
Diclofenac Disrupts the Circadian Clock and through Complex Cross-Talks Aggravates Immune-Mediated Liver Injury—A Repeated Dose Study in Minipigs for 28 Days



Abstract
1. Introduction
2. Results
2.1. Treatment Related Clinical Signs
2.2. Serum Biochemistry
2.3. Hematology
2.4. Serum Electrolyte Analysis
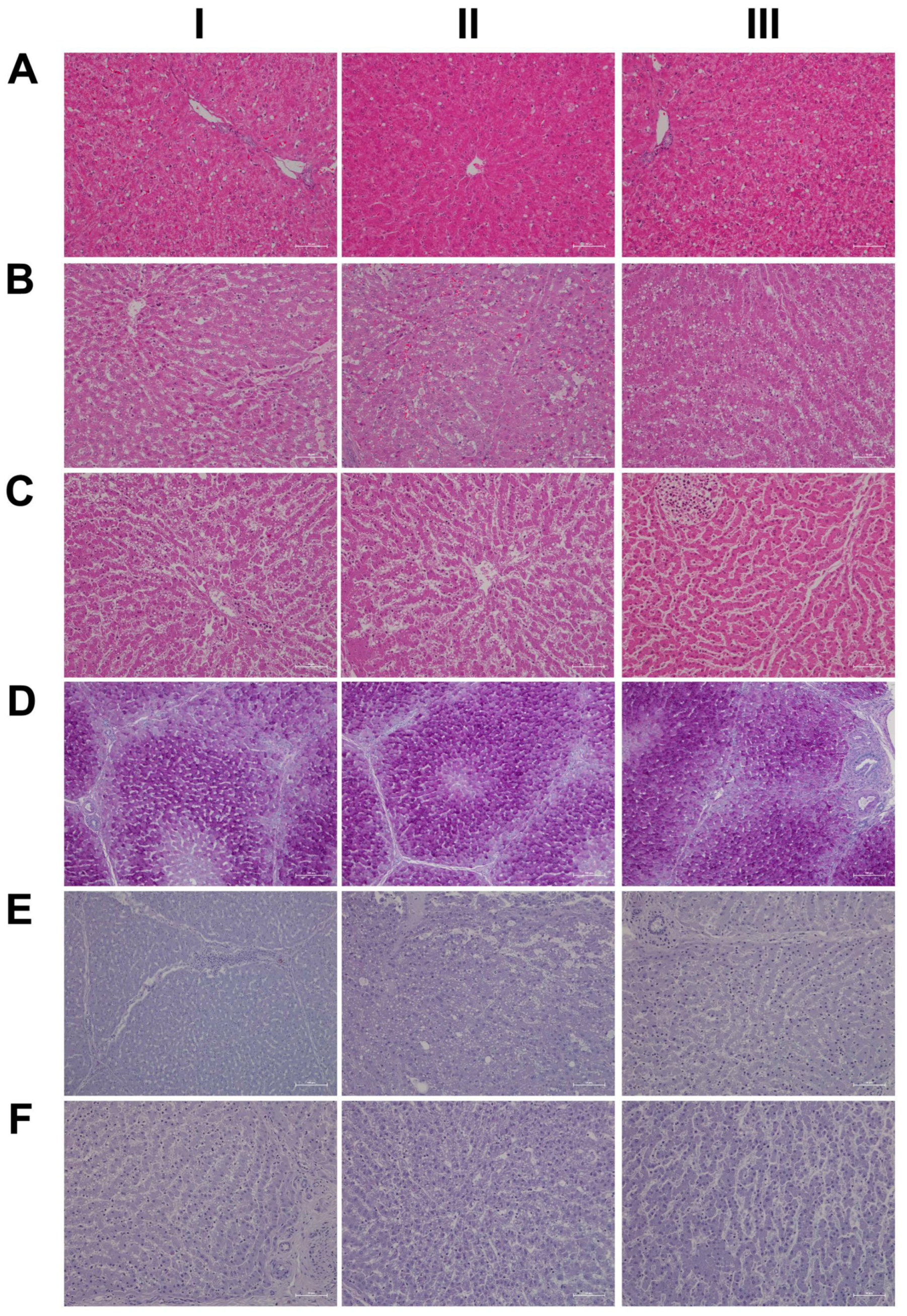
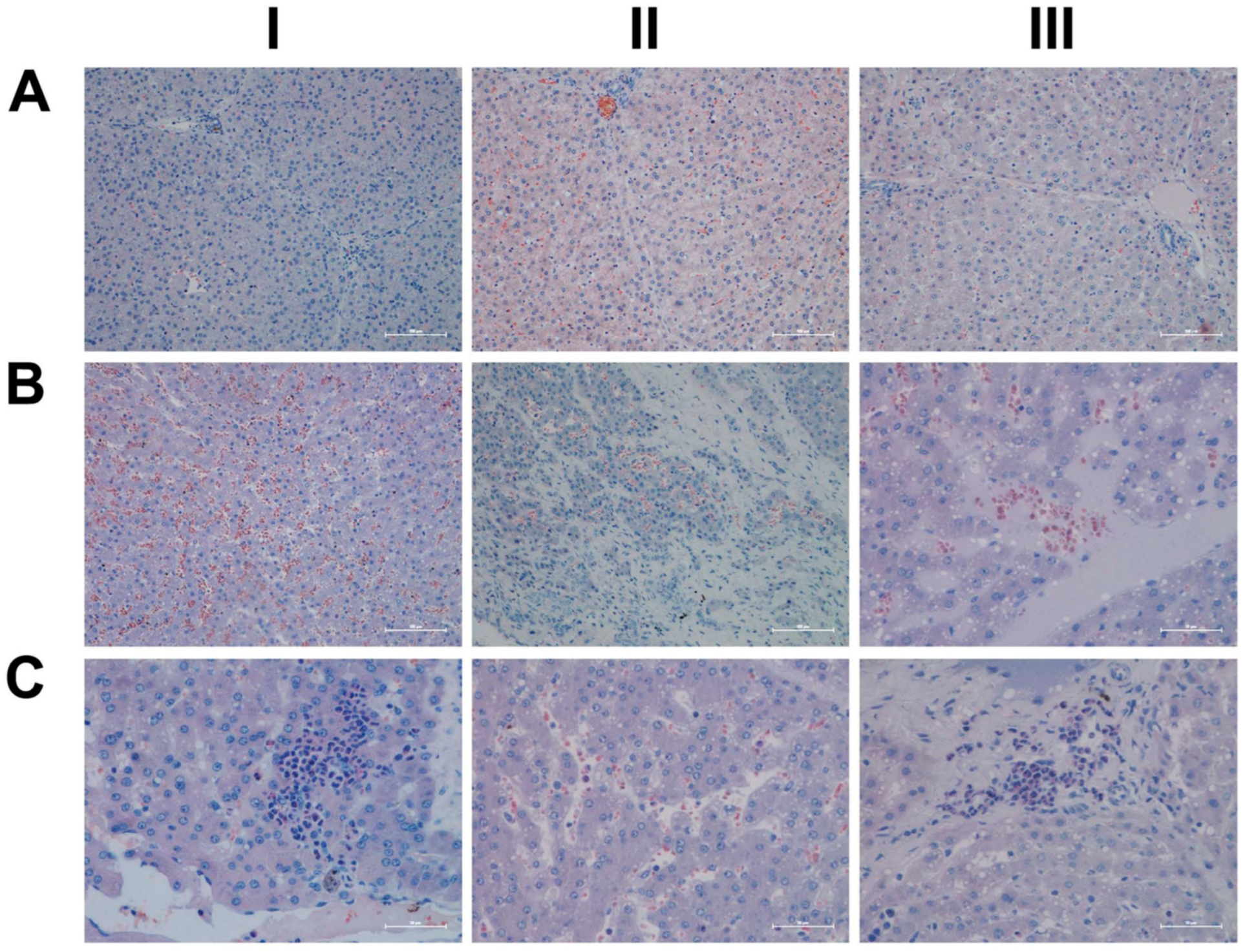
2.5. Histopathology
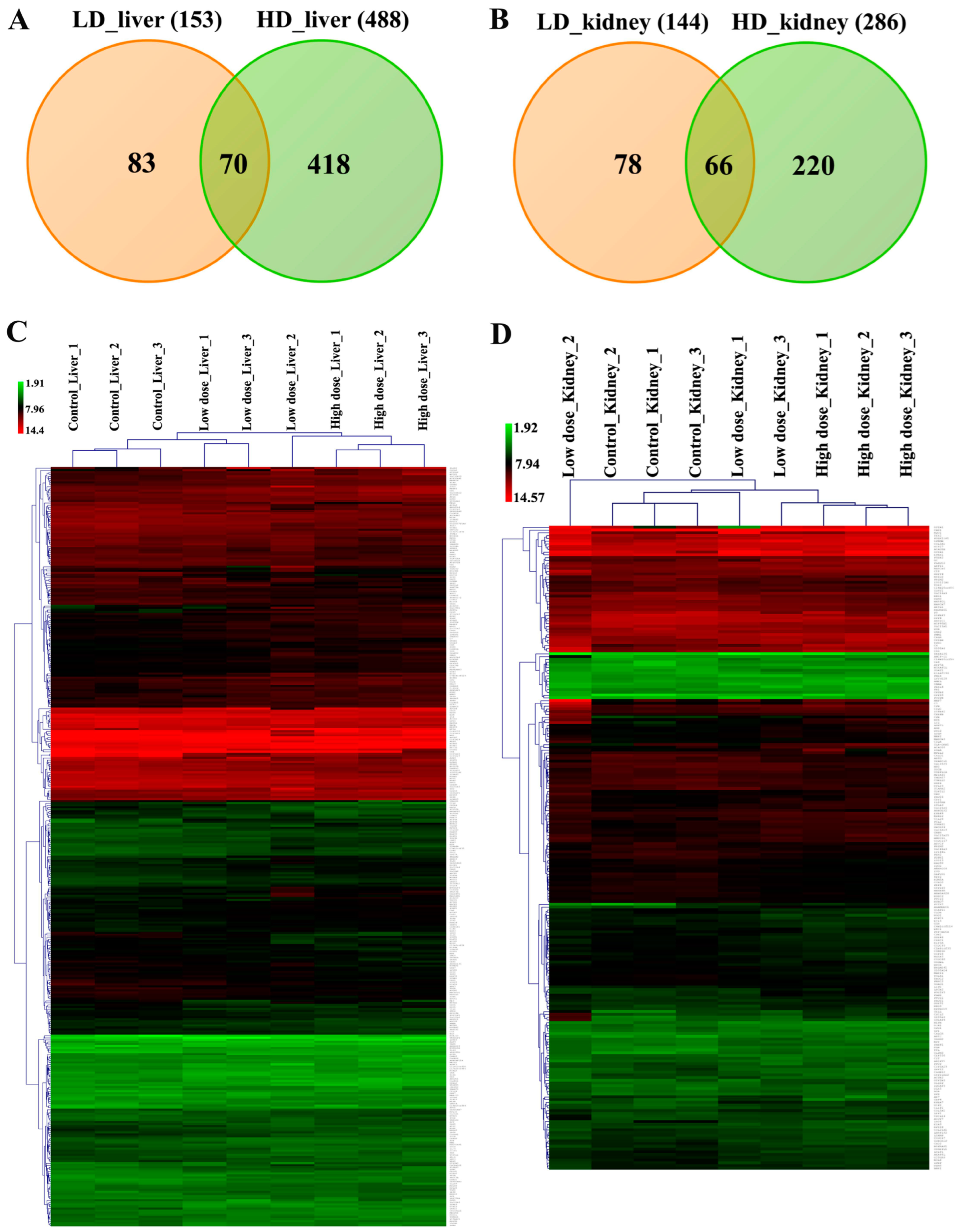
2.6. Genomic Responses in Liver and Kidney to Diclofenac Treatment
2.7. Drug Metabolism and Transporters
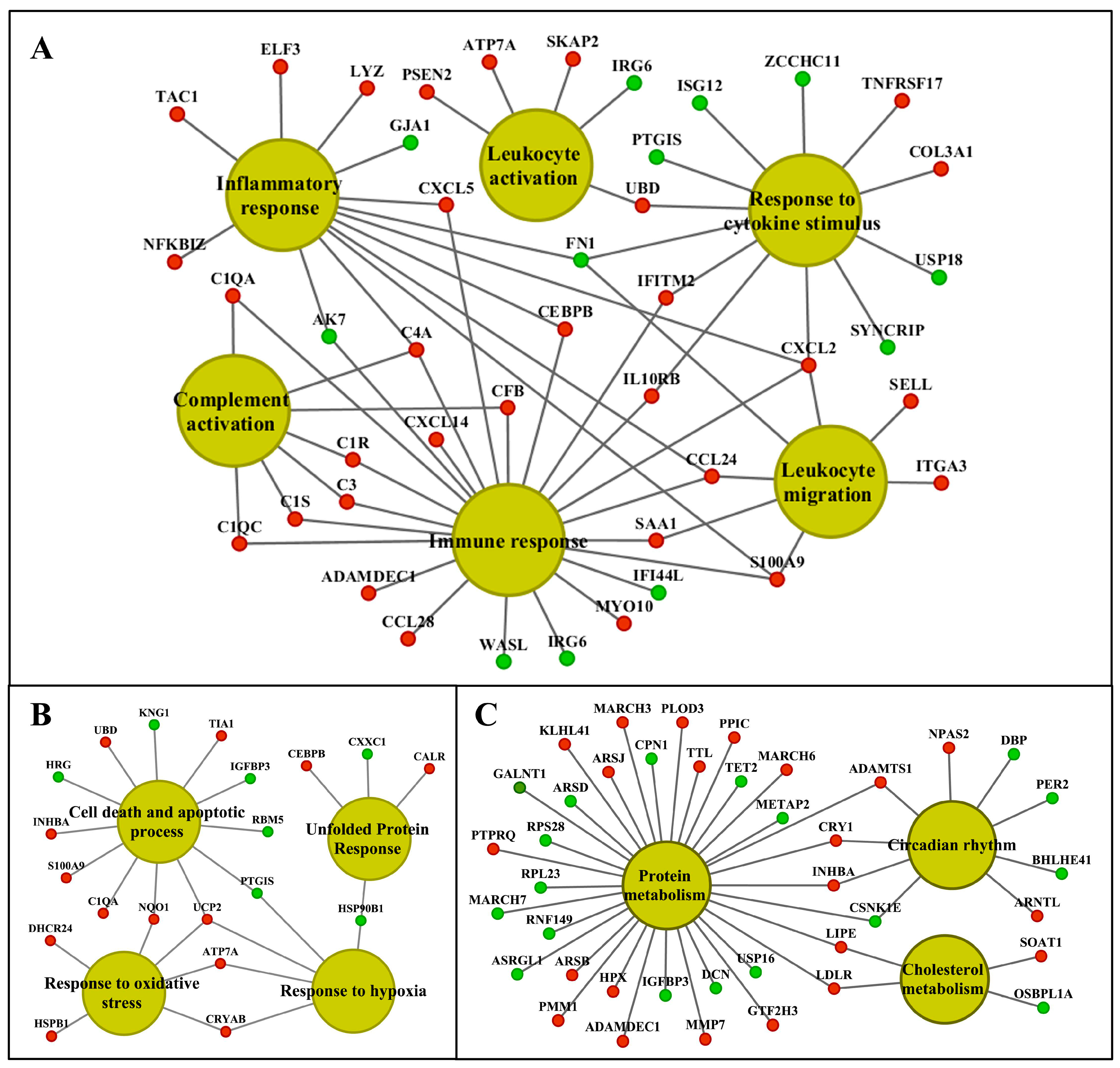
2.8. Functional Enrichment Analysis
2.9. Commonly Regulated Genes in Liver and Kidney
2.10. Molecular Networks in Liver and Kidney
2.11. Master Regulators and Their Associated Networks
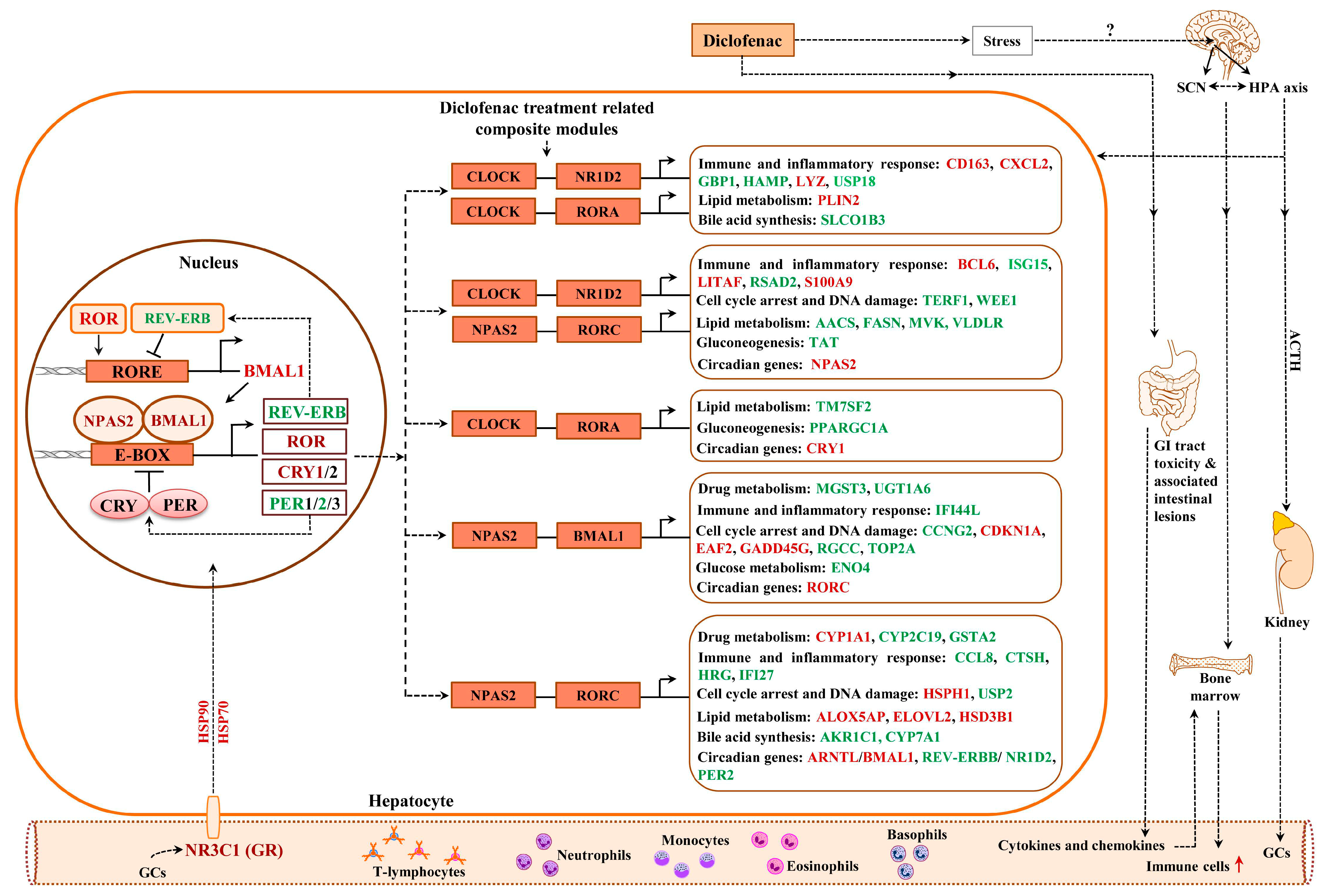
2.12. Enriched Transcription Factor Binding Sites for the Liver Clock
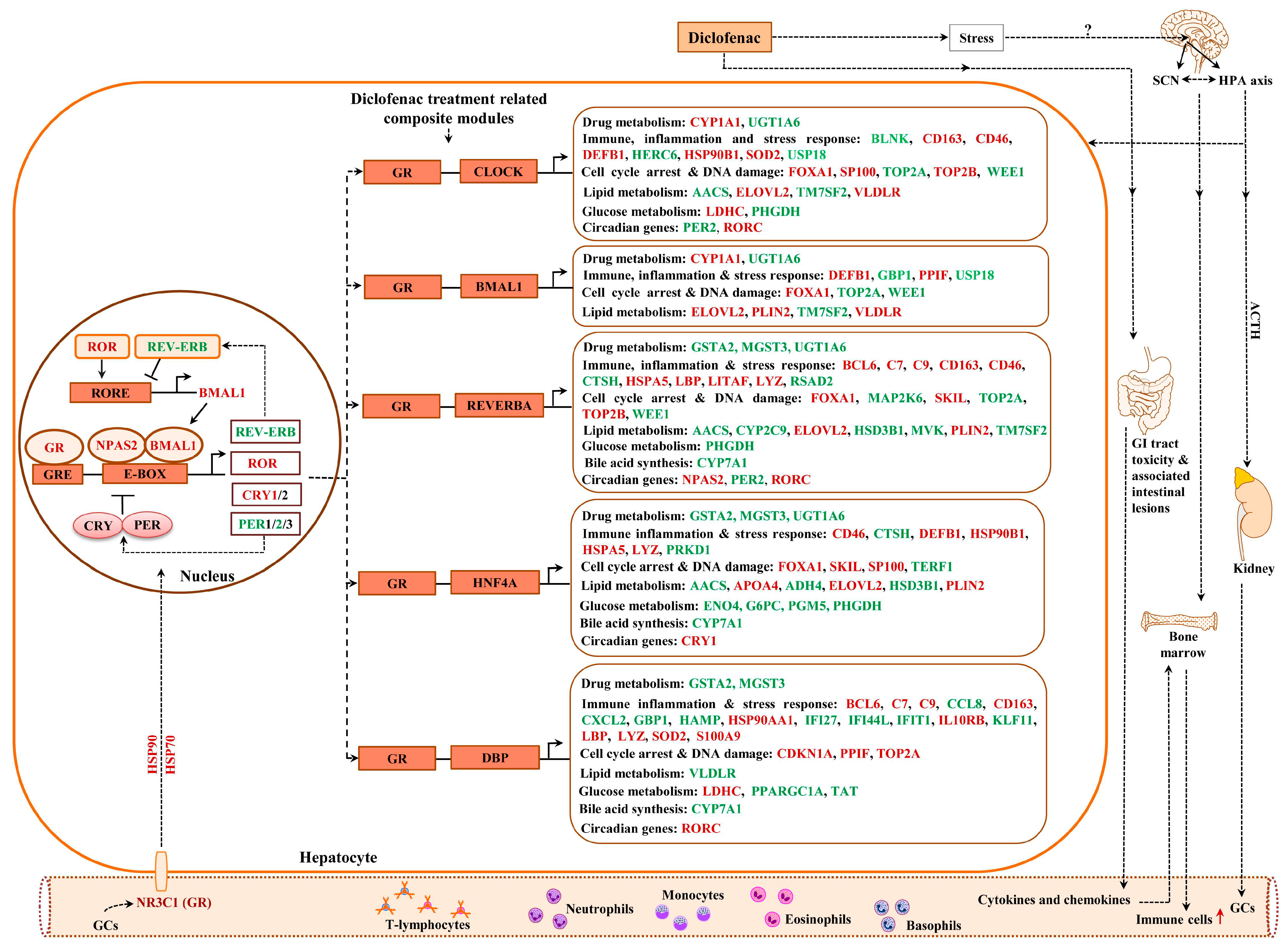
2.13. Composite Modules of the Circadian Clock and Glucocorticoid Receptor
2.14. GR Signaling
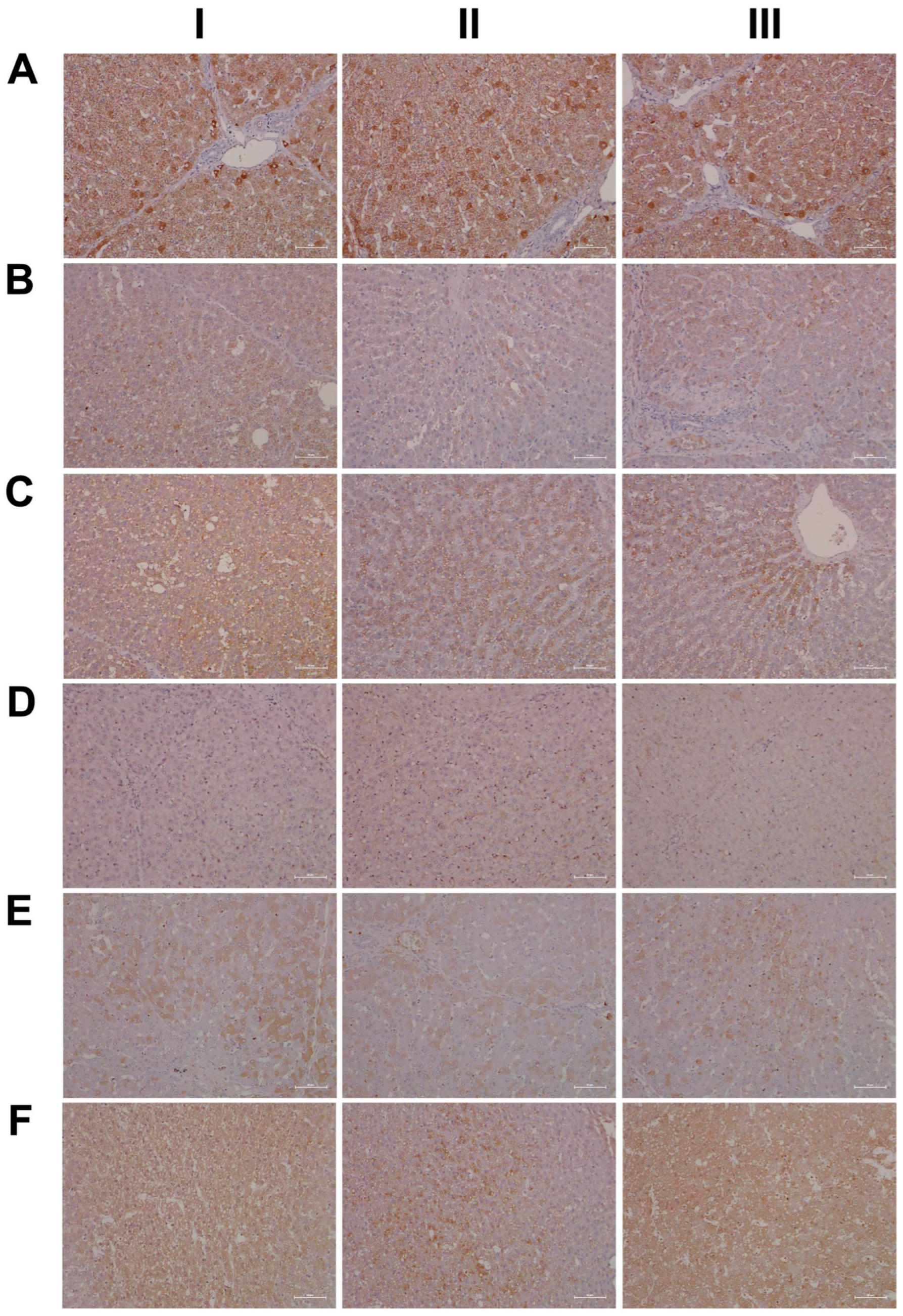
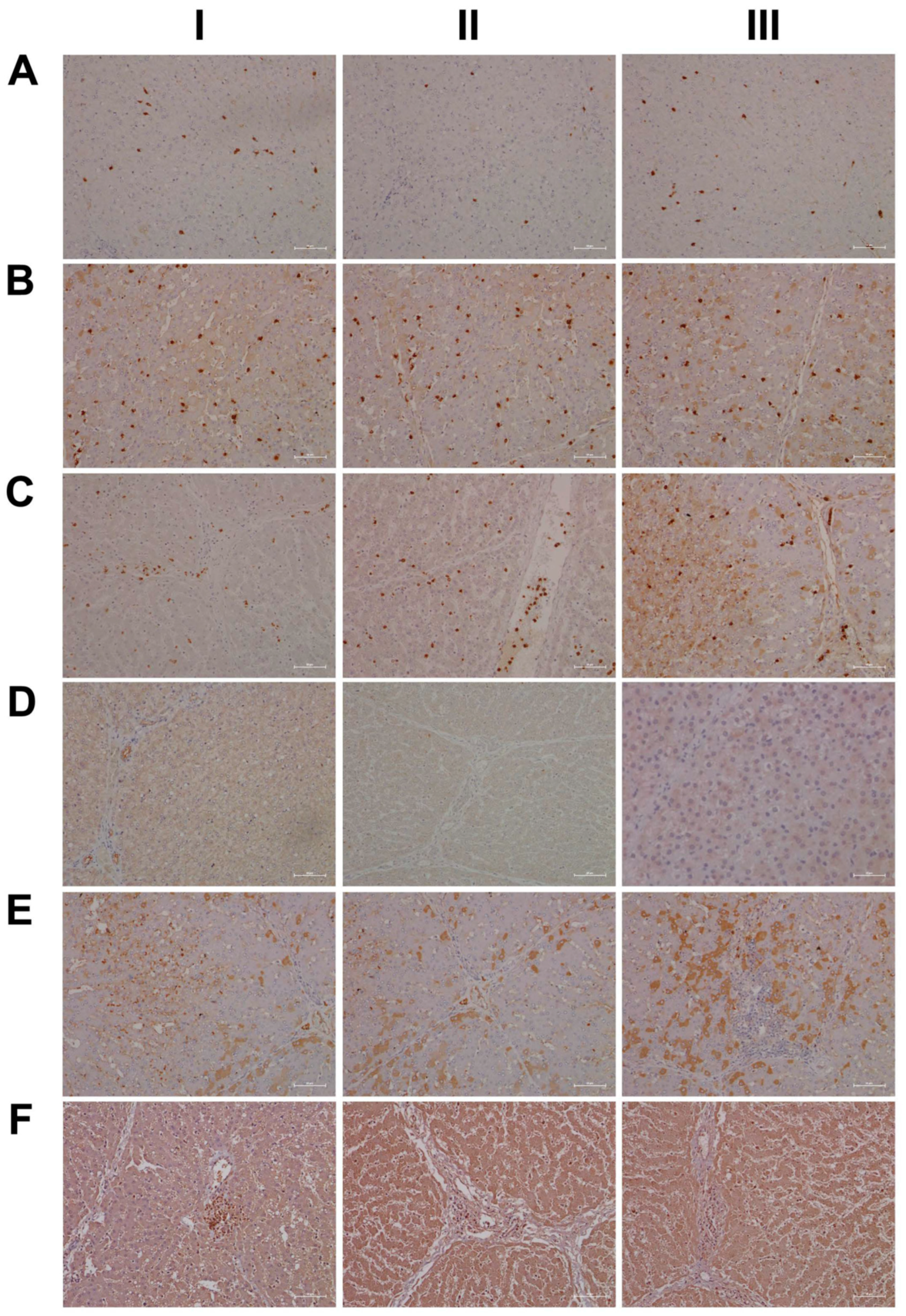
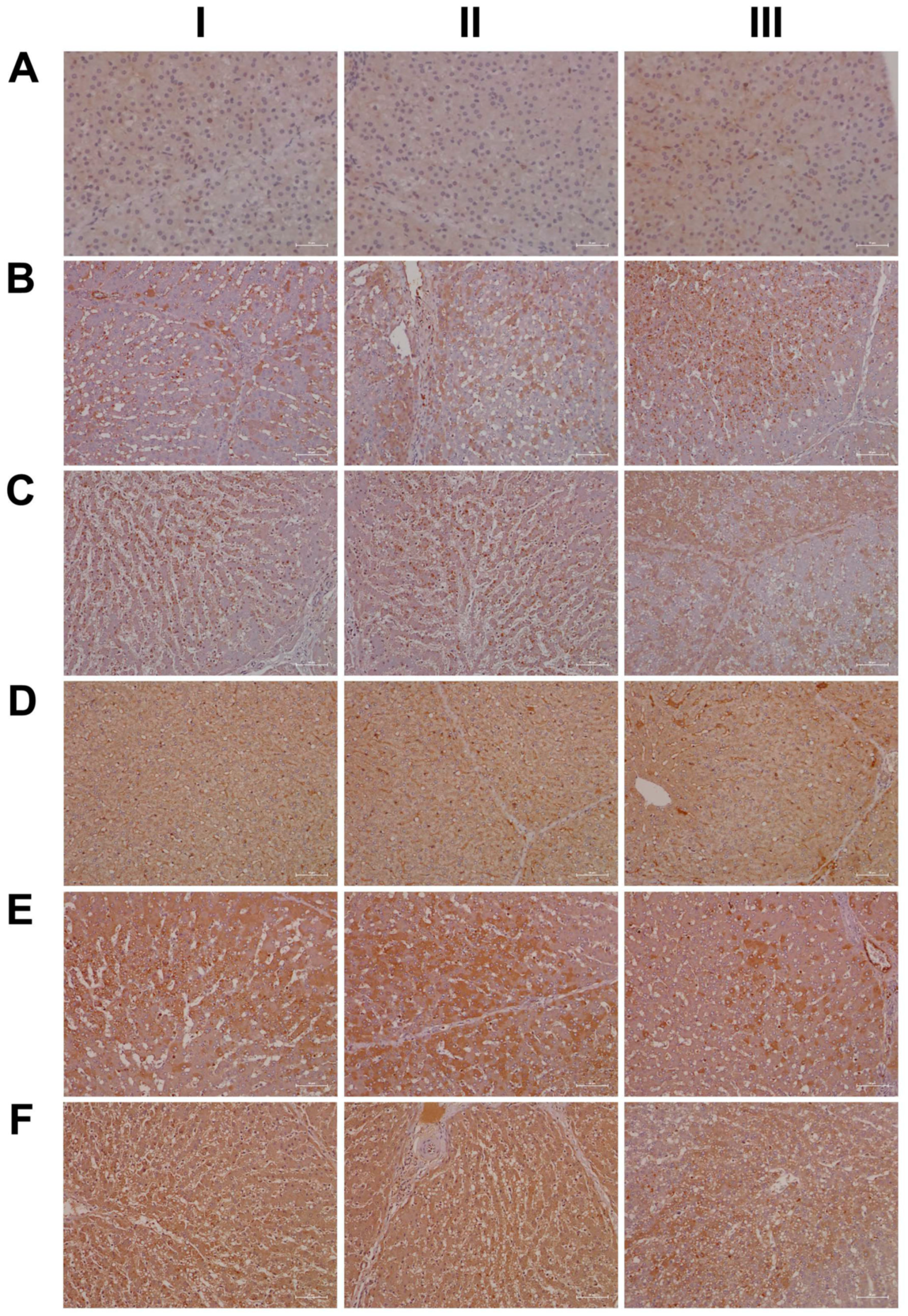
2.15. Immunohistochemistry Confirms Regulation of Liver Clock Components
2.16. Regulatory Gene Networks
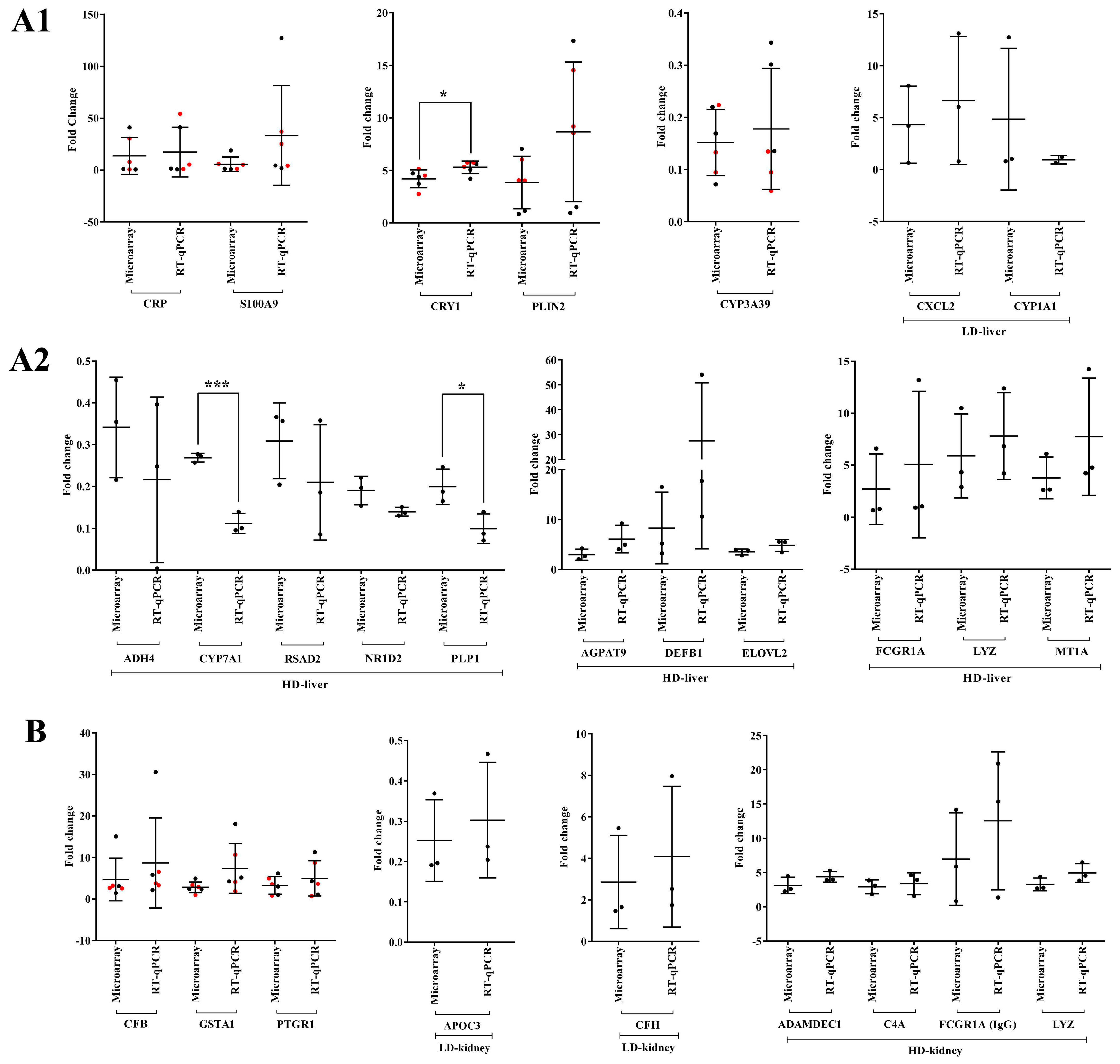
2.17. Validation of Transcriptional Responses by RT-qPCR
3. Discussion
3.1. Diclofenac Reactive Metabolites
3.2. Diclofenac-Induced Expression of Marker Genes of Inflammation
3.3. Diclofenac Regulates Components of the Circadian Clock
3.4. Diclofenac Induces Cell Cycle Arrest and Apoptosis
3.5. Diclofenac-Induced Hepatic Steatosis
3.6. Transcription Factor—And Master Regulatory Gene Networks
4. Materials and Methods
4.1. Animals
4.2. Drug Treatment
4.3. Clinical Pathology
4.4. RNA Extraction
4.5. Microarray Experiments and Data Analysis
4.6. RT-qPCR Validation of Microarray Data
4.7. Histopathology
4.8. Immunohistochemistry
4.9. Bioinformatics
4.9.1. Identification of Orthologous Genes
4.9.2. Enriched Biological Processes and Gene Ontology Mapping
4.9.3. Gene/Protein Interaction Network Construction
4.9.4. Identification of Upstream Master Regulatory Molecules
4.9.5. Promoter Sequence Analysis
4.9.6. Composite Modules
5. Conclusions
Study Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AACS | acetoacetyl-coenzyme A synthetase |
| ACTH | adrenocorticopic hormone |
| ADH4 | alcohol dehydrogenase 4 |
| ADR | adverse drug reaction |
| AhR | aryl hydrocarbon receptor |
| ALB | albumin |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| ANGPTL4 | angiopoietin-like 4 |
| ARID5A | AT rich interactive domain 5A |
| AST | aspartate aminotransferase |
| BMCA | B-cell maturation antigen |
| BUN | blood urea nitrogen |
| C/EBPβ | CCAAT/enhancer-binding protein beta |
| CAE | chloroacetate esterase |
| CARPA | complement activation related pseudoallergy |
| CAV1 | caveolin-1 |
| CBG | corticosteroid binding globulin |
| CCL | C-C motif chemokine ligand |
| CDK | cyclin-dependent kinase |
| CBG | corticosteroid binding globulin |
| cGMP | cyclic guanosine monophosphate |
| CHOL | cholesterol |
| C/EBPB | CCAAT/enhancer-binding protein-homologous protein beta |
| CPS | carbamoyl phosphate synthetase |
| CREA | creatinine |
| CRF | corticotropin-releasing factor |
| CRY1 | cryptochrome 1 |
| CXCL | C-X-C motif chemokine ligand |
| CYP | cytochrome P450 |
| CPS1 | carbamoyl-phosphate synthetase 1 |
| DEFB1 | ß-defensin |
| DEGs | differentially expressed genes |
| DILI | drug induced liver injury |
| DPP4 | dipeptidyl-peptidase 4 |
| ECM | extracellular matrix |
| EDTA-2K | dipotassium ethylenediaminetetraacetic acid (anticoagulant) |
| ELOVL2 | ELOVL fatty acid elongase 2 |
| ER stress | endoplasmic reticulum stress |
| EvG | Elastika van Gieson (stain) |
| FDR | false discovery rate |
| GGT | gamma glutamyl transferase |
| GLU | glucose |
| GO | gene ontology |
| GR | glucocorticoid receptor |
| GRE | glucocorticoid response elements |
| H&E | Hematoxylin and Eosin |
| HCT | hematocrit |
| HepPar1 | Hepatocyte Paraffin 1 a hepatocyte specific antigen antibody |
| HGB | hemoglobin |
| HIF1A | hypoxia inducible factor 1 alpha subunit |
| HPA | axis- hypothalamus-pituitary-adrenal neuroendocrine axis |
| HRG | histidine rich glycoprotein |
| IFN | interferon |
| IGF | insulin-like growth factor |
| IGFBP | insulin-like growth factor binding protein |
| KLF | krüppel-like family transcription factor |
| LITAF | lipopolysaccharide-induced tumor necrosis factor-alpha factor |
| LPS | lipopolysaccharide |
| MAC | membrane attack complex |
| MCH | mean corpuscular hemoglobin |
| MCHC | mean corpuscular hemoglobin concentration |
| MCP | monocyte chemotactic protein |
| MCV | mean corpuscular volume |
| MEF2 | myocyte enhancer factor-2 |
| MeV | Multi Experimental Viewer |
| MIP | macrophage inflammatory protein |
| MMP7 | matrix metallopeptidase 7 |
| MPO | myeloperoxidase |
| NFKBIZ | nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor zeta |
| NK | natural killer cells |
| NMDA | N-methyl-D-aspartate |
| NSAID | nonsteroidal anti-inflammatory drug |
| PAS | Periodic acid-Schiff reaction |
| PER2 | period circadian regulator 2 |
| PLIN2 | perilipin 2 |
| PLT | platelet |
| PML | polymorphonuclear leukocytes |
| PPAR | peroxisome proliferator-activated receptor |
| PPI | protein-protein interaction |
| PWM | positional weight matrices |
| RBC | red blood cells |
| RET | reticulocyte |
| RGC-32 | response gene to complement 32 |
| ROS | reactive oxygen species |
| RT-PCR | reverse transcriptase-polymerase chain reaction |
| SELL | L-selectin |
| SIRT1 | sirtuin 1 |
| SOD | superoxide dismutase 1 |
| SPF | specific pathogen free |
| TBIL | total bilirubin |
| TFBS | transcription factor binding site |
| TG | triglycerides |
| TNF | tumor necrosis factor |
| TP | total protein |
| TSS | transcription start sites |
| VLDLR | very low-density lipoprotein receptor |
| WBC | white blood cells |
References
- Zuckner, J. International Experience with Diclofenac in Rheumatoid Arthritis. Am. J. Med. 1986, 80, 39–42. [Google Scholar] [CrossRef]
- van Laar, M.; Pergolizzi, J.V., Jr.; Mellinghoff, H.U.; Merchante, I.M.; Nalamachu, S.; O’Brien, J.; Perrot, S.; Raffa, R.B. Pain Treatment in Arthritis-Related Pain: Beyond NSAIDs. Open Rheumatol. J. 2012, 6, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Hickey, E.J.; Raje, R.R.; Reid, V.E.; Gross, S.M.; Ray, S.D. Diclofenac Induced in Vivo Nephrotoxicity may Involve Oxidative Stress-Mediated Massive Genomic DNA Fragmentation and Apoptotic Cell Death. Free Radic. Biol. Med. 2001, 31, 139–152. [Google Scholar] [CrossRef]
