
Gut Microbial-Derived Metabolites as Immune Modulators of T Helper 17 and Regulatory T Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Gut Microbiota-Derived Metabolites: Effect on Th17/Treg Balance and Disease
2.1. Produced by Bacteria from Dietary Components
2.1.1. Short-Chain Fatty Acids
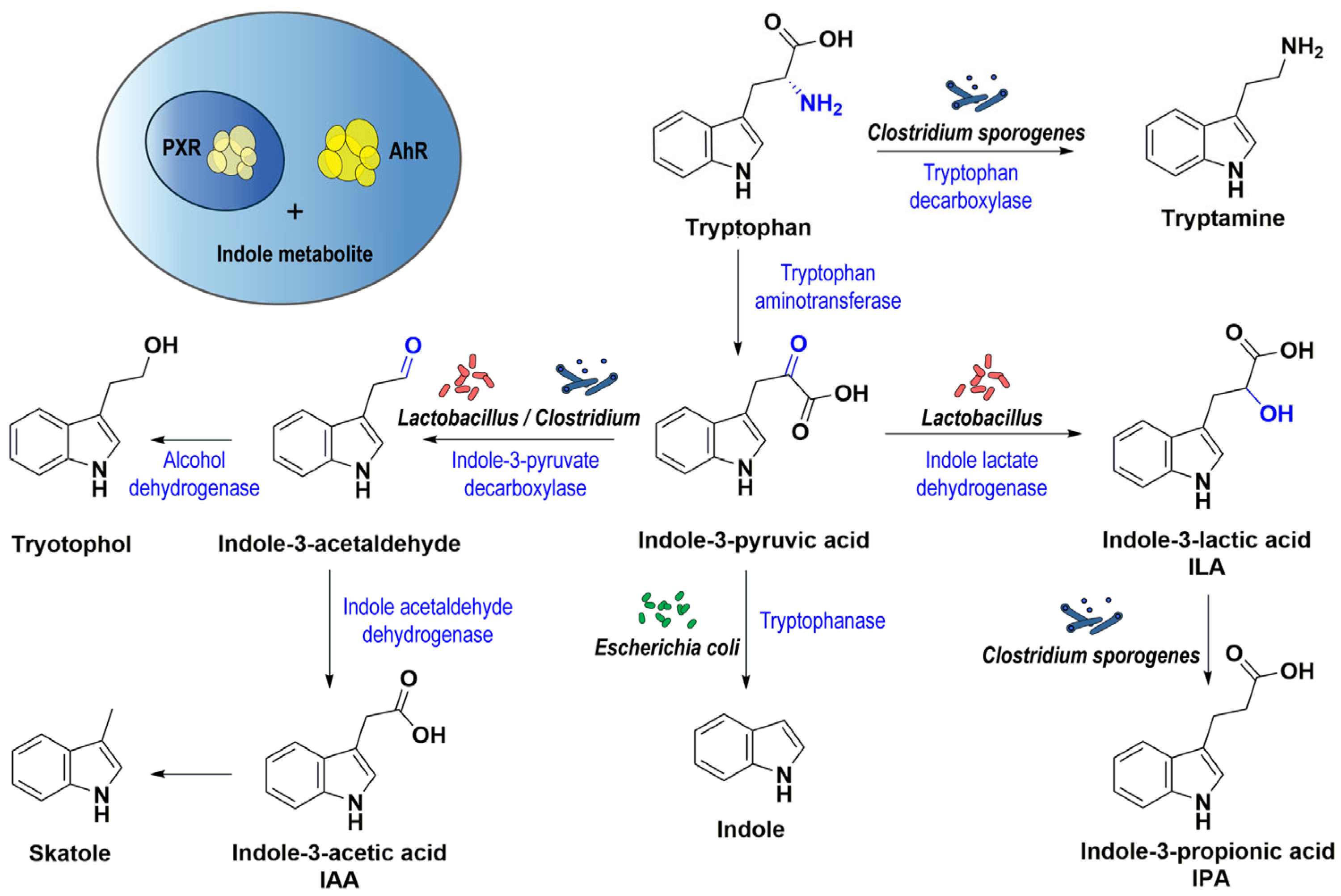
2.1.2. Indole Metabolites
2.1.3. Polyamines
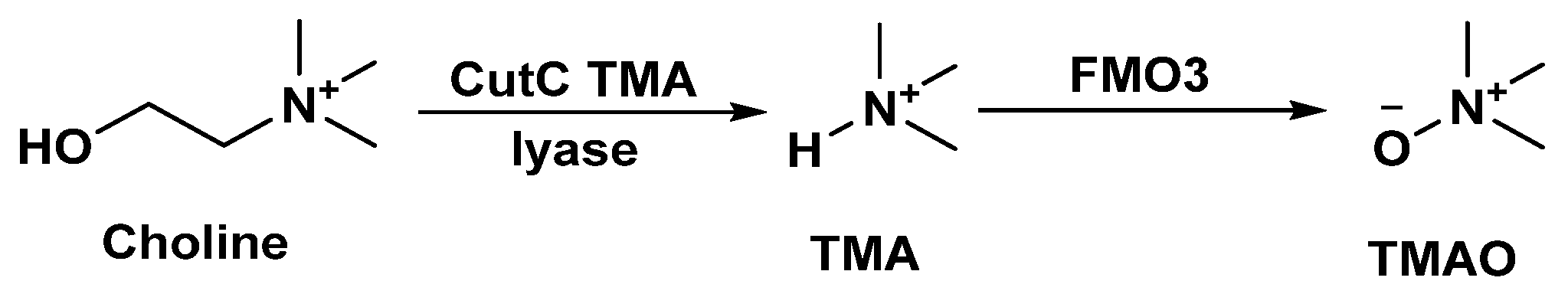
2.1.4. Microbial Choline Metabolism
2.2. Produced by the Host and Modified by Gut Microbiota
Secondary Bile Acids
3. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Schenten, D.; Medzhitov, R. The control of adaptive immune responses by the innate immune system. Adv. Immunol. 2011, 109, 87–124. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Leach, S.T.; Barres, R.; Hesson, L.B.; Grimm, M.C.; Simar, D. The Microbiota and Epigenetic Regulation of T Helper 17/Regulatory T Cells: In Search of a Balanced Immune System. Front. Immunol. 2017, 8, 417. [Google Scholar] [CrossRef] [PubMed]
- Dong, C. Cytokine Regulation and Function in T Cells. Annu. Rev. Immunol. 2021, 39, 51–76. [Google Scholar] [CrossRef]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef]
- Montes, M.; Zhang, X.; Berthelot, L.; Laplaud, D.A.; Brouard, S.; Jin, J.; Rogan, S.; Armao, D.; Jewells, V.; Soulillou, J.P.; et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin. Immunol. 2009, 130, 133–144. [Google Scholar] [CrossRef]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef]
- Huang, H.; Fang, M.; Jostins, L.; Umicevic Mirkov, M.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; Crins, F.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Fossiez, F.; Page, G.; Frappart, L.; Miossec, P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Leipe, J.; Grunke, M.; Dechant, C.; Reindl, C.; Kerzendorf, U.; Schulze-Koops, H.; Skapenko, A. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010, 62, 2876–2885. [Google Scholar] [CrossRef]
- Corthay, A. How do regulatory T cells work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- Curotto de Lafaille, M.A.; Lafaille, J.J. Natural and adaptive foxp3+ regulatory T cells: More of the same or a division of labor? Immunity 2009, 30, 626–635. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Wing, J.B.; Sakaguchi, S. Transcriptional and Epigenetic Control of Regulatory T Cell Development. Prog. Mol. Biol. Transl. Sci. 2015, 136, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gang, X.; Yang, S.; Cui, M.; Sun, L.; Li, Z.; Wang, G. The Alterations in and the Role of the Th17/Treg Balance in Metabolic Diseases. Front. Immunol. 2021, 12, 678355. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef]
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, M.; Rout, A.; Kingsley, T.; Kirchoff, R.; Singh, A.; Verma, V.; Kant, R.; Chaudhary, R. Role of gut microbiota in cardiovascular diseases. World J. Cardiol. 2020, 12, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef]
