
Effects of Defective Unloading and Recycling of PCNA Revealed by the Analysis of ELG1 Mutants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
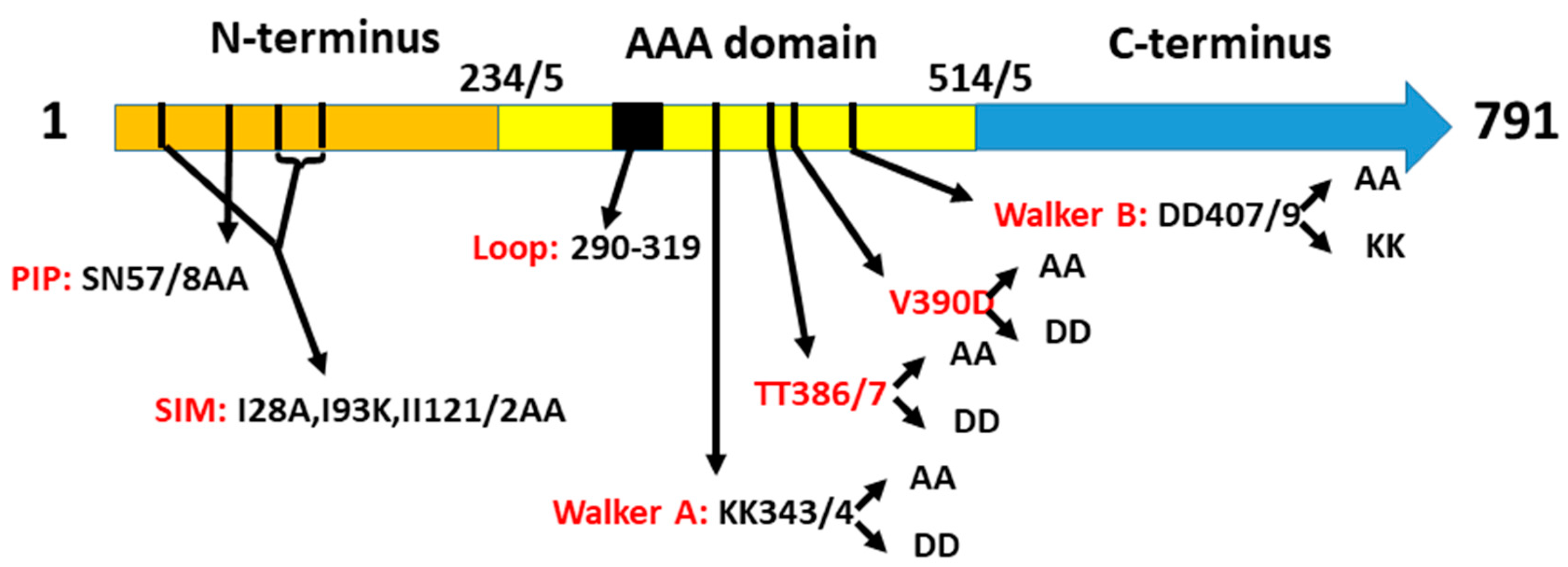
2.1. A Collection of elg1 Mutants
- (1)
- A sequence at the N-terminus of Elg1 bears resemblance to the PCNA-interacting peptide (PIP) of Rfc1 [29]. We mutated amino acids N55 and S57 to alanine (elg1-NS55,57AA; this allele is hereafter referred to as elg1-PIP).
- (2)
- Three SUMO-interacting motifs were found in the same region, and point mutations in them (I28A I93K II121/2AA) reduced the interaction between Elg1 and SUMO [29]. We refer to this allele as elg1-SIM.
- (3)
- The third region mutated in the N terminus was an unstructured loop region, spanning from aa 290 to aa 319 and containing a hydrophobic patch. This site is a possible protein–protein interaction site and is conserved throughout all Elg1 orthologs but is absent in other components of the RFC complex and in the other Rfc1-like proteins [30]. We deleted this loop and replaced it by a short peptide. This allele is referred to as elg1-loop.
