineation of Neuroprotective Effects and Possible Benefits of AntioxidantsTherapy for the Treatment of Alzheimer’s Diseases by Targeting Mitochondrial-Derived Reactive Oxygen Species: Bench to Bedside. Mol. Neurobiol. 2022, 59, 657–680. [Google Scholar] [CrossRef]
- Gabandé-Rodríguez, E.; Keane, L.; Capasso, M. Microglial phagocytosis in aging and Alzheimer’s disease. J. Neurosci. Res. 2020, 98, 284–298. [Google Scholar] [CrossRef]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 12, e12008. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Sienski, G.; Bonner, J.M.; Lin, Y.T.; Seo, J.; Baru, V.; Haque, A.; Milo, B.; Akay, L.A.; Graziosi, A.; et al. PICALM Rescues Endocytic Defects Caused by the Alzheimer’s Disease Risk Factor APOE4. Cell Rep. 2020, 33, 108224. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Osorio, C.; Kanukuntla, T.; Diaz, E.; Jafri, N.; Cummings, M.; Sfera, A. The Post-amyloid Era in Alzheimer’s Disease: Trust Your Gut Feeling. Front. Aging Neurosci. 2019, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Mangialasche, F.; Ngandu, T.; Solomon, A.; Kivipelto, M. Multidomain Interventions to Prevent Cognitive Impairment, Alzheimer’s Disease, and Dementia: From FINGER to World-Wide FINGERS. J. Prev. Alzheimers Dis. 2020, 7, 29–36. [Google Scholar] [CrossRef]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Zhang, L.; Young, J.I.; Gomez, L.; Silva, T.C.; Schmidt, M.A.; Cai, J.; Chen, X.; Martin, E.R.; Wang, L. Sex-specific DNA methylation differences in Alzheimer’s disease pathology. Acta Neuropathol. Commun. 2021, 9, 77. [Google Scholar] [CrossRef]
- Ekundayo, T.C.; Olasehinde, T.A.; Okaiyeto, K.; Okoh, A.I. Microbial Pathogenesis and Pathophysiology of Alzheimer’s Disease: A Systematic Assessment of Microorganisms’ Implications in the Neurodegenerative Disease. Front. Neurosci. 2021, 15, 648484. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.; Nicoletti, F.; Gaetano, A.; Scaccianoce, S. Risk Factors for Alzheimer’s Disease: Focus on Stress. Front. Pharmacol. 2019, 10, 976. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Arslan, M.E.; Barboza, J.N.; Kahraman, C.Y.; de Sousa, D.P.; Mardinoğlu, A. Therapeutic Potential of Ferulic Acid in Alzheimer’s Disease. Curr. Drug Deliv. 2022, 19, 860–873. [Google Scholar] [CrossRef]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the Amyloid Hypothesis in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2019, 68, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. J. Alzheimers Assoc. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Cohen, A.D.; Landau, S.M.; Snitz, B.E.; Klunk, W.E.; Blennow, K.; Zetterberg, H. Fluid and PET biomarkers for amyloid pathology in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 97, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, M.; Gopinath, P.; Govindaraju, T. Role of Post-translational Modifications in Alzheimer’s Disease. Chembiochem A Eur. J. Chem. Biol. 2020, 21, 1052–1079. [Google Scholar] [CrossRef]
- Janowicz, P.W.; Leinenga, G.; Götz, J.; Nisbet, R.M. Ultrasound-mediated blood-brain barrier opening enhances delivery of therapeutically relevant formats of a tau-specific antibody. Sci. Rep. 2019, 9, 9255. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Et Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef]
- Seynnaeve, D.; Vecchio, M.D.; Fruhmann, G.; Verelst, J.; Cools, M.; Beckers, J.; Mulvihill, D.P.; Winderickx, J.; Franssens, V. Recent Insights on Alzheimer’s Disease Originating from Yeast Models. Int. J. Mol. Sci. 2018, 19, 1947. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Vo, V.G. Role of Body-Fluid Biomarkers in Alzheimer’s Disease Diagnosis. Diagnostics 2020, 10, 326. [Google Scholar] [CrossRef] [PubMed]