- Nilsson, N.E.; Kotarsky, K.; Owman, C.; Olde, B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem. Biophys. Res. Commun. 2003, 303, 1047–1052. [Google Scholar] [CrossRef]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Liu, B.; Li, Z. Regulatory T cells and Toll-like receptors: What is the missing link? Int. Immunopharmacol. 2009, 9, 528–533. [Google Scholar] [CrossRef]
- Hu, M.; Alashkar Alhamwe, B.; Santner-Nanan, B.; Miethe, S.; Harb, H.; Renz, H.; Potaczek, D.P.; Nanan, R.K. Short-Chain Fatty Acids Augment Differentiation and Function of Human Induced Regulatory T Cells. Int. J. Mol. Sci. 2022, 23, 5740. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Kashiwagi, I.; Morita, R.; Schichita, T.; Komai, K.; Saeki, K.; Matsumoto, M.; Takeda, K.; Nomura, M.; Hayashi, A.; Kanai, T.; et al. Smad2 and Smad3 Inversely Regulate TGF-beta Autoinduction in Clostridium butyricum-Activated Dendritic Cells. Immunity 2015, 43, 65–79. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Beguet-Crespel, F.; Marinelli, L.; Jamet, A.; Ledue, F.; Blottiere, H.M.; Lapaque, N. Butyrate produced by gut commensal bacteria activates TGF-beta1 expression through the transcription factor SP1 in human intestinal epithelial cells. Sci. Rep. 2018, 8, 9742. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Huda-Faujan, N.; Abdulamir, A.S.; Fatimah, A.B.; Anas, O.M.; Shuhaimi, M.; Yazid, A.M.; Loong, Y.Y. The impact of the level of the intestinal short chain Fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem. J. 2010, 4, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Zhu, F.; Hart, J.; Roalstad, S.; Graves, J.; Lynch, S.; Waubant, E.; US Network of Pediatric MS Centers. Gut microbiota in early pediatric multiple sclerosis: A case-control study. Eur. J. Neurol. 2016, 23, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Barone, M.; La Rosa, F.; Piancone, F.; Mendozzi, L.; d’Arma, A.; Rossi, V.; Pugnetti, L.; Roda, G.; et al. Alterations in Circulating Fatty Acid Are Associated with Gut Microbiota Dysbiosis and Inflammation in Multiple Sclerosis. Front. Immunol. 2020, 11, 1390. [Google Scholar] [CrossRef]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef]
- Ohigashi, S.; Sudo, K.; Kobayashi, D.; Takahashi, O.; Takahashi, T.; Asahara, T.; Nomoto, K.; Onodera, H. Changes of the intestinal microbiota, short chain fatty acids, and fecal pH in patients with colorectal cancer. Dig. Dis. Sci. 2013, 58, 1717–1726. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Barreiro, L.; Eixarch, H.; Cornejo, T.; Costa, C.; Castillo, M.; Mestre, L.; Guaza, C.; Martinez-Cuesta, M.D.C.; Tanoue, T.; Honda, K.; et al. Selected Clostridia Strains from the Human Microbiota and their Metabolite, Butyrate, Improve Experimental Autoimmune Encephalomyelitis. Neurotherapeutics 2021, 18, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Hui, W.; Yu, D.; Cao, Z.; Zhao, X. Butyrate inhibit collagen-induced arthritis via Treg/IL-10/Th17 axis. Int. Immunopharmacol. 2019, 68, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Du, H.X.; Yue, S.Y.; Niu, D.; Liu, C.; Zhang, L.G.; Chen, J.; Chen, Y.; Guan, Y.; Hua, X.L.; Li, C.; et al. Gut Microflora Modulates Th17/Treg Cell Differentiation in Experimental Autoimmune Prostatitis via the Short-Chain Fatty Acid Propionate. Front. Immunol. 2022, 13, 915218. [Google Scholar] [CrossRef]
