

Developing Novel Experimental Models of m-TORopathic Epilepsy and Related Neuropathologies: Translational Insights from Zebrafish
 , , , , , and
, , , , , and
Abstract
1. Introduction
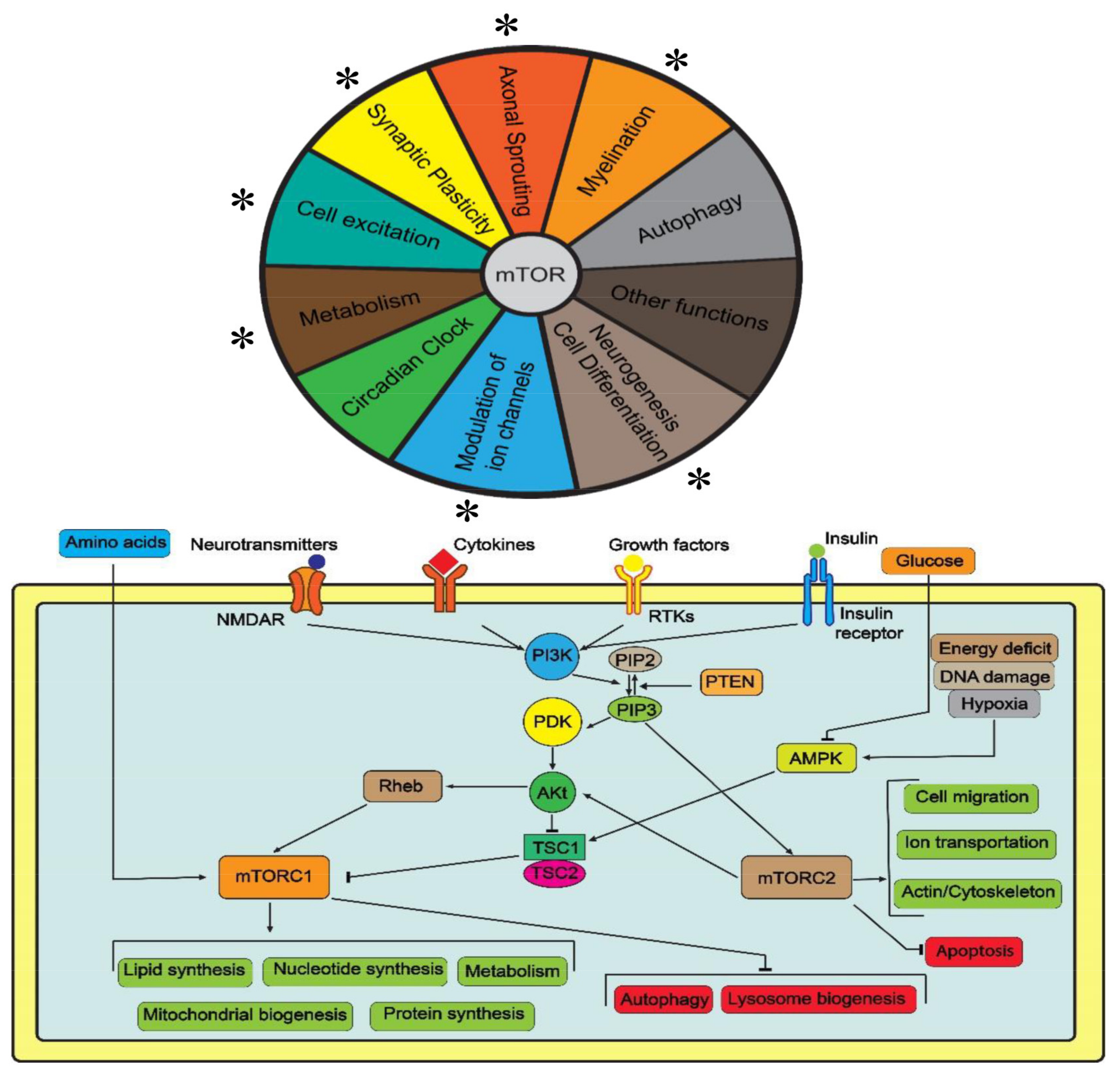
2. The Role of mTOR in the Nervous System
2.1. Autophagy
2.2. Axonal Sprouting and Synaptic Plasticity
2.3. Neurogenesis, Cell Differentiation and Myelination
2.4. Regulation of Ion Channels and Other Functions
3. Clinical mTORopathies
4. Recurrent Behavioral Abnormalities, mTOR and Epilepsy
5. The Key Role of mTOR in Epilepsy
6. Animal Models of mTORopahic Epilepsy
6.1. Selected Rodent Models
6.2. General Overview of Zebrafish Models of Epilepsy
6.3. Zebrafish Models of mTORopathic Epilepsy
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- El-Hashemite, N.; Zhang, H.; Henske, E.P.; Kwiatkowski, D.J. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet 2003, 361, 1348–1349. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Rogers, S.; Cesare, A.J. The mTOR pathway: Implications for DNA replication. Prog. Biophys. Mol. Biol. 2019, 147, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Andrews, M.G.; Subramanian, L.; Kriegstein, A.R. mTOR signaling regulates the morphology and migration of outer radial glia in developing human cortex. eLife 2020, 9, e58737. [Google Scholar] [CrossRef]
- Trejo, J.L.; Pons, S. Phosphatidylinositol-3-OH kinase regulatory subunits are differentially expressed during development of the rat cerebellum. J. Neurobiol. 2001, 47, 39–50. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, S.; Zhou, H.; Tao, X.; Ni, Y.; Pei, D.; Kang, S.; Yan, W.; Lu, J. Role of mTOR-Regulated Autophagy in Synaptic Plasticity Related Proteins Downregulation and the Reference Memory Deficits Induced by Anesthesia/Surgery in Aged Mice. Front. Aging Neurosci. 2021, 13, 628541. [Google Scholar] [CrossRef]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef]
- Sharma, A.; Mehan, S. Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int. 2021, 147, 105067. [Google Scholar] [CrossRef]
- Russo, E.; Citraro, R.; Constanti, A.; De Sarro, G. The mTOR signaling pathway in the brain: Focus on epilepsy and epileptogenesis. Mol. Neurobiol. 2012, 46, 662–681. [Google Scholar] [CrossRef]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Jang, S.Y.; Shin, Y.K.; Park, S.Y.; Park, J.Y.; Rha, S.H.; Kim, J.K.; Lee, H.J.; Park, H.T. Autophagy is involved in the reduction of myelinating Schwann cell cytoplasm during myelin maturation of the peripheral nerve. PLoS ONE 2015, 10, e0116624. [Google Scholar] [CrossRef]
- Nie, D.; Di Nardo, A.; Han, J.M.; Baharanyi, H.; Kramvis, I.; Huynh, T.; Dabora, S.; Codeluppi, S.; Pandolfi, P.P.; Pasquale, E.B.; et al. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat. Neurosci. 2010, 13, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, B.A.; Hake, H.S.; Ishiwata, T.; Farmer, C.E.; Loetz, E.C.; Fleshner, M.; Bland, S.T.; Greenwood, B.N. Exercise increases mTOR signaling in brain regions involved in cognition and emotional behavior. Behav. Brain Res. 2017, 323, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Magri, L.; Cambiaghi, M.; Cominelli, M.; Alfaro-Cervello, C.; Cursi, M.; Pala, M.; Bulfone, A.; Garcia-Verdugo, J.M.; Leocani, L.; Minicucci, F.; et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 2011, 9, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Lei, J.; Elsayed, N.A.; Lee, J.Y.; Shin, N.; Na, Q.; Chudnovets, A.; Jia, B.; Wang, X.; Burd, I. The effect of intrauterine inflammation on mTOR signaling in mouse fetal brain. Dev. Neurobiol. 2020, 80, 149–159. [Google Scholar] [CrossRef]
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.E.; McLane, L.E.; Bercury, K.K.; Macklin, W.B.; Wood, T.L. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J. Neurosci. 2014, 34, 4453–4465. [Google Scholar] [CrossRef]
- Figlia, G.; Gerber, D.; Suter, U. Myelination and mTOR. Glia 2018, 66, 693–707. [Google Scholar] [CrossRef]
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. 2009, 29, 6860–6870. [Google Scholar] [CrossRef]
- Cao, R. mTOR Signaling, Translational Control, and the Circadian Clock. Front. Genet. 2018, 9, 367. [Google Scholar] [CrossRef]