- Roth, R.A.; Ganey, P.E. Intrinsic Versus Idiosyncratic Drug-Induced Hepatotoxicity—Two Villains Or One? J. Pharmacol. Exp. Ther. 2010, 332, 692–697. [Google Scholar] [CrossRef]
- Mendes, R.T.; Stanczyk, C.P.; Sordi, R.; Otuki, M.F.; dos Santos, F.A.; Fernandes, D. Selective Inhibition of Cyclooxygenase-2: Risks and Benefits. Rev. Bras. Reumatol. 2012, 52, 767–782. [Google Scholar]
- Boelsterli, U.A. Diclofenac-Induced Liver Injury: A Paradigm of Idiosyncratic Drug Toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.E.; Vincent, A.S.; Halliwell, B.; Wong, K.P. Action of Diclofenac on Kidney Mitochondria and Cells. Biochem. Biophys. Res. Commun. 2006, 348, 494–500. [Google Scholar] [CrossRef] [PubMed]
- van Swelm, R.P.; Laarakkers, C.M.; Pertijs, J.C.; Verweij, V.; Masereeuw, R.; Russel, F.G. Urinary Proteomic Profiling Reveals Diclofenac-Induced Renal Injury and Hepatic Regeneration in Mice. Toxicol. Appl. Pharmacol. 2013, 269, 141–149. [Google Scholar] [CrossRef]
- Banks, A.T.; Zimmerman, H.J.; Ishak, K.G.; Harter, J.G. Diclofenac-Associated Hepatotoxicity: Analysis of 180 Cases Reported to the Food and Drug Administration as Adverse Reactions. Hepatology 1995, 22, 820–827. [Google Scholar] [CrossRef]
- Laine, L.; Goldkind, L.; Curtis, S.P.; Connors, L.G.; Yanqiong, Z.; Cannon, C.P. How Common is Diclofenac-Associated Liver Injury? Analysis of 17,289 Arthritis Patients in a Long-Term Prospective Clinical Trial. Am. J. Gastroenterol. 2009, 104, 356. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Bjornsson, E.S.; Bergmann, O.M.; Bjornsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, Presentation, and Outcomes in Patients with Drug-Induced Liver Injury in the General Population of Iceland. Gastroenterology 2013, 144, 1419–1425; quiz e19–e20. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.E.; Dickie, A.P.; Schreiter, K.; Wehr, R.; Wilson, E.M.; Bial, J.; Scheer, N.; Wilson, I.D.; Riley, R.J. The Pharmacokinetics and Metabolism of Diclofenac in Chimeric Humanized and Murinized FRG Mice. Arch. Toxicol. 2018, 92, 1953–1967. [Google Scholar] [CrossRef] [PubMed]
- Bort, R.; Ponsoda, X.; Jover, R.; Gomez-Lechon, M.J.; Castell, J.V. Diclofenac Toxicity to Hepatocytes: A Role for Drug Metabolism in Cell Toxicity. J. Pharmacol. Exp. Ther. 1999, 288, 65–72. [Google Scholar] [PubMed]
- Owumi, S.E.; Dim, U.J. Biochemical Alterations in Diclofenac-Treated Rats: Effect of Selenium on Oxidative Stress, Inflammation, and Hematological Changes. Toxicol. Res. Appl. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Gomez-Lechon, M.J.; Ponsoda, X.; O’Connor, E.; Donato, T.; Castell, J.V.; Jover, R. Diclofenac Induces Apoptosis in Hepatocytes by Alteration of Mitochondrial Function and Generation of ROS. Biochem. Pharmacol. 2003, 66, 2155–2167. [Google Scholar] [CrossRef]
- Aithal, G.P. Hepatotoxicity Related to Antirheumatic Drugs. Nat. Rev. Rheumatol. 2011, 7, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Aithal, G.P.; Leathart, J.B.; Swainsbury, R.A.; Dang, T.S.; Day, C.P. Genetic Susceptibility to Diclofenac-Induced Hepatotoxicity: Contribution of UGT2B7, CYP2C8, and ABCC2 Genotypes. Gastroenterology 2007, 132, 272–281. [Google Scholar] [CrossRef]
- Lee, E.H.; Oh, J.H.; Selvaraj, S.; Park, S.M.; Choi, M.S.; Spanel, R.; Yoon, S.; Borlak, J. Immunogenomics Reveal Molecular Circuits of Diclofenac Induced Liver Injury in Mice. Oncotarget 2016, 7, 14983. [Google Scholar] [CrossRef]
- Selvaraj, S.; Oh, J.H.; Spanel, R.; Langer, F.; Han, H.Y.; Lee, E.H.; Yoon, S.; Borlak, J. The Pathogenesis of Diclofenac Induced Immunoallergic Hepatitis in a Canine Model of Liver Injury. Oncotarget 2017, 8, 107763–107824. [Google Scholar] [CrossRef]
- Yano, A.; Higuchi, S.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Involvement of Immune-Related Factors in Diclofenac-Induced Acute Liver Injury in Mice. Toxicology 2012, 293, 107–114. [Google Scholar] [CrossRef]
- Bode, G.; Clausing, P.; Gervais, F.; Loegsted, J.; Luft, J.; Nogues, V.; Sims, J.; Steering Group of the RETHINK Project. The Utility of the Minipig as an Animal Model in Regulatory Toxicology. J. Pharmacol. Toxicol. Methods 2010, 62, 196–220. [Google Scholar] [CrossRef]
- Rubic-Schneider, T.; Christen, B.; Brees, D.; Kammuller, M. Minipigs in Translational Immunosafety Sciences: A Perspective. Toxicol. Pathol. 2016, 44, 315–324. [Google Scholar] [CrossRef]
- Singh, V.K.; Thrall, K.D.; Hauer-Jensen, M. Minipigs as Models in Drug Discovery. Expert Opin. Drug Discov. 2016, 11, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, A.; Bailey, S.M.; Staels, B.; Baumert, T.F. The Circadian Clock and Liver Function in Health and Disease. J. Hepatol. 2019, 71, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, N.; Ye, B.; Ju, W.; Orser, B.; Fox, J.E.; Wheeler, M.B.; Wang, Q.; Lu, W.Y. Non-Steroidal Anti-Inflammatory Drugs Increase Insulin Release from Beta Cells by Inhibiting ATP-Sensitive Potassium Channels. Br. J. Pharmacol. 2007, 151, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.I.; Torres-López, J.E.; Castañeda-Hernández, G.; Rosas, R.; Vidal-Cantú, G.C.; Granados-Soto, V. Pharmacological Evidence for the Activation of K(+) Channels by Diclofenac. Eur. J. Pharmacol. 2002, 438, 85–91. [Google Scholar] [CrossRef]
- Khoja, S.; Nitzahn, M.; Truong, B.; Lambert, J.; Willis, B.; Allegri, G.; Rufenacht, V.; Haberle, J.; Lipshutz, G.S. A Constitutive Knockout of Murine Carbamoyl Phosphate Synthetase 1 Results in Death with Marked Hyperglutaminemia and Hyperammonemia. J. Inherit. Metab. Dis. 2019, 42, 1044–1053. [Google Scholar] [CrossRef]
- Ranucci, G.; Rigoldi, M.; Cotugno, G.; Bernabei, S.M.; Liguori, A.; Gasperini, S.; Goffredo, B.M.; Martinelli, D.; Monti, L.; Francalanci, P.; et al. Chronic Liver Involvement in Urea Cycle Disorders. J. Inherit. Metab. Dis. 2019, 42, 1118–1127. [Google Scholar] [CrossRef]
- Butler, S.L.; Dong, H.; Cardona, D.; Jia, M.; Zheng, R.; Zhu, H.; Crawford, J.M.; Liu, C. The Antigen for Hep Par 1 Antibody is the Urea Cycle Enzyme Carbamoyl Phosphate Synthetase 1. Lab. Investig. 2008, 88, 78–88. [Google Scholar] [CrossRef]
- Sahini, N.; Borlak, J. Genomics of Human Fatty Liver Disease Reveal Mechanistically Linked Lipid Droplet-Associated Gene Regulations in Bland Steatosis and Nonalcoholic Steatohepatitis. Transl. Res. 2016, 177, 41–69. [Google Scholar] [CrossRef]
- Sahini, N.; Borlak, J. Recent Insights into the Molecular Pathophysiology of Lipid Droplet Formation in Hepatocytes. Prog. Lipid Res. 2014, 54, 86–112. [Google Scholar] [CrossRef]