- (4)
- By homology modeling of the Elg1 protein on the solved crystal structure of the RFC complex together with PCNA, it was determined that threonines 386/7 in Elg1 correspond to asparagines 694 and 695 of the human Rfc1, which were shown to be at the interface of the two proteins [30]. These residues were mutated to either aspartic acid (hereafter referred to as elg1-TT386/7DD) or to alanine (elg1-TT386/7AA).
- (5)
- These mutants were also combined with the SIM mutation, creating SIM+AA and SIM+DD alleles.
- (6)
- Elg1 and all members of the AAA+ family contain Walker A and Walker B motifs [31]. The current models suggest that ATP binding is crucial for the association of Elg1-RLC with PCNA for its unloading, and that the hydrolysis of ATP is important for the subsequent detachment of Elg1-RFC from PCNA, allowing its recycling [23,32]. We mutated the two lysines (at positions 343, 344) either to aspartic acid (elg1-KK343/4DD) or to alanine (elg1-KK343/4AA). These are referred to hereafter as WalkerA-DD (WA-DD) and Walker A-AA (WA-AA).
- (7)
- We also mutated two aspartic acids at positions 407,409 that lie within the Walker B motif, which are conserved between Elg1 and its human ortholog ATAD5 [23]. These residues are thought to be crucial for ATP binding and/or hydrolysis. They were mutated either to lysine (elg1-DD407/409KK) or to alanine (elg1-DD407/9AA), hereafter referred to as Walker B-KK (WB-KK) or Walker B-AA (WB-AA).
- (8)
- We also created a double Walker A + Walker B mutant: elg1-KK343/4DD+DD407/409AA, hereafter referred to as elg1-WAB.
- (9)
- Finally, we mutated position 390 from valine to aspartic acid (elg1-V390D) and to alanine (elg1-V390A). These mutations disrupt the similarity in Elg1 at positions 381-390 (LLDFTTTHYV) to a small patch of the DNA repair and telomeric protein Yku80 (aa 450-456 LLDrTTTsgV). Moreover, the valine is conserved throughout Elg1 orthologs [23].