- Roa, J.; Tena-Sempere, M. Energy balance and puberty onset: Emerging role of central mTOR signaling. Trends Endocrinol. Metab. 2010, 21, 519–528. [Google Scholar] [CrossRef]
- Dang, L.T.; Glanowska, K.M.; Iffland Ii, P.H.; Barnes, A.E.; Baybis, M.; Liu, Y.; Patino, G.; Vaid, S.; Streicher, A.M.; Parker, W.E.; et al. Multimodal Analysis of STRADA Function in Brain Development. Front. Cell. Neurosci. 2020, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Muhlebner, A.; Bongaarts, A.; Sarnat, H.B.; Scholl, T.; Aronica, E. New insights into a spectrum of developmental malformations related to mTOR dysregulations: Challenges and perspectives. J. Anat. 2019, 235, 521–542. [Google Scholar] [CrossRef] [PubMed]
- Scholl, T.; Muhlebner, A.; Ricken, G.; Gruber, V.; Fabing, A.; Samueli, S.; Groppel, G.; Dorfer, C.; Czech, T.; Hainfellner, J.A.; et al. Impaired oligodendroglial turnover is associated with myelin pathology in focal cortical dysplasia and tuberous sclerosis complex. Brain Pathol. 2017, 27, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Gruber, V.; Scholl, T.; Samueli, S.; Groppel, G.; Muhlebner, A.; Hainfellner, J.A.; Feucht, M. Pathophysiology of neurodevelopmental mTOR pathway-associated epileptic conditions: Current status of biomedical research. Clin. Neuropathol. 2019, 38, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Munoz, J.; Villarejo-Zori, B.; Largo-Barrientos, P.; Boya, P. Towards a better understanding of the neuro-developmental role of autophagy in sickness and in health. Cell Stress 2021, 5, 99–118. [Google Scholar] [CrossRef]
- Lim, J.S.; Kim, W.I.; Kang, H.C.; Kim, S.H.; Park, A.H.; Park, E.K.; Cho, Y.W.; Kim, S.; Kim, H.M.; Kim, J.A.; et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 2015, 21, 395–400. [Google Scholar] [CrossRef]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef]
- Hsieh, D.T.; Whiteway, S.L.; Rohena, L.O.; Thiele, E.A. Tuberous sclerosis complex: Five new things. Neurol. Clin. Pract. 2016, 6, 339–347. [Google Scholar] [CrossRef]
- Gruber, V.E.; Lang, J.; Endmayr, V.; Diehm, R.; Pimpel, B.; Glatter, S.; Anink, J.J.; Bongaarts, A.; Luinenburg, M.J.; Reinten, R.J.; et al. Impaired myelin production due to an intrinsic failure of oligodendrocytes in mTORpathies. Neuropathol. Appl. Neurobiol. 2021, 47, 812–825. [Google Scholar] [CrossRef]
- Kabat, J.; Krol, P. Focal cortical dysplasia—Review. Pol. J. Radiol. 2012, 77, 35–43. [Google Scholar] [CrossRef]
- Kaleka, G.; McCormick, M.E.; Krishnan, A. Beta-Propeller Protein-Associated Neurodegeneration (BPAN) Detected in a Child with Epileptic Spasms. Cureus 2019, 11, e5404. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Hanif, M.; Sarfraz, Z.; Nepal, G.; Naz, S.; Mukherjee, D.; Ruxmohan, S. Hemimegalencephaly: A rare congenital malformation of cortical development. Clin. Case Rep. 2021, 9, e05238. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Skibo, J.; Kalume, F.; Ni, J.; Rankin, S.; Lu, Y.; Dobyns, W.B.; Mills, G.B.; Zhao, J.J.; Baker, S.J.; et al. Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy. eLife 2015, 4, e12703. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Puig, R.; Canals, F.; Colome, N.; Merlino, G.; Recio, J.A. Uncoupling of the LKB1-AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF. PLoS ONE 2009, 4, e4771. [Google Scholar] [CrossRef]
- Hu, X.; Pandolfi, P.P.; Li, Y.; Koutcher, J.A.; Rosenblum, M.; Holland, E.C. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia 2005, 7, 356–368. [Google Scholar] [CrossRef]
- Abel, T.W.; Baker, S.J.; Fraser, M.M.; Tihan, T.; Nelson, J.S.; Yachnis, A.T.; Bouffard, J.P.; Mena, H.; Burger, P.C.; Eberhart, C.G. Lhermitte-Duclos disease: A report of 31 cases with immunohistochemical analysis of the PTEN/AKT/mTOR pathway. J. Neuropathol. Exp. Neurol. 2005, 64, 341–349. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, X.; Zhou, T. TBCK influences cell proliferation, cell size and mTOR signaling pathway. PLoS ONE 2013, 8, e71349. [Google Scholar] [CrossRef]
- Sarnat, H.B.; Flores-Sarnat, L. Infantile tauopathies: Hemimegalencephaly; tuberous sclerosis complex; focal cortical dysplasia 2; ganglioglioma. Brain Dev. 2015, 37, 553–562. [Google Scholar] [CrossRef]
- Muotri, A.R. The Human Model: Changing Focus on Autism Research. Biol. Psychiatry 2016, 79, 642–649. [Google Scholar] [CrossRef]