- Bradbury, M.W. Lipid Metabolism and Liver Inflammation. I. Hepatic Fatty Acid Uptake: Possible Role in Steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G194–G198. [Google Scholar] [CrossRef]
- Reddy, J.K.; Rao, M.S. Lipid Metabolism and Liver Inflammation. II. Fatty Liver Disease and Fatty Acid Oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef]
- Liu, Y.J.; Li, H.; Tian, Y.; Han, J.; Wang, X.Y.; Li, X.Y.; Tian, C.; Zhang, P.H.; Hao, Y.; Gao, F.; et al. PCTR1 Ameliorates Lipopolysaccharide-Induced Acute Inflammation and Multiple Organ Damage Via Regulation of Linoleic Acid Metabolism by Promoting FADS1/FASDS2/ELOV2 Expression and Reducing PLA2 Expression. Lab. Investig. 2020, 100, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Hashimoto, R.; Shibata, M.; Shimada, H.; Iwaki, M. Involvement of Reactive Metabolites of Diclofenac in Cytotoxicity in Sandwich-Cultured Rat Hepatocytes. Int. J. Toxicol. 2017, 36, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1alpha Protects Against Oxidative Stress by Directly Targeting Mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.; Soehnlein, O.; Viola, J.R. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front. Immunol. 2018, 9, 2739. [Google Scholar] [CrossRef] [PubMed]
- Vlaicu, S.I.; Tatomir, A.; Anselmo, F.; Boodhoo, D.; Chira, R.; Rus, V.; Rus, H. RGC-32 and Diseases: The First 20 Years. Immunol. Res. 2019, 67, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Borniger, J.C.; Walker Ii, W.H.; Surbhi; Emmer, K.M.; Zhang, N.; Zalenski, A.A.; Muscarella, S.L.; Fitzgerald, J.A.; Smith, A.N.; Braam, C.J.; et al. A Role for Hypocretin/Orexin in Metabolic and Sleep Abnormalities in a Mouse Model of Non-Metastatic Breast Cancer. Cell Metab. 2018, 28, 118–129.e5. [Google Scholar] [CrossRef]
- Hirano, A.; Hsu, P.K.; Zhang, L.; Xing, L.; McMahon, T.; Yamazaki, M.; Ptacek, L.J.; Fu, Y.H. DEC2 Modulates Orexin Expression and Regulates Sleep. Proc. Natl. Acad. Sci. USA 2018, 115, 3434–3439. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The Biology of the Glucocorticoid Receptor: New Signaling Mechanisms in Health and Disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.J.; Durrington, H.J.; Gibbs, J.E.; Blaikley, J.; Loudon, A.S.; Ray, D.W.; Sabroe, I. A Matter of Time: Study of Circadian Clocks and their Role in Inflammation. J. Leukoc. Biol. 2016, 99, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Hergenhan, S.; Holtkamp, S.; Scheiermann, C. Molecular Interactions between Components of the Circadian Clock and the Immune System. J. Mol. Biol. 2020, 432, 3700–3713. [Google Scholar] [CrossRef] [PubMed]
- Vieira, E.; Mirizio, G.G.; Barin, G.R.; de Andrade, R.V.; Nimer, N.F.S.; La Sala, L. Clock Genes, Inflammation and the Immune System-Implications for Diabetes, Obesity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9743. [Google Scholar] [CrossRef] [PubMed]
- Reinke, H.; Asher, G. Circadian Clock Control of Liver Metabolic Functions. Gastroenterology 2016, 150, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Z.; Yang, P.; Xia, L.; Zhou, M.; Wang, S.; Du, J.; Zhang, J. PER1 Prevents Excessive Innate Immune Response during Endotoxin-Induced Liver Injury through Regulation of Macrophage Recruitment in Mice. Cell Death Dis. 2016, 7, e2176. [Google Scholar] [CrossRef]
- Narasimamurthy, R.; Hatori, M.; Nayak, S.K.; Liu, F.; Panda, S.; Verma, I.M. Circadian Clock Protein Cryptochrome Regulates the Expression of Proinflammatory Cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 12662–12667. [Google Scholar] [CrossRef]
- Cermakian, N.; Westfall, S.; Kiessling, S. Circadian Clocks and Inflammation: Reciprocal Regulation and Shared Mediators. Arch. Immunol. Ther. Exp. 2014, 62, 303–318. [Google Scholar] [CrossRef]
- Dubrovsky, Y.V.; Samsa, W.E.; Kondratov, R.V. Deficiency of Circadian Protein CLOCK Reduces Lifespan and Increases Age-Related Cataract Development in Mice. Aging 2010, 2, 936–944. [Google Scholar] [CrossRef]
- Spengler, M.L.; Kuropatwinski, K.K.; Comas, M.; Gasparian, A.V.; Fedtsova, N.; Gleiberman, A.S.; Gitlin, I.I.; Artemicheva, N.M.; Deluca, K.A.; Gudkov, A.V.; et al. Core Circadian Protein CLOCK is a Positive Regulator of NF-kappaB- Mediated Transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E2457–E2465. [Google Scholar] [CrossRef]
- DeBruyne, J.P.; Weaver, D.R.; Dallmann, R. The Hepatic Circadian Clock Modulates Xenobiotic Metabolism in Mice. J. Biol. Rhythms 2014, 29, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Fribourgh, J.L.; Partch, C.L. Assembly and Function of bHLH-PAS Complexes. Proc. Natl. Acad. Sci. USA 2017, 114, 5330–5332. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.X.; Tang, X.; Zhu, J.Y.; Luo, L.; Ma, X.Y.; Cheng, S.W.; Zhang, W.; Tang, W.Q.; Ma, W.; Yang, X.; et al. Correction to: Cytochrome P450 1A1 Enhances Inflammatory Responses and Impedes Phagocytosis of Bacteria in Macrophages during Sepsis. Cell Commun. Signal. 2020, 18, 74. [Google Scholar] [CrossRef] [PubMed]
- Dickmeis, T. Glucocorticoids and the Circadian Clock. J. Endocrinol. 2009, 200, 3–22. [Google Scholar] [CrossRef]
- Yeager, M.P.; Pioli, P.A.; Guyre, P.M. Cortisol Exerts Bi-Phasic Regulation of Inflammation in Humans. Dose Response 2011, 9, 332–347. [Google Scholar] [CrossRef]
- Meyer, E.J.; Nenke, M.A.; Rankin, W.; Lewis, J.G.; Torpy, D.J. Corticosteroid-Binding Globulin: A Review of Basic and Clinical Advances. Horm. Metab. Res. 2016, 48, 359–371. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Libert, C. On the Trail of the Glucocorticoid Receptor: Into the Nucleus and Back. Traffic 2012, 13, 364–374. [Google Scholar] [CrossRef]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef]
- Binder, E.B.; Nemeroff, C.B. The CRF System, Stress, Depression and Anxiety-Insights from Human Genetic Studies. Mol. Psychiatry 2010, 15, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.A.; Chouridou, E.; Koureta, M.; Lambropoulou, M.; Kolios, G.; Chatzaki, E. The Corticotropin Releasing Factor System in the Liver: Expression, Actions and Possible Implications in Hepatic Physiology and Pathology. Hormones 2013, 12, 236–245. [Google Scholar] [CrossRef]