- Cubinkova, V.; Valachova, B.; Uhrinova, I.; Brezovakova, V.; Smolek, T.; Jadhav, S.; Zilka, N. Alternative hypotheses related to Alzheimer’s disease. Bratisl. Lek. Listy 2018, 119, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Rodríguez-Pérez, M.; Gómez-Torres, Ó.; Pintado-Losa, C.; Burgos-Ramos, E. Hydroxytyrosol improves mitochondrial energetics of a cellular model of Alzheimer’s disease. Nutr. Neurosci. 2022, 25, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimers Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Leuzy, A.; Mattsson-Carlgren, N.; Palmqvist, S.; Janelidze, S.; Dage, J.L.; Hansson, O. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol. Med. 2022, 14, e14408. [Google Scholar] [CrossRef]
- Chrem Mendez, P.; Surace, E.; Bérgamo, Y.; Calandri, I.; Vázquez, S.; Sevlever, G.; Allegri, R.F. Biomarkers for Alzheimer’s disease. Where we stand and where we are headed. Medicina 2019, 79, 546–551. [Google Scholar]
- Dallé, E.; Mabandla, M.V.; Daniels, W.M.U. Dielectric Constant and Conductivity of Blood Plasma: Possible Novel Biomarkers for Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2020, 2020, 5756382. [Google Scholar] [CrossRef]
- Dhiman, K.; Blennow, K.; Zetterberg, H.; Martins, R.N.; Gupta, V.B. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell. Mol. Life Sci. CMLS 2019, 76, 1833–1863. [Google Scholar] [CrossRef]
- Paolacci, L.; Giannandrea, D.; Mecocci, P.; Parnetti, L. Biomarkers for Early Diagnosis of Alzheimer’s Disease in the Oldest Old: Yes or No? J. Alzheimers Dis. JAD 2017, 58, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; Scheltens, P.; et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Shaikh, M.F.; Piperi, C. Flotillin: A Promising Biomarker for Alzheimer’s Disease. J. Pers. Med. 2020, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Gleerup, H.S.; Hasselbalch, S.G.; Simonsen, A.H. Biomarkers for Alzheimer’s Disease in Saliva: A Systematic Review. Dis. Markers 2019, 2019, 4761054. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Verma, S. Use of ocular biomarkers as a potential tool for early diagnosis of Alzheimer’s disease. Indian J. Ophthalmol. 2020, 68, 555–561. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; Del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef]
- Denner, J.; Tanzi, R.; Jacobson, S. Animal Models of Alzheimer’s Disease Should Be Controlled for Roseolovirus. J. Alzheimers Dis. JAD 2020, 77, 543–545. [Google Scholar] [CrossRef]
- Kosel, F.; Pelley, J.M.S.; Franklin, T.B. Behavioural and psychological symptoms of dementia in mouse models of Alzheimer’s disease-related pathology. Neurosci. Biobehav. Rev. 2020, 112, 634–647. [Google Scholar] [CrossRef]
- Nakai, T.; Yamada, K.; Mizoguchi, H. Alzheimer’s Disease Animal Models: Elucidation of Biomarkers and Therapeutic Approaches for Cognitive Impairment. Int. J. Mol. Sci. 2021, 22, 5549. [Google Scholar] [CrossRef]
- Ni, R. Magnetic Resonance Imaging in Animal Models of Alzheimer’s Disease Amyloidosis. Int. J. Mol. Sci. 2021, 22, 12768. [Google Scholar] [CrossRef]
- Scheffer, S.; Hermkens, D.M.A.; van der Weerd, L.; de Vries, H.E.; Daemen, M. Vascular Hypothesis of Alzheimer Disease: Topical Review of Mouse Models. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling neurodegeneration in Caenorhabditiselegans. Dis. Model. Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef] [PubMed]