- Pagani, M.; Barsotti, N.; Bertero, A.; Trakoshis, S.; Ulysse, L.; Locarno, A.; Miseviciute, I.; De Felice, A.; Canella, C.; Supekar, K.; et al. mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat. Commun. 2021, 12, 6084. [Google Scholar] [CrossRef]
- Yenkoyan, K.; Grigoryan, A.; Fereshetyan, K.; Yepremyan, D. Advances in understanding the pathophysiology of autism spectrum disorders. Behav. Brain Res. 2017, 331, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Hoeffer, C.A.; Sanchez, E.; Hagerman, R.J.; Mu, Y.; Nguyen, D.V.; Wong, H.; Whelan, A.M.; Zukin, R.S.; Klann, E.; Tassone, F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012, 11, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E. Epilepsy in fragile X syndrome. Dev. Med. Child Neurol. 2002, 44, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Ali Sher, A.; Del Bigio, M.R.; Sztainberg, Y.; Siu, V.M.; et al. MECP2 Mutation Interrupts Nucleolin-mTOR-P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef]
- Bordi, M.; Darji, S.; Sato, Y.; Mellen, M.; Berg, M.J.; Kumar, A.; Jiang, Y.; Nixon, R.A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019, 10, 563. [Google Scholar] [CrossRef]
- Crino, P.B. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 2011, 17, 734–742. [Google Scholar] [CrossRef]
- Wong, M. A critical review of mTOR inhibitors and epilepsy: From basic science to clinical trials. Expert Rev. Neurother. 2013, 13, 657–669. [Google Scholar] [CrossRef]
- Liu, J.; Reeves, C.; Michalak, Z.; Coppola, A.; Diehl, B.; Sisodiya, S.M.; Thom, M. Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol. Commun. 2014, 2, 71. [Google Scholar] [CrossRef]
- Ostendorf, A.P.; Wong, M. mTOR inhibition in epilepsy: Rationale and clinical perspectives. CNS Drugs 2015, 29, 91–99. [Google Scholar] [CrossRef]
- Temkin, N.R.; Jarell, A.D.; Anderson, G.D. Antiepileptogenic agents: How close are we? Drugs 2001, 61, 1045–1055. [Google Scholar] [CrossRef]
- Meng, X.F.; Yu, J.T.; Song, J.H.; Chi, S.; Tan, L. Role of the mTOR signaling pathway in epilepsy. J. Neurol. Sci. 2013, 332, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Raab-Graham, K.F.; Haddick, P.C.; Jan, Y.N.; Jan, L.Y. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science 2006, 314, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Barbaro, M.F.; Baraban, S.C. A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci. Lett. 2006, 401, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Wong, M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed. J. 2013, 36, 40–50. [Google Scholar] [CrossRef]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Xu, L.; Gutmann, D.H.; Wong, M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol. 2008, 63, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, M.C.; Sunnen, C.N.; Lugo, J.N.; Anderson, A.E.; D’Arcangelo, G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis. Models Mech. 2009, 2, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Blundell, J.; Ogawa, S.; Kwon, C.H.; Zhang, W.; Sinton, C.; Powell, C.M.; Parada, L.F. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 2009, 29, 1773–1783. [Google Scholar] [CrossRef]
- Choi, J.H.; Bertram, P.G.; Drenan, R.; Carvalho, J.; Zhou, H.H.; Zheng, X.F. The FKBP12-rapamycin-associated protein (FRAP) is a CLIP-170 kinase. EMBO Rep. 2002, 3, 988–994. [Google Scholar] [CrossRef]
- Jiang, X.; Yeung, R.S. Regulation of microtubule-dependent protein transport by the TSC2/mammalian target of rapamycin pathway. Cancer Res. 2006, 66, 5258–5269. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, H.E.; Hauptman, J.S. The Putative Role of mTOR Inhibitors in Non-tuberous Sclerosis Complex-Related Epilepsy. Front. Neurol. 2021, 12, 639319. [Google Scholar] [CrossRef] [PubMed]
- Galanopoulou, A.S.; Gorter, J.A.; Cepeda, C. Finding a better drug for epilepsy: The mTOR pathway as an antiepileptogenic target. Epilepsia 2012, 53, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Kim, S.Y.; Cha, J.H.; Choi, Y.S.; Sung, K.W.; Oh, S.T.; Kim, O.N.; Chung, J.W.; Chun, M.H.; Lee, S.B.; et al. Upregulation of gp130 and STAT3 activation in the rat hippocampus following transient forebrain ischemia. Glia 2003, 41, 237–246. [Google Scholar] [CrossRef]