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080. [Google Scholar] [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 1992, 103, 51–56. [Google Scholar] [CrossRef]
- Harig, J.M.; Soergel, K.H.; Komorowski, R.A.; Wood, C.M. Treatment of diversion colitis with short-chain-fatty acid irrigation. N. Engl. J. Med. 1989, 320, 23–28. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef]
- DeMoss, R.D.; Moser, K. Tryptophanase in diverse bacterial species. J. Bacteriol. 1969, 98, 167–171. [Google Scholar] [CrossRef]
- Gao, K.; Pi, Y.; Mu, C.L.; Peng, Y.; Huang, Z.; Zhu, W.Y. Antibiotics-induced modulation of large intestinal microbiota altered aromatic amino acid profile and expression of neurotransmitters in the hypothalamus of piglets. J. Neurochem. 2018, 146, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Pi, Y.; Mu, C.L.; Farzi, A.; Liu, Z.; Zhu, W.Y. Increasing carbohydrate availability in the hindgut promotes hypothalamic neurotransmitter synthesis: Aromatic amino acids linking the microbiota-brain axis. J. Neurochem. 2019, 149, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Ding, C.; Zhao, W.; Xu, L.; Tian, H.; Gong, J.; Zhu, M.; Li, J.; Li, N. Antibiotics-induced depletion of mice microbiota induces changes in host serotonin biosynthesis and intestinal motility. J. Transl. Med. 2017, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Aragozzini, F.; Ferrari, A.; Pacini, N.; Gualandris, R. Indole-3-lactic acid as a tryptophan metabolite produced by Bifidobacterium spp. Appl. Environ. Microbiol. 1979, 38, 544–546. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef]
- Konopelski, P.; Ufnal, M. Indoles-Gut Bacteria Metabolites of Tryptophan with Pharmacotherapeutic Potential. Curr. Drug Metab. 2018, 19, 883–890. [Google Scholar] [CrossRef]
- Pernomian, L.; Duarte-Silva, M.; de Barros Cardoso, C.R. The Aryl Hydrocarbon Receptor (AHR) as a Potential Target for the Control of Intestinal Inflammation: Insights from an Immune and Bacteria Sensor Receptor. Clin. Rev. Allergy Immunol. 2020, 59, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal. Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef]
- Qin, Y.; Gao, C.; Luo, J. Metabolism Characteristics of Th17 and Regulatory T Cells in Autoimmune Diseases. Front. Immunol. 2022, 13, 828191. [Google Scholar] [CrossRef]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 866–877. [Google Scholar] [CrossRef]
- Shen, J.; Yang, L.; You, K.; Chen, T.; Su, Z.; Cui, Z.; Wang, M.; Zhang, W.; Liu, B.; Zhou, K.; et al. Indole-3-Acetic Acid Alters Intestinal Microbiota and Alleviates Ankylosing Spondylitis in Mice. Front. Immunol. 2022, 13, 762580. [Google Scholar] [CrossRef]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell. Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Negatu, D.A.; Yamada, Y.; Xi, Y.; Go, M.L.; Zimmerman, M.; Ganapathy, U.; Dartois, V.; Gengenbacher, M.; Dick, T. Gut Microbiota Metabolite Indole Propionic Acid Targets Tryptophan Biosynthesis in Mycobacterium tuberculosis. mBio 2019, 10, e02781-18. [Google Scholar] [CrossRef]