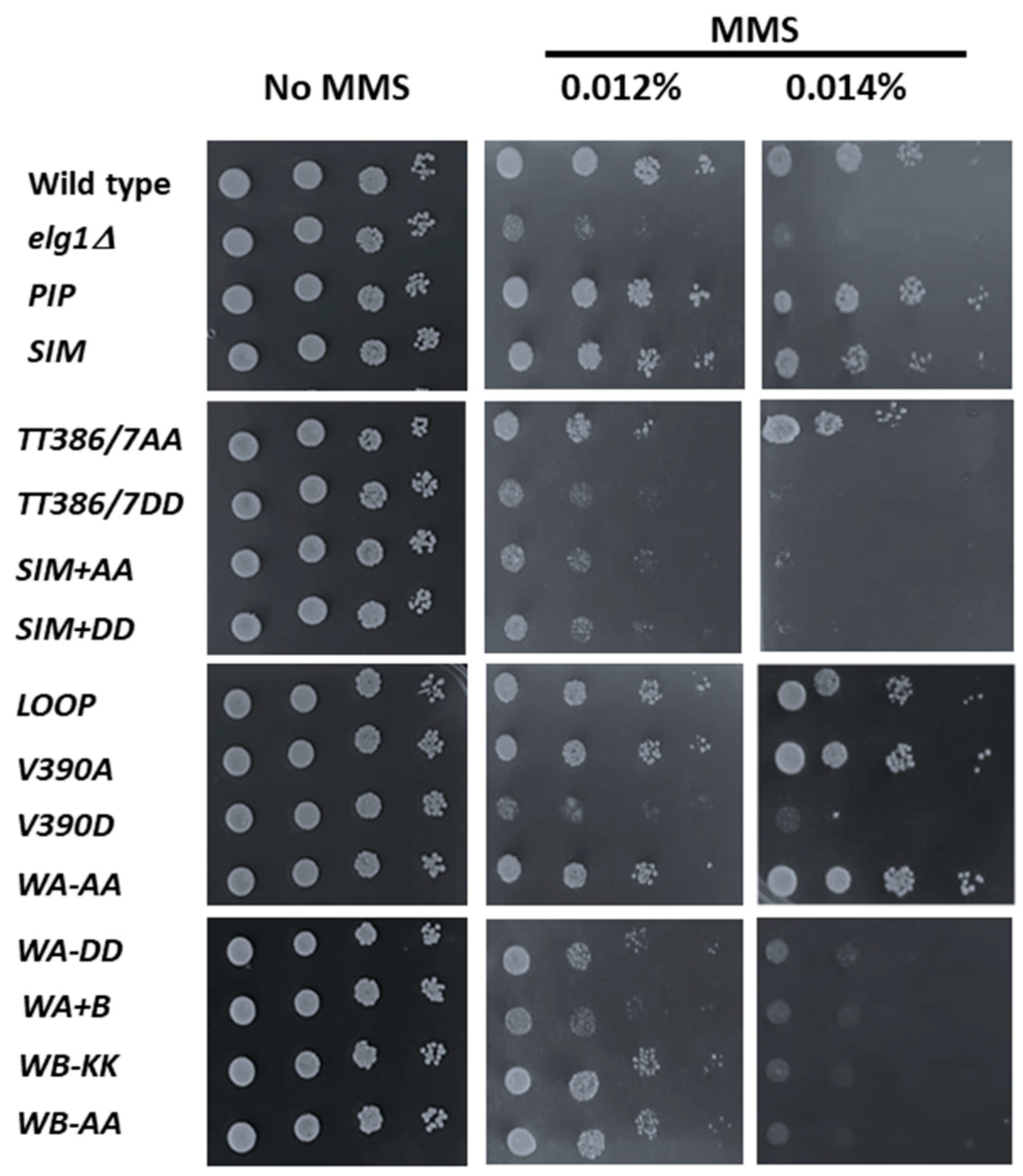
2.2. Sensitivity to DNA Damage
2.3. Mutation Rate
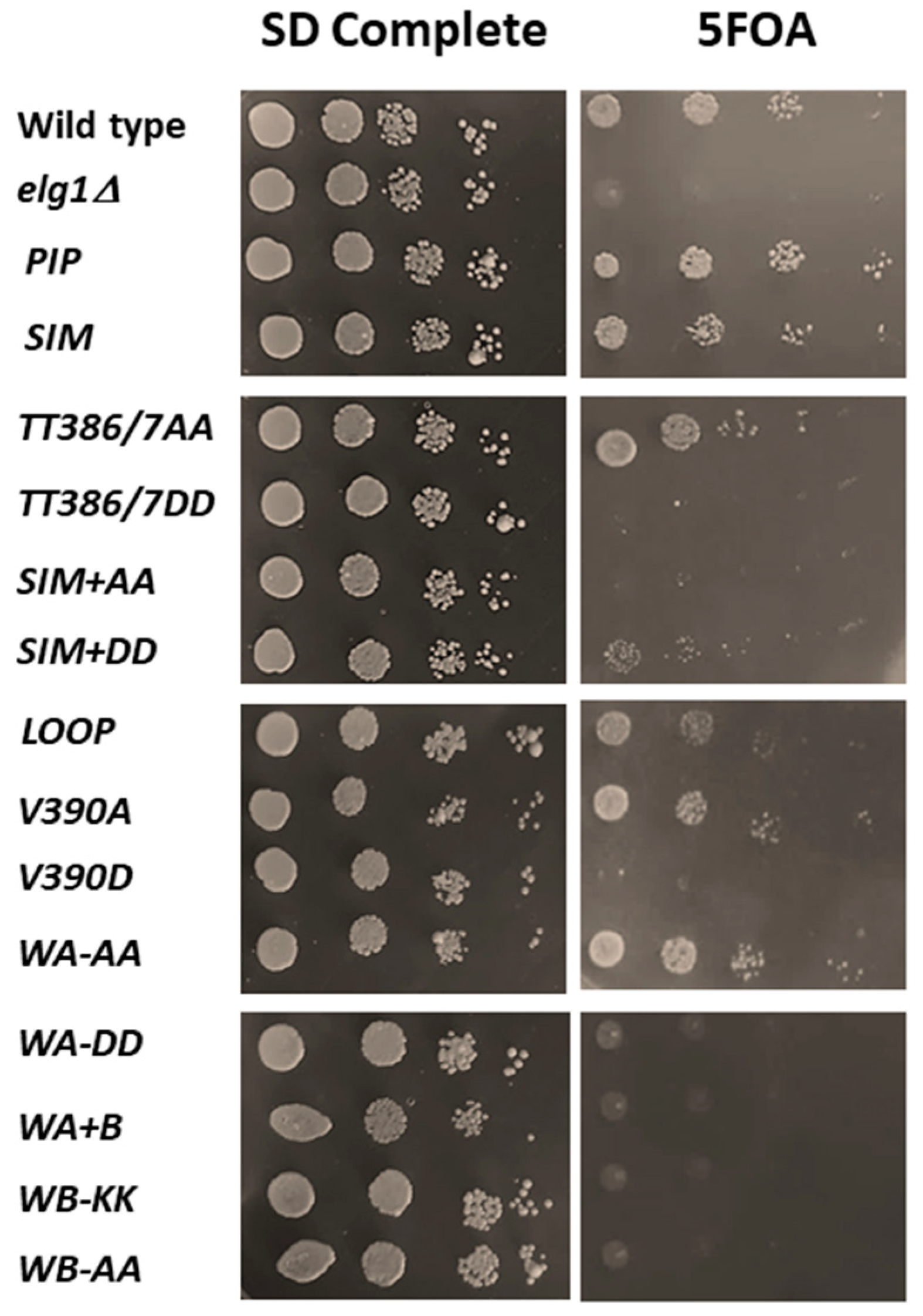
2.4. Rescue of the Synthetic Lethality of Elg1Δ Srs2Δ Double Mutants
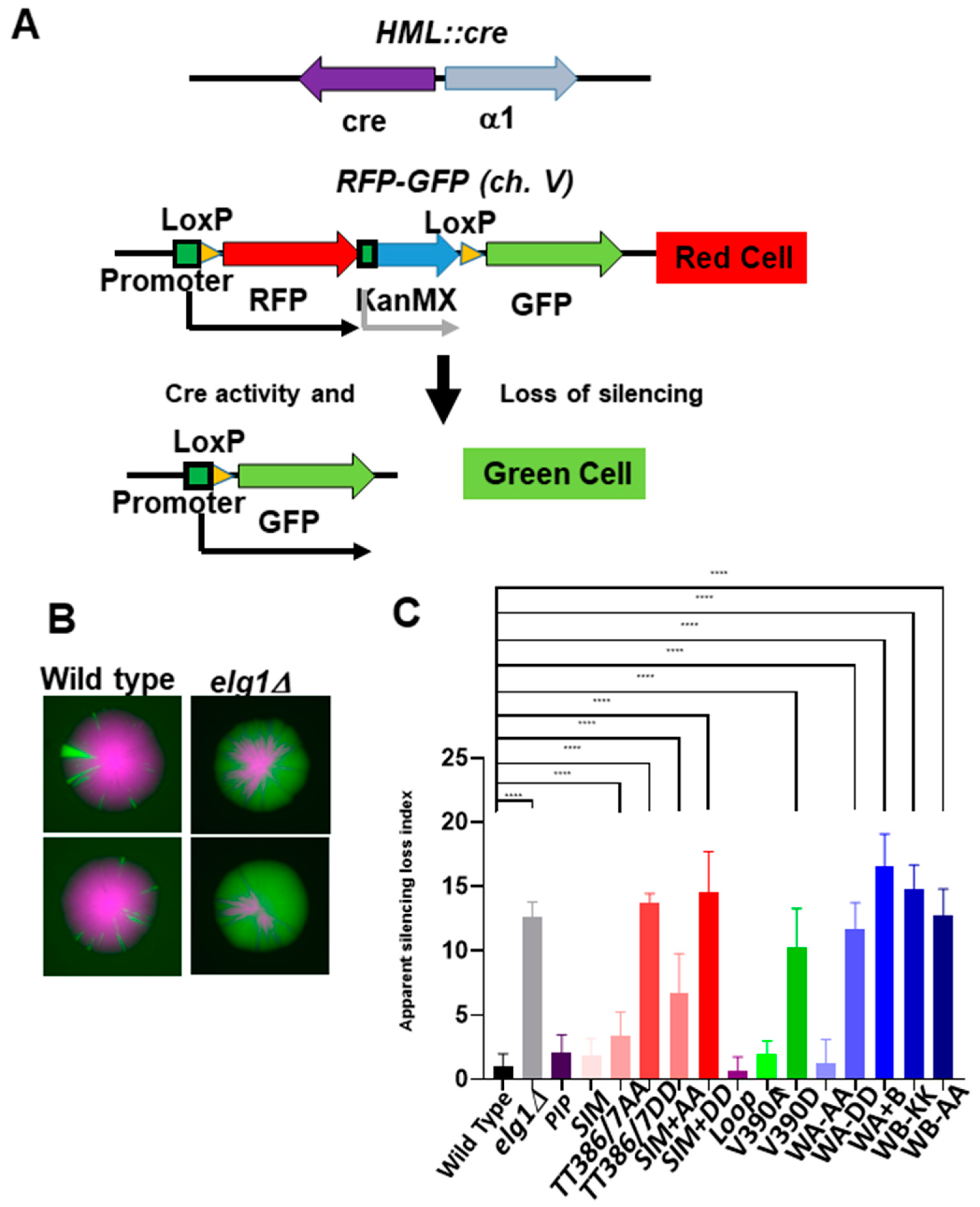
2.5. Gene Silencing in elg1 Mutants
2.6. PCNA Accumulation Levels in the elg1 Mutants
3. Discussion
4. Materials and Methods
4.1. Chromatin Fractionation Assay
4.2. Quantification of Silencing Loss Using Flow Cytometry
4.3. Staining for Flow Cytometry
4.4. Mutation Measurement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed–fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef] [PubMed]
- Eyre-Walker, A.; Keightley, P.D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 2007, 8, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Arbel, M.; Liefshitz, B.; Kupiec, M. DNA damage bypass pathways and their effect on mutagenesis in yeast. FEMS Microbiol. Rev. 2020, 45, fuaa038. [Google Scholar] [CrossRef]
- Bellí, G.; Colomina, N.; Castells-Roca, L.; Lorite, N.P. Post-Translational Modifications of PCNA: Guiding for the Best DNA Damage Tolerance Choice. J. Fungi 2022, 8, 621. [Google Scholar] [CrossRef] [PubMed]
- McClure, A.W.; Canal, B.; Diffley, J.F.X. A DNA replication fork-centric view of the budding yeast DNA damage response. DNA Repair 2022, 119, 103393. [Google Scholar] [CrossRef]
- Bar-Ziv, R.; Voichek, Y.; Barkai, N. Chromatin dynamics during DNA replication. Genome Res. 2016, 26, 1245–1256. [Google Scholar] [CrossRef]
- Yuan, G.C.; Liu, Y.J.; Dion, M.F.; Slack, M.D.; Wu, L.F.; Altschuler, S.J.; Rando, O.J. Molecular biology: Genome-scale identification of nucleosome positions in S. cerevisiae. Science 2005, 309, 1112178. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.D.; Widom, J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2000, 296, 979–987. [Google Scholar] [CrossRef]