- Eeg-Olofsson, O. Virological and immunological aspects of seizure disorders. Brain Dev. 2003, 25, 9–13. [Google Scholar] [CrossRef]
- Fotheringham, J.; Donati, D.; Akhyani, N.; Fogdell-Hahn, A.; Vortmeyer, A.; Heiss, J.D.; Williams, E.; Weinstein, S.; Bruce, D.A.; Gaillard, W.D.; et al. Association of human herpesvirus-6B with mesial temporal lobe epilepsy. PLoS Med. 2007, 4, e180. [Google Scholar] [CrossRef]
- Getts, D.R.; Balcar, V.J.; Matsumoto, I.; Muller, M.; King, N.J. Viruses and the immune system: Their roles in seizure cascade development. J. Neurochem. 2008, 104, 1167–1176. [Google Scholar] [CrossRef]
- Misra, U.K.; Tan, C.T.; Kalita, J. Viral encephalitis and epilepsy. Epilepsia 2008, 49, 13–18. [Google Scholar] [CrossRef]
- Azkona, G.; Sanchez-Pernaute, R. Mice in translational neuroscience: What R we doing? Prog. Neurobiol. 2022, 217, 102330. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G.F.; Gonzalez-Sulser, A.; Abbott, C.M. Modelling epilepsy in the mouse: Challenges and solutions. Dis. Models Mech. 2021, 14, dmm047449. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Babiec, L.; Jesko, H.; Gassowska-Dobrowolska, M.; Cieslik, M.; Matuszewska, M.; Frontczak-Baniewicz, M.; Zajdel, K.; Adamczyk, A. Synaptic Alterations in a Transgenic Model of Tuberous Sclerosis Complex: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2021, 22, 10058. [Google Scholar] [CrossRef] [PubMed]
- Cambiaghi, M.; Magri, L.; Cursi, M. Importance of EEG in validating the chronic effects of drugs: Suggestions from animal models of epilepsy treated with rapamycin. Seizure 2015, 27, 30–39. [Google Scholar] [CrossRef]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef]
- Rosa, J.G.S.; Lima, C.; Lopes-Ferreira, M. Zebrafish Larvae Behavior Models as a Tool for Drug Screenings and Pre-Clinical Trials: A Review. Int. J. Mol. Sci. 2022, 23, 6647. [Google Scholar] [CrossRef]
- Saleem, S.; Kannan, R.R. Zebrafish: An emerging real-time model system to study Alzheimer’s disease and neurospecific drug discovery. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 2018, 11, 294. [Google Scholar] [CrossRef]
- Norton, W.; Bally-Cuif, L. Adult zebrafish as a model organism for behavioural genetics. BMC Neurosci. 2010, 11, 90. [Google Scholar] [CrossRef]
- Kundap, U.P.; Kumari, Y.; Othman, I.; Shaikh, M.F. Zebrafish as a Model for Epilepsy-Induced Cognitive Dysfunction: A Pharmacological, Biochemical and Behavioral Approach. Front. Pharmacol. 2017, 8, 515. [Google Scholar] [CrossRef]
- Yaksi, E.; Jamali, A.; Diaz Verdugo, C.; Jurisch-Yaksi, N. Past, present and future of zebrafish in epilepsy research. FEBS J. 2021, 288, 7243–7255. [Google Scholar] [CrossRef]
- Mussulini, B.H.; Leite, C.E.; Zenki, K.C.; Moro, L.; Baggio, S.; Rico, E.P.; Rosemberg, D.B.; Dias, R.D.; Souza, T.M.; Calcagnotto, M.E.; et al. Seizures induced by pentylenetetrazole in the adult zebrafish: A detailed behavioral characterization. PLoS ONE 2013, 8, e54515. [Google Scholar] [CrossRef]
- Burrows, D.R.W.; Samarut, E.; Liu, J.; Baraban, S.C.; Richardson, M.P.; Meyer, M.P.; Rosch, R.E. Imaging epilepsy in larval zebrafish. Eur. J. Paediatr. Neurol. 2020, 24, 70–80. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Wang, J.; Hu, G.; Liu, Z.; Yan, D.; Serikuly, N.; Alpyshov, E.T.; Demin, K.A.; Galstyan, D.S.; et al. Behavioral and physiological effects of acute and chronic kava exposure in adult zebrafish. Neurotoxicol. Teratol. 2020, 79, 106881. [Google Scholar] [CrossRef]
- Wong, K.; Stewart, A.; Gilder, T.; Wu, N.; Frank, K.; Gaikwad, S.; Suciu, C.; Dileo, J.; Utterback, E.; Chang, K.; et al. Modeling seizure-related behavioral and endocrine phenotypes in adult zebrafish. Brain Res. 2010, 1348, 209–215. [Google Scholar] [CrossRef]
- Stewart, A.M.; Desmond, D.; Kyzar, E.; Gaikwad, S.; Roth, A.; Riehl, R.; Collins, C.; Monnig, L.; Green, J.; Kalueff, A.V. Perspectives of zebrafish models of epilepsy: What, how and where next? Brain Res. Bull. 2012, 87, 135–143. [Google Scholar] [CrossRef]
- Gawel, K.; Langlois, M.; Martins, T.; van der Ent, W.; Tiraboschi, E.; Jacmin, M.; Crawford, A.D.; Esguerra, C.V. Seizing the moment: Zebrafish epilepsy models. Neurosci. Biobehav. Rev. 2020, 116, 1–20. [Google Scholar] [CrossRef]