- Aoki, R.; Aoki-Yoshida, A.; Suzuki, C.; Takayama, Y. Indole-3-Pyruvic Acid, an Aryl Hydrocarbon Receptor Activator, Suppresses Experimental Colitis in Mice. J. Immunol. 2018, 201, 3683–3693. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef]
- Konopelski, P.; Mogilnicka, I. Biological Effects of Indole-3-Propionic Acid, a Gut Microbiota-Derived Metabolite, and Its Precursor Tryptophan in Mammals’ Health and Disease. Int. J. Mol. Sci. 2022, 23, 1222. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lee, J.H.; Lee, J. Diverse roles of microbial indole compounds in eukaryotic systems. Biol. Rev. Camb. Philos. Soc. 2021, 96, 2522–2545. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, R.; Ilha, M.; Vaittinen, M.; Kaminska, D.; Mannisto, V.; Karja, V.; Tuomainen, M.; Hanhineva, K.; Romeo, S.; Pajukanta, P.; et al. Indole-3-Propionic Acid, a Gut-Derived Tryptophan Metabolite, Associates with Hepatic Fibrosis. Nutrients 2021, 13, 3509. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Mare, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Sommella, E.; Salviati, E.; Campiglia, P.; Ganguli, K.; Djebali, K.; Zhu, W.; Walker, W.A. Indole-3-lactic acid, a metabolite of tryptophan, secreted by Bifidobacterium longum subspecies infantis is anti-inflammatory in the immature intestine. Pediatr. Res. 2020, 88, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Fuchs, D.; Widner, B.; Glover, C.; Henderson, D.C.; Allen-Mersh, T.G. Serum tryptophan decrease correlates with immune activation and impaired quality of life in colorectal cancer. Br. J. Cancer 2002, 86, 1691–1696. [Google Scholar] [CrossRef]
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 2021, 12, 5674. [Google Scholar] [CrossRef]
- Holbert, C.E.; Cullen, M.T.; Casero, R.A., Jr.; Stewart, T.M. Polyamines in cancer: Integrating organismal metabolism and antitumour immunity. Nat. Rev. Cancer 2022, 22, 467–480. [Google Scholar] [CrossRef]
- Kurihara, S. Polyamine metabolism and transport in gut microbes. Biosci. Biotechnol. Biochem. 2022, 86, 957–966. [Google Scholar] [CrossRef]
- Tofalo, R.; Cocchi, S.; Suzzi, G. Polyamines and Gut Microbiota. Front. Nutr. 2019, 6, 16. [Google Scholar] [CrossRef]
- Stewart, T.M.; Holbert, C.E.; Casero, R.A., Jr. Helping the helpers: Polyamines help maintain helper T-cell lineage fidelity. Immunometabolism 2022, 4, e00002. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Huang, J.F.; Cheng, Y.; Dai, M.Y.; Zhu, W.F.; Yang, X.W.; Gonzalez, F.J.; Li, F. Polyamine metabolism links gut microbiota and testicular dysfunction. Microbiome 2021, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Carriche, G.M.; Almeida, L.; Stuve, P.; Velasquez, L.; Dhillon-LaBrooy, A.; Roy, U.; Lindenberg, M.; Strowig, T.; Plaza-Sirvent, C.; Schmitz, I.; et al. Regulating T-cell differentiation through the polyamine spermidine. J. Allergy Clin. Immunol. 2021, 147, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Pugin, B.; Barcik, W.; Westermann, P.; Heider, A.; Wawrzyniak, M.; Hellings, P.; Akdis, C.A.; O’Mahony, L. A wide diversity of bacteria from the human gut produces and degrades biogenic amines. Microb. Ecol. Health Dis. 2017, 28, 1353881. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Baixauli, F.; Sanin, D.E.; Edwards-Hicks, J.; Villa, M.; Kabat, A.M.; Kaminski, M.M.; Stanckzak, M.; Weiss, H.J.; Grzes, K.M.; et al. Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell 2021, 184, 4186–4202 e4120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zheng, C.; Cao, J.; Cao, G.; Shou, P.; Lin, L.; Velletri, T.; Jiang, M.; Chen, Q.; Han, Y.; et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ. 2016, 23, 1850–1861. [Google Scholar] [CrossRef]
- Alexander, M.; Ang, Q.Y.; Nayak, R.R.; Bustion, A.E.; Sandy, M.; Zhang, B.; Upadhyay, V.; Pollard, K.S.; Lynch, S.V.; Turnbaugh, P.J. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe 2022, 30, 17–30. [Google Scholar] [CrossRef]
- Hesterberg, R.S.; Cleveland, J.L.; Epling-Burnette, P.K. Role of Polyamines in Immune Cell Functions. Med. Sci. 2018, 6, 22. [Google Scholar] [CrossRef]
- Wu, R.; Chen, X.; Kang, S.; Wang, T.; Gnanaprakasam, J.R.; Yao, Y.; Liu, L.; Fan, G.; Burns, M.R.; Wang, R. De novo synthesis and salvage pathway coordinately regulate polyamine homeostasis and determine T cell proliferation and function. Sci. Adv. 2020, 6, eabc4275. [Google Scholar] [CrossRef]
- Wagner, A.; Wang, C.; Fessler, J.; DeTomaso, D.; Avila-Pacheco, J.; Kaminski, J.; Zaghouani, S.; Christian, E.; Thakore, P.; Schellhaass, B.; et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 2021, 184, 4168–4185. [Google Scholar] [CrossRef]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, C.C.; Croyal, M.; Ene, L.; Aguesse, A.; Billon-Crossouard, S.; Krempf, M.; Lemoine, S.; Guebre-Egziabher, F.; Juillard, L.; Soulage, C.O. Elevation of Trimethylamine-N-Oxide in Chronic Kidney Disease: Contribution of Decreased Glomerular Filtration Rate. Toxins 2019, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dai, M. Trimethylamine N-Oxide Generated by the Gut Microbiota Is Associated with Vascular Inflammation: New Insights into Atherosclerosis. Mediators Inflamm. 2020, 2020, 4634172. [Google Scholar] [CrossRef]
- Rath, S.; Rox, K.; Kleine Bardenhorst, S.; Schminke, U.; Dorr, M.; Mayerle, J.; Frost, F.; Lerch, M.M.; Karch, A.; Bronstrup, M.; et al. Higher Trimethylamine-N-Oxide Plasma Levels with Increasing Age Are Mediated by Diet and Trimethylamine-Forming Bacteria. mSystems 2021, 6, e0094521. [Google Scholar] [CrossRef]
- Senthong, V.; Kiatchoosakun, S.; Wongvipaporn, C.; Phetcharaburanin, J.; Tatsanavivat, P.; Sritara, P.; Phrommintikul, A. Gut microbiota-generated metabolite, trimethylamine-N-oxide, and subclinical myocardial damage: A multicenter study from Thailand. Sci. Rep. 2021, 11, 14963. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Yang, F.; Zhao, R.; Pan, X.; Liang, J.; Tian, L.; Li, X.; Liu, L.; Xing, Y.; et al. Gut Microbiota-Dependent Marker TMAO in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front. Pharmacol. 2019, 10, 1360. [Google Scholar] [CrossRef]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef]