- Chen, H.; Shi, H.; Liu, Y.; Ren, X.; He, S.; Chang, X.; Yin, Y. Activation of Corticotropin-Releasing Factor Receptor 1 Aggravates Dextran Sodium Sulphate-Induced Colitis in Mice by Promoting M1 Macrophage Polarization. Mol. Med. Rep. 2018, 17, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Idiosyncratic DILI: Analysis of 46,266 Cases Assessed for Causality by RUCAM and Published from 2014 to Early 2019. Front. Pharmacol. 2019, 10, 730. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Liguori, M.J.; Sparkenbaugh, E.M.; Waring, J.F.; Blomme, E.A.; Ganey, P.E.; Roth, R.A. Gene Expression Profiles in Livers from Diclofenac-Treated Rats Reveal Intestinal Bacteria-Dependent and -Independent Pathways Associated with Liver Injury. J. Pharmacol. Exp. Ther. 2008, 327, 634–644. [Google Scholar] [CrossRef]
- Soucek, P.; Zuber, R.; Anzenbacherova, E.; Anzenbacher, P.; Guengerich, F.P. Minipig Cytochrome P450 3A, 2A and 2C Enzymes have Similar Properties to Human Analogs. BMC Pharmacol. 2001, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.; Yang, J.; Liu, Y.; Wei, H. Tissue Distribution of CYP3A29 mRNA Expression in Bama Miniature Pig by Quantitative Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR). Xenobiotica 2009, 39, 423–429. [Google Scholar] [CrossRef]
- Puccinelli, E.; Gervasi, P.G.; Longo, V. Xenobiotic Metabolizing Cytochrome P450 in Pig, a Promising Animal Model. Curr. Drug Metab. 2011, 12, 507–525. [Google Scholar] [CrossRef]
- Liu, X.; Wen, J.; Chen, R.; Zhang, T.; Jiang, J.; Deng, Y. T-2 Toxin Induces the Expression of Porcine CYP3A22 Via the Upregulation of the Transcription Factor, NF-Y. Biochim. Biophys. Acta 2016, 1860, 2191–2201. [Google Scholar] [CrossRef]
- Jover, R.; Bort, R.; Gomez-Lechon, M.J.; Castell, J.V. Down-Regulation of Human CYP3A4 by the Inflammatory Signal Interleukin-6: Molecular Mechanism and Transcription Factors Involved. FASEB J. 2002, 16, 1799–1801. [Google Scholar] [CrossRef]
- Aitken, A.E.; Morgan, E.T. Gene-Specific Effects of Inflammatory Cytokines on Cytochrome P450 2C, 2B6 and 3A4 mRNA Levels in Human Hepatocytes. Drug Metab. Dispos. 2007, 35, 1687–1693. [Google Scholar] [CrossRef]
- Miyamoto, G.; Zahid, N.; Uetrecht, J.P. Oxidation of Diclofenac to Reactive Intermediates by Neutrophils, Myeloperoxidase, and Hypochlorous Acid. Chem. Res. Toxicol. 1997, 10, 414–419. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile Acids: Regulation of Synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Li, T.; Owsley, E.; Matozel, M.; Hsu, P.; Novak, C.M.; Chiang, J.Y. Transgenic Expression of Cholesterol 7alpha-Hydroxylase in the Liver Prevents High-Fat Diet-Induced Obesity and Insulin Resistance in Mice. Hepatology 2010, 52, 678–690. [Google Scholar] [CrossRef]
- Sarkar, U.; Ravindra, K.C.; Large, E.; Young, C.L.; Rivera-Burgos, D.; Yu, J.; Cirit, M.; Hughes, D.J.; Wishnok, J.S.; Lauffenburger, D.A.; et al. Integrated Assessment of Diclofenac Biotransformation, Pharmacokinetics, and Omics-Based Toxicity in a Three-Dimensional Human Liver-Immunocompetent Coculture System. Drug Metab. Dispos. 2017, 45, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Roma, M.G.; Toledo, F.D.; Boaglio, A.C.; Basiglio, C.L.; Crocenzi, F.A.; Sanchez Pozzi, E.J. Ursodeoxycholic Acid in Cholestasis: Linking Action Mechanisms to Therapeutic Applications. Clin. Sci. 2011, 121, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Kanai, S.; Kitani, K. Glycoursodeoxycholate is as Effective as Tauroursodeoxycholate in Preventing the Taurocholate-Induced Cholestasis in the Rat. Res. Commun. Chem. Pathol. Pharmacol. 1983, 42, 423–430. [Google Scholar]
- Heemskerk, S.; Pickkers, P.; Bouw, M.P.; Draisma, A.; van der Hoeven, J.G.; Peters, W.H.; Smits, P.; Russel, F.G.; Masereeuw, R. Upregulation of Renal Inducible Nitric Oxide Synthase during Human Endotoxemia and Sepsis is Associated with Proximal Tubule Injury. Clin. J. Am. Soc. Nephrol. 2006, 1, 853–862. [Google Scholar] [CrossRef]
- Semple, F.; Dorin, J.R. Beta-Defensins: Multifunctional Modulators of Infection, Inflammation and More? J. Innate Immun. 2012, 4, 337–348. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From Bacterial Killing to Immune Modulation: Recent Insights into the Functions of Lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef] [PubMed]
- Klag, T.; Thomas, M.; Ehmann, D.; Courth, L.; Mailander-Sanchez, D.; Weiss, T.S.; Dayoub, R.; Abshagen, K.; Vollmar, B.; Thasler, W.E.; et al. Beta-Defensin 1 is Prominent in the Liver and Induced during Cholestasis by Bilirubin and Bile Acids Via Farnesoid X Receptor and Constitutive Androstane Receptor. Front. Immunol. 2018, 9, 1735. [Google Scholar] [CrossRef]
- Gujral, J.S.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Neutrophils Aggravate Acute Liver Injury during Obstructive Cholestasis in Bile Duct-Ligated Mice. Hepatology 2003, 38, 355–363. [Google Scholar] [CrossRef]
- Bautista, A.P. Chronic Alcohol Intoxication Induces Hepatic Injury through Enhanced Macrophage Inflammatory Protein-2 Production and Intercellular Adhesion Molecule-1 Expression in the Liver. Hepatology 1997, 25, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Stachlewitz, R.F.; Liguori, M.J.; Blomme, E.A.; Waring, J.F.; Luyendyk, J.P.; Maddox, J.F.; Ganey, P.E.; Roth, R.A. Modest Inflammation Enhances Diclofenac Hepatotoxicity in Rats: Role of Neutrophils and Bacterial Translocation. J. Pharmacol. Exp. Ther. 2006, 319, 1191–1199. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ho, Y.S.; Fisher, M.A.; Lawson, J.A.; Farhood, A. Glutathione Peroxidase-Deficient Mice are More Susceptible to Neutrophil-Mediated Hepatic Parenchymal Cell Injury during Endotoxemia: Importance of an Intracellular Oxidant Stress. Hepatology 1999, 29, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, S.K.; Jaeschke, H. Role of Neutrophils in the Pathogenesis of Acute Inflammatory Liver Injury. Toxicol. Pathol. 2007, 35, 757–766. [Google Scholar] [CrossRef]
- Deng, X.; Luyendyk, J.P.; Ganey, P.E.; Roth, R.A. Inflammatory Stress and Idiosyncratic Hepatotoxicity: Hints from Animal Models. Pharmacol. Rev. 2009, 61, 262–282. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Gottsche, B.; Mueller-Eckhardt, C. Autoantibodies and Drug- Or Metabolite-Dependent Antibodies in Patients with Diclofenac-Induced Immune Haemolysis. Br. J. Haematol. 1991, 77, 546–549. [Google Scholar] [CrossRef]
- Romano, A.; Quaratino, D.; Papa, G.; Di Fonso, M.; Artesani, M.C.; Venuti, A. Delayed Hypersensitivity to Diclofenac: A Report on Two Cases. Ann. Allergy Asthma Immunol. 1998, 81, 373–375. [Google Scholar] [CrossRef]
- Harrer, A.; Lang, R.; Grims, R.; Braitsch, M.; Hawranek, T.; Aberer, W.; Vogel, L.; Schmid, W.; Ferreira, F.; Himly, M. Diclofenac Hypersensitivity: Antibody Responses to the Parent Drug and Relevant Metabolites. PLoS ONE 2010, 5, e13707. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef]