- Baraban, S.C.; Dinday, M.T.; Castro, P.A.; Chege, S.; Guyenet, S.; Taylor, M.R. A large-scale mutagenesis screen to identify seizure-resistant zebrafish. Epilepsia 2007, 48, 1151–1157. [Google Scholar] [CrossRef]
- Barreiros, M.O.; Barbosa, F.G.; Dantas, D.O.; Santos, D.; Ribeiro, S.; Santos, G.C.O.; Barros, A.K. Zebrafish automatic monitoring system for conditioning and behavioral analysis. Sci. Rep. 2021, 11, 9330. [Google Scholar] [CrossRef]
- Bozhko, D.V.; Myrov, V.O.; Kolchanova, S.M.; Polovian, A.I.; Galumov, G.K.; Demin, K.A.; Zabegalov, K.N.; Strekalova, T.; de Abreu, M.S.; Petersen, E.V.; et al. Artificial intelligence-driven phenotyping of zebrafish psychoactive drug responses. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 112, 110405. [Google Scholar] [CrossRef] [PubMed]
- Cachat, J.; Stewart, A.; Utterback, E.; Hart, P.; Gaikwad, S.; Wong, K.; Kyzar, E.; Wu, N.; Kalueff, A.V. Three-dimensional neurophenotyping of adult zebrafish behavior. PLoS ONE 2011, 6, e17597. [Google Scholar] [CrossRef] [PubMed]
- Turrini, L.; Sorelli, M.; de Vito, G.; Credi, C.; Tiso, N.; Vanzi, F.; Pavone, F.S. Multimodal Characterization of Seizures in Zebrafish Larvae. Biomedicines 2022, 10, 951. [Google Scholar] [CrossRef] [PubMed]
- Rosch, R.E.; Hunter, P.R.; Baldeweg, T.; Friston, K.J.; Meyer, M.P. Calcium imaging and dynamic causal modelling reveal brain-wide changes in effective connectivity and synaptic dynamics during epileptic seizures. PLoS Comput. Biol. 2018, 14, e1006375. [Google Scholar] [CrossRef]
- Siebel, A.M.; Menezes, F.P.; da Costa Schaefer, I.; Petersen, B.D.; Bonan, C.D. Rapamycin suppresses PTZ-induced seizures at different developmental stages of zebrafish. Pharmacol. Biochem. Behav. 2015, 139, 163–168. [Google Scholar] [CrossRef]
- Mazumder, A.G.; Kumari, S.; Singh, D. Anticonvulsant action of a selective phosphatidylinositol-3-kinase inhibitor LY294002 in pentylenetetrazole-mediated convulsions in zebrafish. Epilepsy Res. 2019, 157, 106207. [Google Scholar] [CrossRef]
- Griffin, A.; Carpenter, C.; Liu, J.; Paterno, R.; Grone, B.; Hamling, K.; Moog, M.; Dinday, M.T.; Figueroa, F.; Anvar, M.; et al. Phenotypic analysis of catastrophic childhood epilepsy genes. Commun. Biol. 2021, 4, 680. [Google Scholar] [CrossRef]
- Kim, S.H.; Speirs, C.K.; Solnica-Krezel, L.; Ess, K.C. Zebrafish model of tuberous sclerosis complex reveals cell-autonomous and non-cell-autonomous functions of mutant tuberin. Dis. Models Mech. 2011, 4, 255–267. [Google Scholar] [CrossRef]
- Scheldeman, C.; Mills, J.D.; Siekierska, A.; Serra, I.; Copmans, D.; Iyer, A.M.; Whalley, B.J.; Maes, J.; Jansen, A.C.; Lagae, L.; et al. mTOR-related neuropathology in mutant tsc2 zebrafish: Phenotypic, transcriptomic and pharmacological analysis. Neurobiol. Dis. 2017, 108, 225–237. [Google Scholar] [CrossRef]
- Kedra, M.; Banasiak, K.; Kisielewska, K.; Wolinska-Niziol, L.; Jaworski, J.; Zmorzynska, J. TrkB hyperactivity contributes to brain dysconnectivity, epileptogenesis, and anxiety in zebrafish model of Tuberous Sclerosis Complex. Proc. Natl. Acad. Sci. USA 2020, 117, 2170–2179. [Google Scholar] [CrossRef]
- Serra, I.; Scheldeman, C.; Bazelot, M.; Whalley, B.J.; Dallas, M.L.; de Witte, P.A.M.; Williams, C.M. Cannabidiol modulates phosphorylated rpS6 signalling in a zebrafish model of Tuberous Sclerosis Complex. Behav. Brain Res. 2019, 363, 135–144. [Google Scholar] [CrossRef]
- Morales, P.; Jagerovic, N. Synthetic and Natural Derivatives of Cannabidiol. Adv. Exp. Med. Biol. 2021, 1297, 11–25. [Google Scholar] [CrossRef]
- Pressler, R.M.; Lagae, L. Why we urgently need improved seizure and epilepsy therapies for children and neonates. Neuropharmacology 2020, 170, 107854. [Google Scholar] [CrossRef]
- De Calbiac, H.; Dabacan, A.; Marsan, E.; Tostivint, H.; Devienne, G.; Ishida, S.; Leguern, E.; Baulac, S.; Muresan, R.C.; Kabashi, E.; et al. Depdc5 knockdown causes mTOR-dependent motor hyperactivity in zebrafish. Ann. Clin. Transl. Neurol. 2018, 5, 510–523. [Google Scholar] [CrossRef]
- Swaminathan, A.; Hassan-Abdi, R.; Renault, S.; Siekierska, A.; Riche, R.; Liao, M.; de Witte, P.A.M.; Yanicostas, C.; Soussi-Yanicostas, N.; Drapeau, P.; et al. Non-canonical mTOR-Independent Role of DEPDC5 in Regulating GABAergic Network Development. Curr. Biol. 2018, 28, 1924–1937. [Google Scholar] [CrossRef]