- Lu, X.; Simon, M.D.; Chodaparambil, J.V.; Hansen, J.C.; Shokat, K.M.; Luger, K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol. 2008, 15, 1122–1124. [Google Scholar] [CrossRef]
- Kalashnikova, A.A.; Porter-Goff, M.E.; Muthurajan, U.M.; Luger, K.; Hansen, J.C. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J. R. Soc. Interface 2013, 10, 20121022. [Google Scholar] [CrossRef]
- Reverón-Gómez, N.; González-Aguilera, C.; Stewart-Morgan, K.R.; Petryk, N.; Flury, V.; Graziano, S.; Johansen, J.V.; Jakobsen, J.S.; Alabert, C.; Groth, A. Accurate Recycling of Parental Histones Reproduces the Histone Modification Landscape during DNA Replication. Mol. Cell 2018, 72, 239–249.e5. [Google Scholar] [CrossRef] [PubMed]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2010, 107, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Arbel, M.; Choudhary, K.; Tfilin, O.; Kupiec, M. PCNA loaders and unloaders—One ring that rules them all. Genes 2021, 12, 1812. [Google Scholar] [CrossRef] [PubMed]
- Paunesku, T.; Mittal, S.; Protić, M.; Oryhon, J.; Korolev, S.V.; Joachimiak, A.; Woloschak, G.E. Proliferating cell nuclear antigen (PCNA): Ringmaster of the genome. Int. J. Radiat. Biol. 2001, 77, 1007–1021. [Google Scholar] [CrossRef]
- Madru, C.; Henneke, G.; Raia, P.; Hugonneau-Beaufet, I.; Pehau-Arnaudet, G.; England, P.; Lindahl, E.; Delarue, M.; Carroni, M.; Sauguet, L. Structural basis for the increased processivity of D-family DNA polymerases in complex with PCNA. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Zhang, W.; Qin, Z.; Zhang, X.; Xiao, W. Roles of sequential ubiquitination of PCNA in DNA-damage tolerance. FEBS Lett. 2011, 585, 2786–2794. [Google Scholar] [CrossRef]