- Publicover, J.; Gaggar, A.; Nishimura, S.; Van Horn, C.M.; Goodsell, A.; Muench, M.O.; Reinhardt, R.L.; van Rooijen, N.; Wakil, A.E.; Peters, M.; et al. Age-Dependent Hepatic Lymphoid Organization Directs Successful Immunity to Hepatitis B. J. Clin. Investig. 2013, 123, 3728–3739. [Google Scholar] [CrossRef]
- Li, C.; Kang, D.; Sun, X.; Liu, Y.; Wang, J.; Gao, P. The Effect of C-X-C Motif Chemokine 13 on Hepatocellular Carcinoma Associates with Wnt Signaling. Biomed. Res. Int. 2015, 2015, 345413. [Google Scholar] [CrossRef] [PubMed]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine Receptor CXCR6-Dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shao, Y.; Zhang, S.; Xie, A.; Ye, Y.; Shi, L.; Jin, L.; Pan, X.; Lin, Z.; Li, X.; et al. CXCL16 Deficiency Attenuates Acetaminophen-Induced Hepatotoxicity through Decreasing Hepatic Oxidative Stress and Inflammation in Mice. Acta Biochim. Biophys. Sin. 2017, 49, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Tsou, C.L.; Croft, K.; Charo, I.F. CCR2 Mediates Hematopoietic Stem and Progenitor Cell Trafficking to Sites of Inflammation in Mice. J. Clin. Investig. 2010, 120, 1192–1203. [Google Scholar] [CrossRef]
- Hellier, S.; Frodsham, A.J.; Hennig, B.J.; Klenerman, P.; Knapp, S.; Ramaley, P.; Satsangi, J.; Wright, M.; Zhang, L.; Thomas, H.C.; et al. Association of Genetic Variants of the Chemokine Receptor CCR5 and its Ligands, RANTES and MCP-2, with Outcome of HCV Infection. Hepatology 2003, 38, 1468–1476. [Google Scholar] [CrossRef]
- Viola, A.; Luster, A.D. Chemokines and their Receptors: Drug Targets in Immunity and Inflammation. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 171–197. [Google Scholar] [CrossRef]
- Wasmuth, H.E.; Tacke, F.; Trautwein, C. Chemokines in Liver Inflammation and Fibrosis. Semin. Liver Dis. 2010, 30, 215–225. [Google Scholar] [CrossRef]
- Hori, T.; Naishiro, Y.; Sohma, H.; Suzuki, N.; Hatakeyama, N.; Yamamoto, M.; Sonoda, T.; Mizue, Y.; Imai, K.; Tsutsumi, H.; et al. CCL8 is a Potential Molecular Candidate for the Diagnosis of Graft-Versus-Host Disease. Blood 2008, 111, 4403–4412. [Google Scholar] [CrossRef]
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 Receptor Signaling: Master Regulator of Intestinal Mucosal Homeostasis in Mice and Humans. Adv. Immunol. 2014, 122, 177–210. [Google Scholar]
- Disteldorf, E.M.; Krebs, C.F.; Paust, H.J.; Turner, J.E.; Nouailles, G.; Tittel, A.; Meyer-Schwesinger, C.; Stege, G.; Brix, S.; Velden, J.; et al. CXCL5 Drives Neutrophil Recruitment in TH17-Mediated GN. J. Am. Soc. Nephrol. 2015, 26, 55–66. [Google Scholar] [CrossRef]
- Starnes, T.; Rasila, K.K.; Robertson, M.J.; Brahmi, Z.; Dahl, R.; Christopherson, K.; Hromas, R. The Chemokine CXCL14 (BRAK) Stimulates Activated NK Cell Migration: Implications for the Downregulation of CXCL14 in Malignancy. Exp. Hematol. 2006, 34, 1101–1105. [Google Scholar] [CrossRef]
- Hara, T.; Tanegashima, K. Pleiotropic Functions of the CXC-Type Chemokine CXCL14 in Mammals. J. Biochem. 2012, 151, 469–476. [Google Scholar] [CrossRef]
- Wada, T.; Furuichi, K.; Sakai, N.; Shimizu, M.; Segawa, C.; Kobayashi, K.; Mukaida, N.; Kasahara, T.; Matsushima, K.; Yokoyama, H. Eotaxin Contributes to Renal Interstitial Eosinophilia. Nephrol. Dial. Transplant. 1999, 14, 76–80. [Google Scholar] [CrossRef]
- Badewa, A.P.; Hudson, C.E.; Heiman, A.S. Regulatory Effects of Eotaxin, Eotaxin-2, and Eotaxin-3 on Eosinophil Degranulation and Superoxide Anion Generation. Exp. Biol. Med. 2002, 227, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Coquery, C.M.; Wade, N.S.; Loo, W.M.; Kinchen, J.M.; Cox, K.M.; Jiang, C.; Tung, K.S.; Erickson, L.D. Neutrophils Contribute to Excess Serum BAFF Levels and Promote CD4+ T Cell and B Cell Responses in Lupus-Prone Mice. PLoS ONE 2014, 9, e102284. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.G.; Vilcek, J. TSG-6: An IL-1/TNF-Inducible Protein with Anti-Inflammatory Activity. Cytokine Growth Factor Rev. 1997, 8, 143–156. [Google Scholar] [CrossRef]
- Thurman, J.M.; Holers, V.M. The Central Role of the Alternative Complement Pathway in Human Disease. J. Immunol. 2006, 176, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Farrar, C.A.; Abe, K.; Pratt, J.R.; Marsh, J.E.; Wang, Y.; Stahl, G.L.; Sacks, S.H. Predominant Role for C5b-9 in Renal Ischemia/Reperfusion Injury. J. Clin. Investig. 2000, 105, 1363–1371. [Google Scholar] [CrossRef]
- Danobeitia, J.S.; Djamali, A.; Fernandez, L.A. The Role of Complement in the Pathogenesis of Renal Ischemia-Reperfusion Injury and Fibrosis. Fibrogenesis Tissue Repair 2014, 7, 16. [Google Scholar] [CrossRef]
- Keller, M.; Mazuch, J.; Abraham, U.; Eom, G.D.; Herzog, E.D.; Volk, H.D.; Kramer, A.; Maier, B. A Circadian Clock in Macrophages Controls Inflammatory Immune Responses. Proc. Natl. Acad. Sci. USA 2009, 106, 21407–21412. [Google Scholar] [CrossRef]
- Sukumaran, S.; Almon, R.R.; DuBois, D.C.; Jusko, W.J. Circadian Rhythms in Gene Expression: Relationship to Physiology, Disease, Drug Disposition and Drug Action. Adv. Drug Deliv. Rev. 2010, 62, 904–917. [Google Scholar] [CrossRef]
- Wang, L.; Liangpunsakul, S. Circadian Clock Control of Hepatic Lipid Metabolism: Role of Small Heterodimer Partner (Shp). J. Investig. Med. 2016, 64, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Green, C.B.; Takahashi, J.S.; Bass, J. The Meter of Metabolism. Cell 2008, 134, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Schibler, U. Crosstalk between Components of Circadian and Metabolic Cycles in Mammals. Cell Metab. 2011, 13, 125–137. [Google Scholar] [CrossRef]
- Kakan, X.; Chen, P.; Zhang, J. Clock Gene mPer2 Functions in Diurnal Variation of Acetaminophen Induced Hepatotoxicity in Mice. Exp. Toxicol. Pathol. 2011, 63, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Kakan, X.; Wang, S.; Dong, W.; Jia, A.; Cai, C.; Zhang, J. Deletion of Clock Gene Per2 Exacerbates Cholestatic Liver Injury and Fibrosis in Mice. Exp. Toxicol. Pathol. 2013, 65, 427–432. [Google Scholar] [CrossRef]
- Chen, P.; Li, C.; Pang, W.; Zhao, Y.; Dong, W.; Wang, S.; Zhang, J. The Protective Role of Per2 Against Carbon Tetrachloride-Induced Hepatotoxicity. Am. J. Pathol. 2009, 174, 63–70. [Google Scholar] [CrossRef]
- Bugge, A.; Feng, D.; Everett, L.J.; Briggs, E.R.; Mullican, S.E.; Wang, F.; Jager, J.; Lazar, M.A. Rev-Erbalpha and Rev-Erbbeta Coordinately Protect the Circadian Clock and Normal Metabolic Function. Genes Dev. 2012, 26, 657–667. [Google Scholar] [CrossRef]