- De Meulemeester, A.S.; Heylen, L.; Siekierska, A.; Mills, J.D.; Romagnolo, A.; Van Der Wel, N.N.; Aronica, E.; de Witte, P.A.M. Hyperactivation of mTORC1 in a double hit mutant zebrafish model of tuberous sclerosis complex causes increased seizure susceptibility and neurodevelopmental abnormalities. Front. Cell Dev. Biol. 2022, 10, 952832. [Google Scholar] [CrossRef]
- Wang, T.; Zhou, M.; Zhang, Q.; Zhang, C.; Peng, G. ubtor Mutation Causes Motor Hyperactivity by Activating mTOR Signaling in Zebrafish. Neurosci. Bull. 2021, 37, 1658–1670. [Google Scholar] [CrossRef]
- Lu, J.; Peatman, E.; Tang, H.; Lewis, J.; Liu, Z. Profiling of gene duplication patterns of sequenced teleost genomes: Evidence for rapid lineage-specific genome expansion mediated by recent tandem duplications. BMC Genom. 2012, 13, 246. [Google Scholar] [CrossRef]
- Parker, M.O.; Brock, A.J.; Walton, R.T.; Brennan, C.H. The role of zebrafish (Danio rerio) in dissecting the genetics and neural circuits of executive function. Front. Neural. Circuits 2013, 7, 63. [Google Scholar] [CrossRef]
- Andrews, M.G.; Subramanian, L.; Salma, J.; Kriegstein, A.R. How mechanisms of stem cell polarity shape the human cerebral cortex. Nat. Rev. Neurosci. 2022, 23, 711–724. [Google Scholar] [CrossRef]
- Folgueira, M.; Bayley, P.; Navratilova, P.; Becker, T.S.; Wilson, S.W.; Clarke, J.D. Morphogenesis underlying the development of the everted teleost telencephalon. Neural Dev. 2012, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wong, M. Pentylenetetrazole-induced seizures cause acute, but not chronic, mTOR pathway activation in rat. Epilepsia 2012, 53, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; Shirsath, N.; Lang, V.; Berard, A.; Diehl, S.; Kaufmann, R.; Boehncke, W.-H.; Wolf, P. Inflammation dependent mTORC1 signaling interferes with the switch from keratinocyte proliferation to differentiation. PLoS ONE 2017, 12, e0180853. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Giussani, G. Aging and the epidemiology of epilepsy. Neuroepidemiology 2018, 51, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Hui, J.B.; Silva, J.C.H.; Pelaez, M.C.; Sévigny, M.; Venkatasubramani, J.P.; Plumereau, Q.; Chahine, M.; Proulx, C.D.; Sephton, C.F.; Dutchak, P.A. NPRL2 inhibition of mTORC1 controls sodium channel expression and brain amino acid homeostasis. Eneuro 2022, 9, ENEURO.0317-21.2022. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disorders, % Occurrence of Epilepsy | Affected Genes | Clinical Signs | Pathophysiology | Therapy of Epilepsy |
|---|---|---|---|---|
|
Focal cortical dysplasia type IIB (FCDIIB), 5–25% | MTOR | Intractable seizures | Neuronal migration disruption by hyperactive mTOR, causing more cytomegalic neurons and spontaneous seizures | Antiepileptic drugs, surgical treatment |
| NPRL2, NPRL3, DEPDC5 | Mutational inactivation, mTOR inhibition release and increased mTOR signaling | |||
|
Tuberous sclerosis complex (TSC), 80–90% | TSC1 | Subependymal nodules, giant cell tumors, cortical tubers, epilepsy | Overactivation of the mTOR kinase-dependent pathway | Antiepileptic drugs |
| TSC2 | Benign tumors, epilepsy | Overactivation of the mTOR kinase-dependent pathway | ||
| Hemimegaloencephaly (HME), >90% | AKT1, AKT3, PIK3CA, PI3KR | Enlarged heads of children, epilepsy, partial paralysis and cognitive delay | Mutation-facilitated mTOR signaling | Hemispherectomy |
| Glioneuronal tumors (GNT), >90% | BRAF | Early-onset focal epilepsy, chronic drug-resistant epilepsy | In V600E mutation, LKB1-induced phosphorylated LKB1 activates mTOR * | Surgical resection and radiation |
| Fragile X syndrome, 10–20% | FMR1 | Intellectual disability, autism, seizures, hyper-activity, inattention, craniofacial deficits | FMR1/FMRP negative feedback loop causes mTOR inhibition | Symptomatic therapy (e.g., lithium, gabapentin) |
| Pretzel syndrome, >90% | STRADA | Severe developmental delay, epilepsy | A homozygous deletion of exons of the gene encoding the pseudokinase STRADA, an upstream inhibitor of mTOR | Sirolimus for the absent STRAD-alpha protein |
| Neuro-fibromatosis type 1, 5–10% | NF1 | Neurocutaneous lesions, benign and malignant tumors, autism, occasional seizures | In primary cells lacking NF1 and in human malignancies, the mTOR pathway is severely downregulated | Surgical resection and radiation |