- Giong, H.K.; Subramanian, M.; Yu, K.; Lee, J.S. Non-Rodent Genetic Animal Models for Studying Tauopathy: Review of Drosophila, Zebrafish, and C. elegans Models. Int. J. Mol. Sci. 2021, 22, 8465. [Google Scholar] [CrossRef] [PubMed]
- Naranjo-Galindo, F.J.; Ai, R.; Fang, E.F.; Nilsen, H.L.; SenGupta, T.C. elegans as an Animal Model to Study the Intersection of DNA Repair, Aging and Neurodegeneration. Front. Aging 2022, 3, 916118. [Google Scholar] [CrossRef] [PubMed]
- Kitani-Morii, F.; Friedland, R.P.; Yoshida, H.; Mizuno, T. Drosophila as a Model for Microbiota Studies of Neurodegeneration. J. Alzheimers Dis. JAD 2021, 84, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Tue, N.T.; Dat, T.Q.; Ly, L.L.; Anh, V.D.; Yoshida, H. Insights from Drosophila melanogaster model of Alzheimer’s disease. Front. Biosci. 2020, 25, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Macreadie, I. Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 8014. [Google Scholar] [CrossRef]
- Futai, E. Advanced Yeast Models of Familial Alzheimer Disease Expressing FAD-Linked Presenilin to Screen Mutations and γ-Secretase Modulators. Methods Mol. Biol. 2019, 2049, 403–417. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Laor, D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef]
- Goleva, T.N.; Rogov, A.G.; Korshunova, G.A.; Trendeleva, T.A.; Mamaev, D.V.; Aliverdieva, D.A.; Zvyagilskaya, R.A. SkQThy, a novel and promising mitochondria-targeted antioxidant. Mitochondrion 2019, 49, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Mamaev, D.; Zvyagilskaya, R. Yarrowia lipolytica: A multitalented yeast species of ecological significance. FEMS Yeast Res. 2021, 21, foab008. [Google Scholar] [CrossRef] [PubMed]
- Rogov, A.G.; Goleva, T.N.; Epremyan, K.K.; Kireev, I.I.; Zvyagilskaya, R.A. Propagation of Mitochondria-Derived Reactive Oxygen Species within the Dipodascus magnusii Cells. Antioxidants 2021, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Trendeleva, T.A.; Sukhanova, E.I.; Rogov, A.G.; Zvyagilskaya, R.A.; Seveina, I.I.; Ilyasova, T.M.; Cherepanov, D.A.; Skulachev, V.P. Role of charge screening and delocalization for lipophilic cation permeability of model and mitochondrial membranes. Mitochondrion 2013, 13, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Johri, A. Disentangling Mitochondria in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11520. [Google Scholar] [CrossRef] [PubMed]
- Feniouk, B.A.; Skulachev, V.P. Cellular and Molecular Mechanisms of Action of Mitochondria-Targeted Antioxidants. Curr. Aging Sci. 2017, 10, 41–48. [Google Scholar] [CrossRef]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial Dysfunction and Redox Imbalance as a Diagnostic Marker of “Free Radical Diseases”. Anticancer. Res. 2017, 37, 5373–5381. [Google Scholar] [CrossRef]
- Epremyan, K.K.; Goleva, T.N.; Zvyagilskaya, R.A. Effect of Tau Protein on Mitochondrial Functions. Biochem. Biokhimiia 2022, 87, 689–701. [Google Scholar] [CrossRef]
- Batista, A.F.; Rody, T.; Forny-Germano, L.; Cerdeiro, S.; Bellio, M.; Ferreira, S.T.; Munoz, D.P.; De Felice, F.G. Interleukin-1β mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-β oligomers. J. Neuroinflamm. 2021, 18, 54. [Google Scholar] [CrossRef]