- Jetten, A.M.; Kang, H.S.; Takeda, Y. Retinoic Acid-Related Orphan Receptors Alpha and Gamma: Key Regulators of Lipid/Glucose Metabolism, Inflammation, and Insulin Sensitivity. Front. Endocrinol. 2013, 4, 1. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, S.; Zhou, Z.; Guo, L.; Yu, F.; Wu, B. Bmal1 Regulates Circadian Expression of Cytochrome P450 3a11 and Drug Metabolism in Mice. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Ripperger, J.A.; Shearman, L.P.; Reppert, S.M.; Schibler, U. CLOCK, an Essential Pacemaker Component, Controls Expression of the Circadian Transcription Factor DBP. Genes Dev. 2000, 14, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Mitsui, S.; Yan, L.; Yagita, K.; Miyake, S.; Okamura, H. Role of DBP in the Circadian Oscillatory Mechanism. Mol. Cell. Biol. 2000, 20, 4773–4781. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, M.; Stadler, F.; Tamanini, F.; van der Horst, G.T.; Ripperger, J.A. Flexible Phase Adjustment of Circadian Albumin D Site-Binding Protein (DBP) Gene Expression by CRYPTOCHROME1. Genes Dev. 2010, 24, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, M.; Suter, D.M.; Molina, N.; Naef, F.; Schibler, U. Circadian Dbp Transcription Relies on Highly Dynamic BMAL1-CLOCK Interaction with E Boxes and Requires the Proteasome. Mol. Cell 2012, 48, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Gachon, F.; Olela, F.F.; Schaad, O.; Descombes, P.; Schibler, U. The Circadian PAR-Domain Basic Leucine Zipper Transcription Factors DBP, TEF, and HLF Modulate Basal and Inducible Xenobiotic Detoxification. Cell Metab. 2006, 4, 25–36. [Google Scholar] [CrossRef]
- Saran, A.R.; Dave, S.; Zarrinpar, A. Circadian Rhythms in the Pathogenesis and Treatment of Fatty Liver Disease. Gastroenterology 2020, 158, 1948–1966.e1. [Google Scholar] [CrossRef]
- Udoh, U.S.; Valcin, J.A.; Gamble, K.L.; Bailey, S.M. The Molecular Circadian Clock and Alcohol-Induced Liver Injury. Biomolecules 2015, 5, 2504–2537. [Google Scholar] [CrossRef]
- Ge, W.; Wang, T.; Zhao, Y.; Yang, Y.; Sun, Q.; Yang, X.; Gao, Y.; Xu, X.; Zhang, J. Period1 Mediates Rhythmic Metabolism of Toxins by Interacting with CYP2E1. Cell Death Dis. 2021, 12, 76. [Google Scholar] [CrossRef]
- Xu, R.; Shang, C.; Zhao, J.; Han, Y.; Liu, J.; Chen, K.; Shi, W. Knockdown of Response Gene to Complement 32 (RGC32) Induces Apoptosis and Inhibits Cell Growth, Migration, and Invasion in Human Lung Cancer Cells. Mol. Cell. Biochem. 2014, 394, 109–118. [Google Scholar] [CrossRef]
- Tegla, C.A.; Cudrici, C.D.; Nguyen, V.; Danoff, J.; Kruszewski, A.M.; Boodhoo, D.; Mekala, A.P.; Vlaicu, S.I.; Chen, C.; Rus, V.; et al. RGC-32 is a Novel Regulator of the T-Lymphocyte Cell Cycle. Exp. Mol. Pathol. 2015, 98, 328–337. [Google Scholar] [CrossRef]
- Tang, R.; Zhang, G.; Chen, S.Y. Response Gene to Complement 32 Protein Promotes Macrophage Phagocytosis Via Activation of Protein Kinase C Pathway. J. Biol. Chem. 2014, 289, 22715–22722. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.B.; Luan, J.N.; Chen, S.Y. RGC-32 Deficiency Protects Against Hepatic Steatosis by Reducing Lipogenesis. J. Biol. Chem. 2015, 290, 20387–20395. [Google Scholar] [CrossRef]
- Cui, X.B.; Chen, S.Y. Response Gene to Complement 32 in Vascular Diseases. Front. Cardiovasc. Med. 2018, 5, 128. [Google Scholar] [CrossRef]
- de Almeida, C.J.G. Caveolin-1 and Caveolin-2 can be Antagonistic Partners in Inflammation and Beyond. Front. Immunol. 2017, 8, 1530. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rojo, M.A.; Ramm, G.A. Caveolin-1 Function in Liver Physiology and Disease. Trends Mol. Med. 2016, 22, 889–904. [Google Scholar] [CrossRef]
- Li, M.; Chen, D.; Huang, H.; Wang, J.; Wan, X.; Xu, C.; Li, C.; Ma, H.; Yu, C.; Li, Y. Caveolin1 Protects Against Diet Induced Hepatic Lipid Accumulation in Mice. PLoS ONE 2017, 12, e0178748. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Mari, M.; Herms, A.; Fernandez, A.; Fajardo, A.; Kassan, A.; Giralt, A.; Colell, A.; Balgoma, D.; Barbero, E.; et al. Caveolin-1 Deficiency Causes Cholesterol-Dependent Mitochondrial Dysfunction and Apoptotic Susceptibility. Curr. Biol. 2011, 21, 681–686. [Google Scholar] [CrossRef]
- Yin, Y.; Solomon, G.; Deng, C.; Barrett, J.C. Differential Regulation of p21 by p53 and Rb in Cellular Response to Oxidative Stress. Mol. Carcinog. 1999, 24, 15–24. [Google Scholar] [CrossRef]
- Gartel, A.L.; Tyner, A.L. The Role of the Cyclin-Dependent Kinase Inhibitor p21 in Apoptosis. Mol. Cancer. Ther. 2002, 1, 639–649. [Google Scholar]
- Hiromura, K.; Pippin, J.W.; Fero, M.L.; Roberts, J.M.; Shankland, S.J. Modulation of Apoptosis by the Cyclin-Dependent Kinase Inhibitor p27(Kip1). J. Clin. Investig. 1999, 103, 597–604. [Google Scholar] [CrossRef]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of Alcohol Metabolism in Non-Alcoholic Steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Xia, J.H.; Zhang, Y.Y.; Fan, J.G.; Wang, H.; Yuan, J.; Zhao, Z.Z.; Pan, Q.; Mu, Y.L.; Xin, L.L.; et al. Hyperinsulinemia Shifted Energy Supply from Glucose to Ketone Bodies in Early Nonalcoholic Steatohepatitis from High-Fat High-Sucrose Diet Induced Bama Minipigs. Sci. Rep. 2015, 5, 13980. [Google Scholar] [CrossRef]
- Hayakawa, K.; Nakajima, S.; Hiramatsu, N.; Okamura, M.; Huang, T.; Saito, Y.; Tagawa, Y.; Tamai, M.; Takahashi, S.; Yao, J.; et al. ER Stress Depresses NF-kappaB Activation in Mesangial Cells through Preferential Induction of C/EBP Beta. J. Am. Soc. Nephrol. 2010, 21, 73–81. [Google Scholar] [CrossRef]
- Ishihara, K.; Namura, T.; Murayama, H.; Arai, S.; Totani, M.; Ikemoto, M. Possibility of Formation of the S100A8/A9-Proinflammatory Cytokine Complexes in Vivo in Acute Inflammation and their Functional Roles. Rinsho Byori. Jpn. J. Clin. Pathol. 2009, 57, 324–331. [Google Scholar]
- Fujiu, K.; Manabe, I.; Nagai, R. Renal Collecting Duct Epithelial Cells Regulate Inflammation in Tubulointerstitial Damage in Mice. J. Clin. Investig. 2011, 121, 3425–3441. [Google Scholar] [CrossRef] [PubMed]
- Shikata, K.; Suzuki, Y.; Wada, J.; Hirata, K.; Matsuda, M.; Kawashima, H.; Suzuki, T.; Iizuka, M.; Makino, H.; Miyasaka, M. L-Selectin and its Ligands Mediate Infiltration of Mononuclear Cells into Kidney Interstitium After Ureteric Obstruction. J. Pathol. 1999, 188, 93–99. [Google Scholar] [CrossRef]