- Janke, R.; King, G.A.; Kupiec, M.; Rine, J. Pivotal roles of PCNA loading and unloading in heterochromatin function. Proc. Natl. Acad. Sci. USA 2018, 115, E2030–E2039. [Google Scholar] [CrossRef]
- Lee, K.-y.; Park, S.H. Eukaryotic clamp loaders and unloaders in the maintenance of genome stability. Exp. Mol. Med. 2020, 52, 1948–1958. [Google Scholar] [CrossRef]
- Dovrat, D.; Stodola, J.L.; Burgers, P.M.J.; Aharoni, A. Sequential switching of binding partners on PCNA during in vitro Okazaki fragment maturation. Proc. Natl. Acad. Sci. USA 2014, 111, 14118–14123. [Google Scholar] [CrossRef]
- Thakar, T.; Leung, W.; Nicolae, C.M.; Clements, K.E.; Shen, B.; Bielinsky, A.K.; Moldovan, G.L. Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun. 2020, 11, 2147. [Google Scholar] [CrossRef]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed]
- Motegi, A.; Liaw, H.J.; Lee, K.Y.; Roest, H.P.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.D.; Ding, H.; Hoeijmakers, J.H.J.; et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Ryu, E.; Lee, S.W.; Park, J.; Ha, N.Y.; Ra, J.S.; Kim, Y.J.; Kim, J.; Abdel-Rahman, M.; Park, S.H.; et al. Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat. Commun. 2019, 10, 2420. [Google Scholar] [CrossRef]
- Ben-Aroya, S.; Koren, A.; Liefshitz, B.; Steinlauf, R.; Kupiec, M. ELG1, a yeast gene required for genome stability, forms a complex related to replication factor C. Proc. Natl. Acad. Sci. USA 2003, 100, 9906–9911. [Google Scholar] [CrossRef] [PubMed]
- Sikdar, N.; Banerjee, S.; Lee, K.Y.; Wincovitch, S.; Pak, E.; Nakanishi, K.; Jasin, M.; Dutra, A.; Myung, K. DNA damage responses by human ELGI in S phase are important to maintain genomic integrity. Cell Cycle 2009, 8, 3199–3207. [Google Scholar] [CrossRef]