- Espino de la Fuente-Muñoz, C.; Rosas-Lemus, M.; Moreno-Castilla, P.; Bermúdez-Rattoni, F.; Uribe-Carvajal, S.; Arias, C. Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 8727. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, J.; Ge, X.; Deng, J.; Xu, X.; Zhao, Y.; Wang, H. Activation of ERK-Drp1 signaling promotes hypoxia-induced Aβ accumulation by upregulating mitochondrial fission and BACE1 activity. FEBS Open Bio 2021, 11, 2740–2755. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.E.; Saleh, T.M.; Kalisch, B.E. Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease. Antioxidants 2022, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.; Akram, M.; Shahrukh, M.; Ishrat, T.; Parvez, S. Effects of pramipexole on beta-amyloid(1-42) memory deficits and evaluation of oxidative stress and mitochondrial function markers in the hippocampus of Wistar rat. Neurotoxicology 2022, 92, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxidative Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Zorov, D.B. Pros and Cons of Use of Mitochondria-Targeted Antioxidants. Antioxidants 2019, 8, 316. [Google Scholar] [CrossRef] [PubMed]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [CrossRef]
- Kolosova, N.G.; Tyumentsev, M.A.; Muraleva, N.A.; Kiseleva, E.; Vitovtov, A.O.; Stefanova, N.A. Antioxidant SkQ1 Alleviates Signs of Alzheimer’s Disease-like Pathology in Old OXYS Rats by Reversing Mitochondrial Deterioration. Curr. Alzheimer Res. 2017, 14, 1283–1292. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Ershov, N.I.; Kolosova, N.G. Suppression of Alzheimer’s Disease-Like Pathology Progression by Mitochondria-Targeted Antioxidant SkQ1: A Transcriptome Profiling Study. Oxidative Med. Cell. Longev. 2019, 2019, 3984906. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Muraleva, N.A.; Maksimova, K.Y.; Rudnitskaya, E.A.; Kiseleva, E.; Telegina, D.V.; Kolosova, N.G. An antioxidant specifically targeting mitochondria delays progression of Alzheimer’s disease-like pathology. Aging 2016, 8, 2713–2733. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, V.S.; Mudzhiri, N.M.; Voronkova, A.S.; Baranich, T.I.; Glinkina, V.V.; Illarioshkin, S.N. Mitochondrial Disorders in Alzheimer’s Disease. Biochem. Biokhimiia 2021, 86, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961. [Google Scholar] [CrossRef] [PubMed]
- Wenger, J.; Klinglmayr, E.; Fröhlich, C.; Eibl, C.; Gimeno, A.; Hessenberger, M.; Puehringer, S.; Daumke, O.; Goettig, P. Functional mapping of human dynamin-1-like GTPase domain based on x-ray structure analyses. PLoS ONE 2013, 8, e71835. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-β induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2017, 58, 147–162. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Et Biophys. Acta 2016, 1862, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Mallat, A.; Uchiyama, L.F.; Lewis, S.C.; Fredenburg, R.A.; Terada, Y.; Ji, N.; Nunnari, J.; Tseng, C.C. Discovery and characterization of selective small molecule inhibitors of the mammalian mitochondrial division dynamin, DRP1. Biochem. Biophys. Res. Commun. 2018, 499, 556–562. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e586. [Google Scholar] [CrossRef]
- Rogov, A.G.; Ovchenkova, A.P.; Goleva, T.N.; Kireev, I.I.; Zvyagilskaya, R.A. New yeast models for studying mitochondrial morphology as affected by oxidative stress and other factors. Anal. Biochem. 2018, 552, 24–29. [Google Scholar] [CrossRef]
- Epremyan, K.K.; Goleva, T.N.; Rogov, A.G.; Lavrushkina, S.V.; Zinovkin, R.A.; Zvyagilskaya, R.A. The First Yarrowia lipolytica Yeast Models Expressing Hepatitis B Virus X Protein: Changes in Mitochondrial Morphology and Functions. Microorganisms 2022, 10, 1817. [Google Scholar] [CrossRef] [PubMed]