- Bartneck, M.; Fech, V.; Ehling, J.; Govaere, O.; Warzecha, K.T.; Hittatiya, K.; Vucur, M.; Gautheron, J.; Luedde, T.; Trautwein, C.; et al. Histidine-Rich Glycoprotein Promotes Macrophage Activation and Inflammation in Chronic Liver Disease. Hepatology 2016, 63, 1310–1324. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Simon, S.; Gibert, B.; Kretz-Remy, C.; Nivon, M.; Czekalla, A.; Guillet, D.; Moulin, M.; Diaz-Latoud, C.; Vicart, P. Hsp27 (HspB1) and alphaB-Crystallin (HspB5) as Therapeutic Targets. FEBS Lett. 2007, 581, 3665–3674. [Google Scholar] [CrossRef]
- Gang, G.T.; Hwang, J.H.; Kim, Y.H.; Noh, J.R.; Kim, K.S.; Jeong, J.Y.; Choi, D.E.; Lee, K.W.; Jung, J.Y.; Shong, M.; et al. Protection of NAD(P)H:Quinone Oxidoreductase 1 Against Renal Ischemia/Reperfusion Injury in Mice. Free Radic. Biol. Med. 2014, 67, 139–149. [Google Scholar] [CrossRef]
- Kobayashi, T.; Zadravec, D.; Jacobsson, A. ELOVL2 Overexpression Enhances Triacylglycerol Synthesis in 3T3-L1 and F442A Cells. FEBS Lett. 2007, 581, 3157–3163. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The Role of Lipid Droplets in Metabolic Disease in Rodents and Humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef]
- Jo, H.; Choe, S.S.; Shin, K.C.; Jang, H.; Lee, J.H.; Seong, J.K.; Back, S.H.; Kim, J.B. Endoplasmic Reticulum Stress Induces Hepatic Steatosis Via Increased Expression of the Hepatic very Low-Density Lipoprotein Receptor. Hepatology 2013, 57, 1366–1377. [Google Scholar] [CrossRef]
- Sahini, N.; Selvaraj, S.; Borlak, J. Whole Genome Transcript Profiling of Drug Induced Steatosis in Rats Reveals a Gene Signature Predictive of Outcome. PLoS ONE 2014, 9, e114085. [Google Scholar] [CrossRef]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha Deficiency Causes Multi-System Energy Metabolic Derangements: Muscle Dysfunction, Abnormal Weight Control and Hepatic Steatosis. PLoS Biol. 2005, 3, e101. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty Liver is Associated with Impaired Activity of PPARgamma-Coactivator 1alpha (PGC1alpha) and Mitochondrial Biogenesis in Mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef]
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional Coactivator PGC-1alpha Integrates the Mammalian Clock and Energy Metabolism. Nature 2007, 447, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Noda, K.; Maeda, A.; Matsuoka, M.; Yamasaki, M.; Fukui, T. Acetoacetyl-CoA Synthetase, a Ketone Body-Utilizing Enzyme, is Controlled by SREBP-2 and Affects Serum Cholesterol Levels. Mol. Genet. Metab. 2012, 107, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Q.; Yang, M.; Zhu, B.; Cui, Y.; Xue, Y.; Gong, N.; Cui, A.; Wang, M.; Shen, L.; et al. Mouse KLF11 Regulates Hepatic Lipid Metabolism. J. Hepatol. 2013, 58, 763–770. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Q.; Jiao, T.; Cui, A.; Sun, X.; Fang, W.; Xie, L.; Liu, Y.; Fang, F.; Chang, Y. Involvement of KLF11 in Hepatic Glucose Metabolism in Mice Via Suppressing of PEPCK-C Expression. PLoS ONE 2014, 9, e89552. [Google Scholar] [CrossRef]
- Harvey, P.W.; Sutcliffe, C. Adrenocortical Hypertrophy: Establishing Cause and Toxicological Significance. J. Appl. Toxicol. 2010, 30, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.E.; Liu, Y.; Dentin, R.; Pongsawakul, P.Y.; Liu, A.C.; Hirota, T.; Nusinow, D.A.; Sun, X.; Landais, S.; Kodama, Y.; et al. Cryptochrome Mediates Circadian Regulation of cAMP Signaling and Hepatic Gluconeogenesis. Nat. Med. 2010, 16, 1152–1156. [Google Scholar] [CrossRef]
- Lamia, K.A.; Papp, S.J.; Yu, R.T.; Barish, G.D.; Uhlenhaut, N.H.; Jonker, J.W.; Downes, M.; Evans, R.M. Cryptochromes Mediate Rhythmic Repression of the Glucocorticoid Receptor. Nature 2011, 480, 552–556. [Google Scholar] [CrossRef]
- Harris, A.J.; Thompson, A.R.; Whyte, M.K.; Walmsley, S.R. HIF-Mediated Innate Immune Responses: Cell Signaling and Therapeutic Implications. Hypoxia 2014, 2, 47–58. [Google Scholar]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-Specific Hypoxia-Inducible Factor-1alpha is a Determinant of Lipid Accumulation and Liver Injury in Alcohol-Induced Steatosis in Mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef]
- Nath, B.; Szabo, G. Hypoxia and Hypoxia Inducible Factors: Diverse Roles in Liver Diseases. Hepatology 2012, 55, 622–633. [Google Scholar] [CrossRef]
- Beck, I.M.; Vanden Berghe, W.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in Inflammation: The Interplay of Glucocorticoid Receptor-Based Mechanisms and Kinases and Phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef]
- Baschant, U.; Tuckermann, J. The Role of the Glucocorticoid Receptor in Inflammation and Immunity. J. Steroid Biochem. Mol. Biol. 2010, 120, 69–75. [Google Scholar] [CrossRef]
- Yang, N.; Ray, D.W.; Matthews, L.C. Current Concepts in Glucocorticoid Resistance. Steroids 2012, 77, 1041–1049. [Google Scholar] [CrossRef]
- Tang, X.; Metzger, D.; Leeman, S.; Amar, S. LPS-Induced TNF-Alpha Factor (LITAF)-Deficient Mice Express Reduced LPS-Induced Cytokine: Evidence for LITAF-Dependent LPS Signaling Pathways. Proc. Natl. Acad. Sci. USA 2006, 103, 13777–13782. [Google Scholar] [CrossRef]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; De Stefanis, C.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-Induced TNF-Alpha Factor Mediates Pro-Inflammatory and Pro-Fibrogenic Pattern in Non-Alcoholic Fatty Liver Disease. Oncotarget 2015, 6, 41434–41452. [Google Scholar] [CrossRef]
- Xu, A.; Lam, M.C.; Chan, K.W.; Wang, Y.; Zhang, J.; Hoo, R.L.; Xu, J.Y.; Chen, B.; Chow, W.S.; Tso, A.W.; et al. Angiopoietin-Like Protein 4 Decreases Blood Glucose and Improves Glucose Tolerance but Induces Hyperlipidemia and Hepatic Steatosis in Mice. Proc. Natl. Acad. Sci. USA 2005, 102, 6086–6091. [Google Scholar] [CrossRef]
- Wheatcroft, S.B.; Kearney, M.T. IGF-Dependent and IGF-Independent Actions of IGF-Binding Protein-1 and -2: Implications for Metabolic Homeostasis. Trends Endocrinol. Metab. 2009, 20, 153–162. [Google Scholar] [CrossRef]
- Carter, S.; Li, Z.; Lemieux, I.; Almeras, N.; Tremblay, A.; Bergeron, J.; Poirier, P.; Deshaies, Y.; Despres, J.P.; Picard, F. Circulating IGFBP-2 Levels are Incrementally Linked to Correlates of the Metabolic Syndrome and Independently Associated with VLDL Triglycerides. Atherosclerosis 2014, 237, 645–651. [Google Scholar] [CrossRef]
- Kammel, A.; Saussenthaler, S.; Jahnert, M.; Jonas, W.; Stirm, L.; Hoeflich, A.; Staiger, H.; Fritsche, A.; Haring, H.U.; Joost, H.G.; et al. Early Hypermethylation of Hepatic Igfbp2 Results in its Reduced Expression Preceding Fatty Liver in Mice. Hum. Mol. Genet. 2016, 25, 2588–2599. [Google Scholar]