- Smolikov, S.; Mazor, Y.; Krauskopf, A. ELG1, a regulator of genome stability, has a role in telomere length regulation and in silencing. Proc. Natl. Acad. Sci. USA 2004, 101, 1656–1661. [Google Scholar] [CrossRef]
- Choudhary, K.; Itzkovich, Z.; Alonso-Perez, E.; Bishara, H.; Dunn, B.; Sherlock, G.; Kupiec, M. S. cerevisiae Cells Can Grow without the Pds5 Cohesin Subunit. MBio 2022, 100, 9906–9911. [Google Scholar] [CrossRef]
- Choudhary, K.; Kupiec, M. The cohesin complex of yeasts: Sister chromatid cohesion and beyond. FEMS Microbiol. Rev. 2022, 11, 1–22. [Google Scholar] [CrossRef]
- Parnas, O.; Zipin-Roitman, A.; Pfander, B.; Liefshitz, B.; Mazor, Y.; Ben-Aroya, S.; Jentsch, S.; Kupiec, M. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010, 29, 2611–2622. [Google Scholar] [CrossRef]
- Venclovas, Č.; Colvin, M.E.; Thelen, M.P. Molecular modeling-based analysis of interactions in the RFC-dependent clamp-loading process. Protein Sci. 2008, 11, 0214302. [Google Scholar] [CrossRef]
- Kang, S.; Warner, M.D.; Bell, S.P. Multiple Functions for Mcm2-7 ATPase Motifs during Replication Initiation. Mol. Cell 2014, 55, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.B.; Brown, G.W. The N- and C-termini of Elg1 contribute to the maintenance of genome stability. DNA Repair 2008, 7, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; North, M.; Erixon, K.; Walters, K.; Jenssen, D.; Goldman, A.S.H.; Helleday, T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005, 33, 3799–3811. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, K.; Sebesta, M.; Pacesa, M.; Sau, S.; Bronstein, A.; Parnas, O.; Liefshitz, B.; Venclovas, Č.; Krejci, L.; Kupiec, M. A structure-function analysis of the yeast Elg1 protein reveals the importance of PCNA unloading in genome stability maintenance. Nucleic Acids Res. 2017, 45, 3189–3203. [Google Scholar] [CrossRef]