- Johnson, C.; Prokopienko, A.J.; West, R.E., 3rd; Nolin, T.D.; Stubbs, J.R. Decreased Kidney Function Is Associated with Enhanced Hepatic Flavin Monooxygenase Activity and Increased Circulating Trimethylamine N-Oxide Concentrations in Mice. Drug Metab. Dispos. 2018, 46, 1304–1309. [Google Scholar] [CrossRef]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H.; et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood 2020, 136, 501–515. [Google Scholar] [CrossRef]
- Bain, M.A.; Fornasini, G.; Evans, A.M. Trimethylamine: Metabolic, pharmacokinetic and safety aspects. Curr. Drug Metab. 2005, 6, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Craciun, S.; Balskus, E.P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Steinke, I.; Ghanei, N.; Govindarajulu, M.; Yoo, S.; Zhong, J.; Amin, R.H. Drug Discovery and Development of Novel Therapeutics for Inhibiting TMAO in Models of Atherosclerosis and Diabetes. Front. Physiol. 2020, 11, 567899. [Google Scholar] [CrossRef]
- Tang, W.H. Trimethylamine N-Oxide as a Novel Therapeutic Target in CKD. J. Am. Soc. Nephrol. 2016, 27, 8–10. [Google Scholar] [CrossRef]
- Krueger, S.K.; Williams, D.E. Mammalian flavin-containing monooxygenases: Structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol. Ther. 2005, 106, 357–387. [Google Scholar] [CrossRef]
- Gupta, N.; Buffa, J.A.; Roberts, A.B.; Sangwan, N.; Skye, S.M.; Li, L.; Ho, K.J.; Varga, J.; DiDonato, J.A.; Tang, W.H.W.; et al. Targeted Inhibition of Gut Microbial Trimethylamine N-Oxide Production Reduces Renal Tubulointerstitial Fibrosis and Functional Impairment in a Murine Model of Chronic Kidney Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1239–1255. [Google Scholar] [CrossRef]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef]
- Pathak, P.; Helsley, R.N.; Brown, A.L.; Buffa, J.A.; Choucair, I.; Nemet, I.; Gogonea, C.B.; Gogonea, V.; Wang, Z.; Garcia-Garcia, J.C.; et al. Small molecule inhibition of gut microbial choline trimethylamine lyase activity alters host cholesterol and bile acid metabolism. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1474–H1486. [Google Scholar] [CrossRef]
- Schugar, R.C.; Gliniak, C.M.; Osborn, L.J.; Massey, W.; Sangwan, N.; Horak, A.; Banerjee, R.; Orabi, D.; Helsley, R.N.; Brown, A.L.; et al. Gut microbe-targeted choline trimethylamine lyase inhibition improves obesity via rewiring of host circadian rhythms. Elife 2022, 11, e63998. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.T.; Machalz, D.; Pach, S.; Wolber, G. A benzoxazole derivative as an inhibitor of anaerobic choline metabolism by human gut microbiota. RSC Med. Chem. 2020, 11, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.; Swiderek, K. Discovery of a Histidine-Based Scaffold as an Inhibitor of Gut Microbial Choline Trimethylamine-Lyase. ChemMedChem 2020, 15, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.T.; Deganutti, G.; Reynolds, C.A. Peptidomimetic-based approach toward inhibitors of microbial trimethylamine lyases. Chem. Biol. Drug Des. 2021, 97, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Paik, D.; Yao, L.; Zhang, Y.; Bae, S.; D’Agostino, G.D.; Zhang, M.; Kim, E.; Franzosa, E.A.; Avila-Pacheco, J.; Bisanz, J.E.; et al. Human gut bacteria produce Th17-modulating bile acid metabolites. Nature 2022, 603, 907–912. [Google Scholar] [CrossRef]