| Lhermitte-Duclos disease (LDD), >90% ** | PTEN | Cerebellar gangliocytomas, ataxia, epilepsy | Due to increased PI3K/Akt activation and subsequent activation of the mTOR pathway, PTEN loss increases sensitivity to mTOR inhibitors | Surgical resection |
| TBCK syndrome, 70% | TBCK | Leukoencephalopathy, coarse face, neuropathy, hypotonia, intellectual disability, epilepsy | The mTOR complex’ protein levels significantly drop when TBCK is knocked down, and mTOR signaling is suppressed | Leucine (an mTORC1 activator) |
| Rett syndrome, 60–80% | MECP2 | Speech deficits, uncontrollable movements, early-onset epilepsy | Mutation interrupts nucleolin–mTOR–P70S6K signaling | Antiepileptic drugs, deep brain stimulation |
| Beta-propeller protein-associated neurodegeneration (BPAN), 10% | WDR45 | Intellectual disability, parkinsonism, psychomotor retardation, early-onset epilepsy, autism, dementia | The autophagy-inducing Unc51-like kinase 1 (ULK1)/Atg1-containing complex, controlled by the mTORC1 | Ketogenic diet, vagal stimulation, behavioral therapy |
| Down syndrome, 1–15% *** | Intellectual disability, developmental delay, speech deficits and epilepsy | Hyperactivation of the AKT/mTOR signaling pathway, imbalances in autophagy and mitochondrial turnover | Antiepileptic drugs |
| Disorder Modeled | Affected Genes | Details | Mechanisms/Pathway Affected | Cause of Epilepsy | References |
|---|---|---|---|---|---|
| Tuberous sclerosis (TSC) | TSC2 | Tsc2+/− heterozygous mice (impaired memory and predisposition to seizures) | Overactivation of mTOR signaling due to TSC2 haploinsufficiency | Myelin delamination, larger brains with deficient connectivity | [83] |
| Pretzel syndrome | STRADA | Knockout mice, induced pluripotent stem cell-derived neurons from patients | Insufficient STRADA signaling via AMPK, leading to mTOR overactivation | Lower threshold for action potential, ectopic neurons in the subcortical white matter | [31] |
| Focal cortical dysplasia (FCD) | PIK3CA H1047R, PIK3CA E545K | Despite overt dysplasia, acute inhibition of PI3K signaling reduces epilepsy | Increased PI3K signaling, upregulation of pS473-Akt (mTOR-dependent) | Cortical malformation, hydrocephalic and enlarged brain | [43] |
| FCD type 2B (FCD2B) and TSC | TSC2 | Oligodendroglial knockout in mice | Impeded oligodendrocyte development and myelin sheath formation | Impaired myelination | [39] |
| Fragile X syndrome (FXS) | FMR1 | Knockout mice | Hippocampal mTOR phosphorylated and hyperactive | Affected synaptic plasticity, leading to overconnectivity | [85] |
| Open Questions |
|---|
| Are there non-canonical, mTOR-independent mechanisms in epilepsy seen in mTORopathies? What is the exact role of neuronal vs. neuroglial (and also microglial vs. astrocytic) mechanisms in mTORopathies? Are there disorder-specific neurogenomic, neuroproteomic and neurometabolomic signatures of clinical mTORopathies? If yes, do zebrafish models for these specific mTOR-related disorders display similar ‘omics’ profiles to their human counterparts? Are there epigenetic mechanisms strongly involved in mTORopathic epilepsy? Can neuroinflammation modulate mTOR-related pathogenesis? What is the role of brain-derived neurotrophic factor (BDNF) and its signaling pathway in modulating mTORopathic epilepsy and other mTORopathies? What is the role of neuronal and neuroglial apoptosis in mTORopathic epilepsy? Can early-onset epilepsy (frequently seen in mTORopathies) trigger or promote neuroinflammation and neuronal apoptosis? Can these factors indirectly worsen mTORopathic pathogenesis progression further on? Can various psychological stressors (e.g., early-life, acute or chronic stress) alter the mTOR activity, to contribute to the development or progression of CNS deficits, including epilepsy? Can mTOR inhibitors represent novel therapies for neurodevelopmental disorders? Are there sex differences in mTOR activity in general, and in human, rodent and zebrafish epilepsy in particular? Can valid in vitro models of mTORopathic epilepsy be developed? How does diet (e.g., high-carbohydrate diet that predisposes to inflammation) impact the mTOR activity and, possibly, its role in epilepsy pathogenesis? Are