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Cattoni, D.I.; Nöllmann, M. The fluorescence properties and binding mechanism of SYTOX green, a bright, low photo-damage DNA intercalating agent. Eur. Biophys. J. EBJ 2015, 44, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. A simple and rapid assay of oxidative phosphorylation. Nature 1955, 175, 1120–1121. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Martens, Y.A.; Zhao, N.; Liu, C.C.; Kanekiyo, T.; Yang, A.J.; Goate, A.M.; Holtzman, D.M.; Bu, G. ApoE Cascade Hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron 2022, 110, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Morales-Zavala, F.; Jara-Guajardo, P.; Chamorro, D.; Riveros, A.L.; Chandia-Cristi, A.; Salgado, N.; Pismante, P.; Giralt, E.; Sánchez-Navarro, M.; Araya, E.; et al. In vivo micro computed tomography detection and decrease in amyloid load by using multifunctionalized gold nanorods: A neurotheranostic platform for Alzheimer’s disease. Biomater. Sci. 2021, 9, 4178–4190. [Google Scholar] [CrossRef]
- Gorecki, L.; Uliassi, E.; Bartolini, M.; Janockova, J.; Hrabinova, M.; Hepnarova, V.; Prchal, L.; Muckova, L.; Pejchal, J.; Karasova, J.Z.; et al. Phenothiazine-Tacrine Heterodimers: Pursuing Multitarget Directed Approach in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1698–1715. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A. Role of mitochondria and mitochondria-targeted agents in non-alcoholic fatty liver disease. Clin. Exp. Pharmacol. Physiol. 2018, 45, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T. 36-month LipiDiDiet multinutrient clinical trial in prodromal Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2021, 17, 29–40. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Description |
|---|---|
| Po1f | MatA, leu2-270, ura3-302, xpr2-322, axp-2 |
| pZ-0 | Po1f + pZ URA3, xpr2Δ |
| pZ-eGFP | Po1f + pZ-eGFP URA3 xpr2Δ |
| pZ-Aβ42 | Po1f + pZ-Aβ42 URA3 xpr2Δ |
| pZ-eGFP-Aβ42 | Po1f + pZ-eGFP-Aβ42 URA3 xpr2Δ |
| pZ-Aβ40 | Po1f + pZ-Aβ40 URA3 xpr2Δ |
| pZ-eGFP-Aβ40 | Po1f + pZ-eGFP-Aβ40 URA3 xpr2Δ |
| Primer | Sequence |
|---|---|
| Aβ42-BbsI-Fw1 | TAGAAGACGCAATGGATGCGGAATTTCGC |
| Aβ42-BbsI-Rev1 | TAGAAGACGCGCGCTCACGCAATCACCACG |
| Aβ42-BbsI-Fw3 | TAGAAGACATCAAGATGGATGCGGAATTTCGC |
| Aβ40-BbsI-Rev1 | TAGAAGACGCGCGCTCACACCACGCCGCC |
| eGFP-BbsI-Fw1 | TAGAAGACTAAATGGTGAGCAAGGGCGAGGAG |
| eGFP-BbsI-Rev1 | TAGAAGACGCGCGCTTACTTGTACAGCTCGTCCATG |
| eGFP-BbsI-Rev3 | TAGAAGACCGCTTGTACAGCTCGTCCATGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Epremyan, K.K.; Rogov, A.G.; Goleva, T.N.; Lavrushkina, S.V.; Zinovkin, R.A.; Zvyagilskaya, R.A. Altered Mitochondrial Morphology and Bioenergetics in a New Yeast Model Expressing Aβ42. Int. J. Mol. Sci. 2023, 24, 900. https://doi.org/10.3390/ijms24020900
Epremyan KK, Rogov AG, Goleva TN, Lavrushkina SV, Zinovkin RA, Zvyagilskaya RA. Altered Mitochondrial Morphology and Bioenergetics in a New Yeast Model Expressing Aβ42. International Journal of Molecular Sciences. 2023; 24(2):900. https://doi.org/10.3390/ijms24020900
Chicago/Turabian StyleEpremyan, Khoren K., Anton G. Rogov, Tatyana N. Goleva, Svetlana V. Lavrushkina, Roman A. Zinovkin, and Renata A. Zvyagilskaya. 2023. "Altered Mitochondrial Morphology and Bioenergetics in a New Yeast Model Expressing Aβ42" International Journal of Molecular Sciences 24, no. 2: 900. https://doi.org/10.3390/ijms24020900
APA StyleEpremyan, K. K., Rogov, A. G., Goleva, T. N., Lavrushkina, S. V., Zinovkin, R. A., & Zvyagilskaya, R. A. (2023). Altered Mitochondrial Morphology and Bioenergetics in a New Yeast Model Expressing Aβ42. International Journal of Molecular Sciences, 24(2), 900. https://doi.org/10.3390/ijms24020900