- Goellner, E.M.; Smith, C.E.; Campbell, C.S.; Hombauer, H.; Desai, A.; Putnam, C.D.; Kolodner, R.D. PCNA and Msh2-Msh6 Activate an Mlh1-Pms1 Endonuclease Pathway Required for Exo1-Independent Mismatch Repair. Mol. Cell 2014, 55, 291–304. [Google Scholar] [CrossRef]
- Tran, H.T.; Keen, J.D.; Kricker, M.; Resnick, M.A.; Gordenin, D.A. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 1997, 17, 2859–2865. [Google Scholar] [CrossRef]
- Paul Solomon Devakumar, L.J.; Gaubitz, C.; Lundblad, V.; Kelch, B.A.; Kubota, T. Effective mismatch repair depends on timely control of PCNA retention on DNA by the Elg1 complex. Nucleic Acids Res. 2019, 47, 6826–6841. [Google Scholar] [CrossRef]
- Aylon, Y.; Kupiec, M. The Checkpoint Protein Rad24 of Saccharomyces cerevisiae Is Involved in Processing Double-Strand Break Ends and in Recombination Partner Choice. Mol. Cell. Biol. 2003, 23, 6585–6596. [Google Scholar] [CrossRef]
- Arbel, M.; Bronstein, A.; Sau, S.; Liefshitz, B.; Kupiec, M. Access to pcna by srs2 and elg1 controls the choice between alternative repair pathways in saccharomyces cerevisiae. MBio 2020, 11, e00705-20. [Google Scholar] [CrossRef]
- Gazy, I.; Liefshitz, B.; Bronstein, A.; Parnas, O.; Atias, N.; Sharan, R.; Kupiec, M. A genetic screen for high copy number suppressors of the synthetic lethality between elg1Δ and srs2Δ in yeast. G3 Genes, Genomes, Genet. 2013, 3, 917–926. [Google Scholar] [CrossRef]
- Kubota, T.; Katou, Y.; Nakato, R.; Shirahige, K.; Donaldson, A.D. Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-wide and Requires Okazaki Fragment Ligation. Cell Rep. 2015, 12, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Gali, V.K.; Takahashi, T.S.; Kubota, T. PCNA Retention on DNA into G2/M Phase Causes Genome Instability in Cells Lacking Elg1. Cell Rep. 2016, 16, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shahar, T.R.; Castillo, A.G.; Osborne, M.J.; Borden, K.L.B.; Kornblatt, J.; Verreault, A. Two Fundamentally Distinct PCNA Interaction Peptides Contribute to Chromatin Assembly Factor 1 Function. Mol. Cell. Biol. 2009, 29, 6353–6365. [Google Scholar] [CrossRef] [PubMed]