there overt strain differences in mTOR activity in animal (e.g., mouse and/or zebrafish) models? Are there strain differences in mTORopathic epilepsy in such animal models? Which zebrafish strains are most susceptible to mTORopathic epilepsy? Do inbred (or, rather, outbred) zebrafish strains provide more reliable and reproducible phenotypic data related to mTORopathic epilepsy? What is the potential impact of ‘background’ fish strain on the expression of individual mTOR-related epileptic phenotypes in mutant zebrafish? To what extent can pharmacological manipulations used to induce epilepsy themselves affect mTOR signaling (e.g., pentylenetetrazole, a common chemoconvulsant, transiently activates mTOR activation in rats [122])? Inflammatory cytokines can induce aberrant mTOR activity (e.g., interleukins (IL) IL-1β, IL-17A and TNF-α strongly activate the mTOR kinase, PRAS40 and the downstream targets of mTOR activity 4E-BP1 and the ribosomal protein S6) [123]. Can drugs that upregulate such cytokines be used for the treatment of epilepsy (e.g., reducing glutamate reuptake, leading to elevated glutamate availability, and reducing inhibitory neurotransmission)? Epilepsy is a chronic disorder affecting all ages, but it peaks in the elderly [124]. Age itself can also impact mTOR activity [125]. Does age influence mTOR activity and, consequently, epilepsy in zebrafish and other animal models? How can zebrafish models contribute to probing the role of mutations at the mTOR inhibitor genes in neurodevelopmental disorders (e.g., autism, fragile X syndrome, tuberous sclerosis complex) that present epilepsy episodes as one of their common pathogenic symptoms? Can some novel therapies (e.g., resveratrol inhibiting mTOR through ATP competition [126]) be used as potential treatments for mTORopathic epilepsy? Can conventional antiepileptic drugs exert their auxiliary effects on mTOR signaling? Can there be a synergistic therapeutic action of the two potential mechanisms? Can mTOR-modulating drugs interact with antiepileptic drugs, either potentiating or mitigating their effects? Is there a complex link between other genetic mutations, mTOR activity, ionic channels activity and epilepsy? For example, mutations in the nitrogen permease regulator-like 2 gene (NPRL2) are linked to familial focal epilepsies, and its mutation affects the mTORC1 control of Na+ channel expression and brain amino acid homeostasis, which both can contribute to epilepsy pathogenesis [127]. Are there potential peripheral biochemical biomarkers of mTORopathic epilepsy in zebrafish and other animal models? |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Abreu, M.S.; Demin, K.A.; Kotova, M.M.; Mirzaei, F.; Shariff, S.; Kantawala, B.; Zakharchenko, K.V.; Kolesnikova, T.O.; Dilbaryan, K.; Grigoryan, A.; et al. Developing Novel Experimental Models of m-TORopathic Epilepsy and Related Neuropathologies: Translational Insights from Zebrafish. Int. J. Mol. Sci. 2023, 24, 1530. https://doi.org/10.3390/ijms24021530
de Abreu MS, Demin KA, Kotova MM, Mirzaei F, Shariff S, Kantawala B, Zakharchenko KV, Kolesnikova TO, Dilbaryan K, Grigoryan A, et al. Developing Novel Experimental Models of m-TORopathic Epilepsy and Related Neuropathologies: Translational Insights from Zebrafish. International Journal of Molecular Sciences. 2023; 24(2):1530. https://doi.org/10.3390/ijms24021530
Chicago/Turabian Stylede Abreu, Murilo S., Konstantin A. Demin, Maria M. Kotova, Foad Mirzaei, Sanobar Shariff, Burhan Kantawala, Ksenia V. Zakharchenko, Tatiana O. Kolesnikova, Karen Dilbaryan, Artem Grigoryan, and et al. 2023. "Developing Novel Experimental Models of m-TORopathic Epilepsy and Related Neuropathologies: Translational Insights from Zebrafish" International Journal of Molecular Sciences 24, no. 2: 1530. https://doi.org/10.3390/ijms24021530
APA Stylede Abreu, M. S., Demin, K. A., Kotova, M. M., Mirzaei, F., Shariff, S., Kantawala, B., Zakharchenko, K. V., Kolesnikova, T. O., Dilbaryan, K., Grigoryan, A., Yenkoyan, K. B., & Kalueff, A. V. (2023). Developing Novel Experimental Models of m-TORopathic Epilepsy and Related Neuropathologies: Translational Insights from Zebrafish. International Journal of Molecular Sciences, 24(2), 1530. https://doi.org/10.3390/ijms24021530