- Jin, L.; Martynowski, D.; Zheng, S.; Wada, T.; Xie, W.; Li, Y. Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORgamma. Mol. Endocrinol. 2010, 24, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, P.; Wu, J.; Xue, X.; Song, J.; Sutton, S.W.; Sablad, M.; Yu, J.; Nelen, M.I.; Liu, X.; Castro, G.; et al. Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 12163–12168. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Sefik, E.; Geva-Zatorsky, N.; Oh, S.; Konnikova, L.; Zemmour, D.; McGuire, A.M.; Burzyn, D.; Ortiz-Lopez, A.; Lobera, M.; Yang, J.; et al. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 2015, 349, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Xue, L.N.; Xu, K.Q.; Zhang, W.; Wang, Q.; Wu, J.; Wang, X.Y. Associations between vitamin D receptor polymorphisms and susceptibility to ulcerative colitis and Crohn’s disease: A meta-analysis. Inflamm. Bowel. Dis. 2013, 19, 54–60. [Google Scholar] [CrossRef]
- Yang, B.H.; Hagemann, S.; Mamareli, P.; Lauer, U.; Hoffmann, U.; Beckstette, M.; Fohse, L.; Prinz, I.; Pezoldt, J.; Suerbaum, S.; et al. Foxp3(+) T cells expressing RORgammat represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal. Immunol. 2016, 9, 444–457. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Dorrestein, P.G.E.; Collins, S.; Panitchpakdi, M.; Belda-Ferre, P.; Stewart, A.; Wang, M.; Jarmusch, A.; Avila-Pacheco, J.; Plichta, D.; Aron, A.; et al. A Synthesis-Based Reverse Metabolomics Approach for the Discovery of Chemical Structures from Humans and Animals. Research Square 2021. [Google Scholar] [CrossRef]
- Dvorak, Z.; Kopp, F.; Costello, C.M.; Kemp, J.S.; Li, H.; Vrzalova, A.; Stepankova, M.; Bartonkova, I.; Jiskrova, E.; Poulikova, K.; et al. Targeting the pregnane X receptor using microbial metabolite mimicry. EMBO Mol. Med. 2020, 12, e11621. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo-Barreiro, L.; Zhang, L.; Abdel-Rahman, S.A.; Naik, S.P.; Gabr, M. Gut Microbial-Derived Metabolites as Immune Modulators of T Helper 17 and Regulatory T Cells. Int. J. Mol. Sci. 2023, 24, 1806. https://doi.org/10.3390/ijms24021806
Calvo-Barreiro L, Zhang L, Abdel-Rahman SA, Naik SP, Gabr M. Gut Microbial-Derived Metabolites as Immune Modulators of T Helper 17 and Regulatory T Cells. International Journal of Molecular Sciences. 2023; 24(2):1806. https://doi.org/10.3390/ijms24021806
Chicago/Turabian StyleCalvo-Barreiro, Laura, Longfei Zhang, Somaya A. Abdel-Rahman, Shivani Paritosh Naik, and Moustafa Gabr. 2023. "Gut Microbial-Derived Metabolites as Immune Modulators of T Helper 17 and Regulatory T Cells" International Journal of Molecular Sciences 24, no. 2: 1806. https://doi.org/10.3390/ijms24021806
APA StyleCalvo-Barreiro, L., Zhang, L., Abdel-Rahman, S. A., Naik, S. P., & Gabr, M. (2023). Gut Microbial-Derived Metabolites as Immune Modulators of T Helper 17 and Regulatory T Cells. International Journal of Molecular Sciences, 24(2), 1806. https://doi.org/10.3390/ijms24021806