- Kondratick, C.M.; Litman, J.M.; Shaffer, K.V.; Washington, M.T.; Dieckman, L.M. Crystal structures of PCNA mutant proteins defective in gene silencing suggest a novel interaction site on the front face of the PCNA ring. PLoS ONE 2018, 13, e0193333. [Google Scholar] [CrossRef] [PubMed]
- Liefshitz, B.; Steinlauf, R.; Friedl, A.; Eckardt-Schupp, F.; Kupiec, M. Genetic interactions between mutants of the “error-prone” repair group of Saccharomyces cerevisiae and their effect on recombination and mutagenesis. Mutat. Res.-DNA Repair 1998, 407, 135–145. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itzkovich, Z.; Choudhary, K.; Arbel, M.; Kupiec, M. Effects of Defective Unloading and Recycling of PCNA Revealed by the Analysis of ELG1 Mutants. Int. J. Mol. Sci. 2023, 24, 1568. https://doi.org/10.3390/ijms24021568
Itzkovich Z, Choudhary K, Arbel M, Kupiec M. Effects of Defective Unloading and Recycling of PCNA Revealed by the Analysis of ELG1 Mutants. International Journal of Molecular Sciences. 2023; 24(2):1568. https://doi.org/10.3390/ijms24021568
Chicago/Turabian StyleItzkovich, Ziv, Karan Choudhary, Matan Arbel, and Martin Kupiec. 2023. "Effects of Defective Unloading and Recycling of PCNA Revealed by the Analysis of ELG1 Mutants" International Journal of Molecular Sciences 24, no. 2: 1568. https://doi.org/10.3390/ijms24021568
APA StyleItzkovich, Z., Choudhary, K., Arbel, M., & Kupiec, M. (2023). Effects of Defective Unloading and Recycling of PCNA Revealed by the Analysis of ELG1 Mutants. International Journal of Molecular Sciences, 24(2), 1568. https://doi.org/10.3390/ijms24021568