- Wang, Y.; Liu, L.M.; Wei, L.; Ye, W.W.; Meng, X.Y.; Chen, F.; Xiao, Q.; Chen, J.Y.; Zhou, Y. Angiopoietin-Like Protein 4 Improves Glucose Tolerance and Insulin Resistance but Induces Liver Steatosis in High-Fat-Diet Mice. Mol. Med. Rep. 2016, 14, 3293–3300. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1alpha: A Key Regulator of Energy Metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Londhe, K.B.; Borlak, J. A Cross-Platform Comparison of Genome-Wide Expression Changes of Laser Microdissected Lung Tissue of C-Raf Transgenic Mice using 3’IVT and Exon Array. PLoS ONE 2012, 7, e40778. [Google Scholar] [CrossRef]
- Reimand, J.; Arak, T.; Vilo, J. G:Profiler--a Web Server for Functional Interpretation of Gene Lists (2011 Update). Nucleic Acids Res. 2011, 39, W307–W315. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein Interaction Networks, Integrated Over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Rzhetsky, A.; Iossifov, I.; Koike, T.; Krauthammer, M.; Kra, P.; Morris, M.; Yu, H.; Duboue, P.A.; Weng, W.; Wilbur, W.J.; et al. GeneWays: A System for Extracting, Analyzing, Visualizing, and Integrating Molecular Pathway Data. J. Biomed. Inform. 2004, 37, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.; Reymann, S.; Matys, V.; Nettesheim, P.; Wingender, E.; Borlak, J. A Novel Computational Approach for the Prediction of Networked Transcription Factors of Aryl Hydrocarbon-Receptor-Regulated Genes. Mol. Pharmacol. 2004, 66, 1557–1572. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.; Konovalova, T.; Waleev, T.; Cheremushkin, E.; Kel-Margoulis, O.; Wingender, E. Composite Module Analyst: A Fitness-Based Tool for Identification of Transcription Factor Binding Site Combinations. Bioinformatics 2006, 22, 1190–1197. [Google Scholar] [CrossRef]
- Stegmaier, P.; Voss, N.; Meier, T.; Kel, A.; Wingender, E.; Borlak, J. Advanced Computational Biology Methods Identify Molecular Switches for Malignancy in an EGF Mouse Model of Liver Cancer. PLoS ONE 2011, 6, e17738. [Google Scholar] [CrossRef]
- Helke, K. Pigs in Toxicology: Differences in Metabolism and Background Findings may be Breed Dependent. In Proceedings of the FDA Cellular, Tissue, and Gene Therapies Advisory Committee, 2022 Meeting Presentation, Online, 29–30 June 2022; pp. 1–95. [Google Scholar]
- Helke, K.L.; Nelson, K.N.; Sargeant, A.M.; Jacob, B.; McKeag, S.; Haruna, J.; Vemireddi, V.; Greeley, M.; Brocksmith, D.; Navratil, N.; et al. Pigs in Toxicology: Breed Differences in Metabolism and Background Findings. Toxicol Pathol. 2016, 44, 575–590. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Description | Fold Change (Average) ± SD | |
|---|---|---|---|
| LD_Liver | HD_Liver | ||
| Immune and inflammatory response | |||
| ATG12 | Autophagy related 12 | 1.48 ± 0.2 | 1.53 ± 0.15 |
| BLNK | B-cell linker | −1.57 ± 0.08 | −1.74 ± 0.48 |
| CXCL2 | C-X-C motif chemokine ligand 2 | 4.33 ± 1.26 | 2.56 ± 1.05 |
| GBP1 | Guanylate binding protein 1 | −1.65 ± 0.65 | −2.55 ± 0.54 |
| HSP90AA1 | Heat shock protein 90 alpha family class A member 1 | 1.58 ± 0.2 | 2.01 ± 0.3 |
| HSPA5 | Heat shock protein family A (Hsp70) member 5 | 1.54 ± 0.27 | 1.81 ± 0.65 |
| IFIT1 | Interferon induced protein with tetratricopeptide repeats 1 | −1.75 ± 0.07 | −2.03 ± 0.56 |
| IL10RB | Interleukin 10 receptor, beta | 1.9 ± 0.06 | 2.6 ± 0.28 |
| IRG6 | Inflammatory response protein 6 | −1.82 ± 0.84 | −3.24 ± 1.23 |
| LYZ | Lysozyme | 2.78 ± 1.93 | 5.89 ± 1.04 |
| MAPK14 | Mitogen-activated protein kinase 14 | 1.51 ± 0.27 | 1.53 ± 0.08 |
| MAPK6 | Mitogen-activated protein kinase 6 | 1.52 ± 0.30 | 2.17 ± 0.5 |
| PRKD1 | Protein kinase D1 | −2.04 ± 1.13 | −2.52 ± 0.42 |
| SPP1 | Secreted phosphoprotein 1 | −1.53 ± 0.13 | −1.58 ± 0.67 |
| THY1 | Thy-1 cell surface antigen | −1.68 ± 0.27 | −1.54 ± 0.38 |
| Response to oxidative stress | |||
| NUDT15 | Nudix hydrolase 15 | 1.53 ± 0.25 | 1.62 ± 0.11 |
| SOD2 | Superoxide dismutase 2, mitochondrial | 2.22 ± 0.9 | 1.91 ± 0.53 |
| Response to stress | |||
| PLLP | Plasmolipin | 1.62 ± 0.48 | 1.7 ± 0.41 |
| PTPLAD1 | Protein tyrosine phosphatase-like A domain containing 1 | −1.78 ± 0.19 | −1.87 ± 0.14 |
| RBM3 | RNA binding motif (RNP1, RRM) protein 3 | −1.51 ± 0.15 | −1.93 ± 0.12 |
| SPR | Sepiapterin reductase | −1.61 ± 0.49 | −1.67 ± 0.38 |
| STC1 | Stanniocalcin 1 | 1.54 ± 0.3 | 1.58 ± 0.35 |
| Cell death and apoptosis | |||
| DNPH1 | 2’-deoxynucleoside 5’-phosphate N-hydrolase 1 | −1.52 ± 0.48 | −1.57 ± 0.15 |
| FYN | FYN Proto-Oncogene, Src Family Tyrosine Kinase | −1.52 ± 0.21 | −1.56 ± 0.24 |
| GAS1 | Growth Arrest Specific 1 | −1.53 ± 0.22 | −1.52 ± 0.32 |
| HSPH1 | Heat shock protein family H (Hsp110) member 1 | 1.74 ± 0.4 | 1.77 ± 0.48 |
| KLF11 | Kruppel like factor 11 | −1.77 ± 0.64 | −1.88 ± 0.38 |
| VIL1 | Villin 1 | −1.89 ± 0.35 | −1.79 ± 0.58 |
| Circadian rhythm | |||
| ARNTL/BMAL1 | Aryl hydrocarbon receptor nuclear translocator-like | 2.07 ± 0.46 | 2.69 ± 0.1 |
| CRY1 | Cryptochrome 1 | 4.13 ± 1.23 | 4.28 ± 0.51 |
| DBP | D-box binding PAR bZIP transcription factor | −1.83 ± 0.35 | −2.41 ± 0.65 |
| NR1D2/ REVERBB | Nuclear receptor subfamily 1, group D, member 2 | −2.79 ± 0.38 | −5.25 ± 1.01 |
| Drug metabolism | |||
| CYP3A39 | Cytochrome P450 3A39 | −6.52 ± 2.58 | −6.78 ± 1.82 |
| CYP3A46 | Cytochrome P450 3A46 | −1.95 ± 1.42 | −1.9 ± 0.34 |
| NR1I3 (CAR) | Nuclear Receptor Subfamily 1 Group I Member 3 | 1.42 ± 0.25 | 1.77 ± 0.1 |
| Cell cycle | |||
| CENPF | Centromere protein F | −1.59 ± 0.2 | −1.51 ± 0.28 |
| FOXA1 | Forkhead box A1 | 1.6 ± 0.55 | 1.79 ± 0.37 |
| TOP2A | Topoisomersae II | −1.62 ± 0.49 | −1.77 ± 0.33 |
| USP2 | Ubiquitin specific peptidase 2 | −2.08 ± 2.26 | −1.91 ± 0.42 |
| Lipid metabolism | |||
| ACSL3 | Acyl-CoA synthetase long-chain family member 3 | −1.52 ± 0.83 | −1.66 ± 0.58 |
| DDHD1 | DDHD domain containing 1 | 1.5 ± 0.38 | 1.73 ± 0.27 |
| ECHDC3 | Enoyl-CoA hydratase domain containing 3 | −1.44 ± 0.31 | −1.58 ± 0.42 |
| ELOVL2 | ELOVL fatty acid elongase 2 | 2.66 ± 0.9 | 3.53 ± 0.59 |
| FASN | Fatty Acid Synthase | −1.61 ± 0.22 | −2.12 ± 0.54 |
| FNDC3B | Fibronectin type III domain containing 3B | 1.42 ± 0.2 | 2.09 ± 0.33 |
| KRT8 | Keratin 8 | 1.9 ± 0.38 | 2.6 ± 0.28 |
| KRT18 | Keratin 18 | 1.74 ± 0.62 | 1.67 ± 0.25 |
| LPPR4 | Lipid phosphate phosphatase-related protein type 4 | −2.04 ± 0.41 | −2.38 ± 0.79 |
| PLIN2 | Perilipin 2 | 3.01 ± 1.39 | 4.7 ± 1.15 |
| PLP1 | Proteolipid protein 1 | −2.4 ± 1.2 | −5.01 ± 1.01 |
| PPARGC1A | Peroxisome proliferator activated receptor gamma, coactivator 1 alpha | −1.63 ± 0.28 | −2.11 ± 0.46 |
| SERPINA6 | Serpin peptidase inhibitor, clade A, member 6 | −1.6 ± 0.57 | −1.54 ± 0.33 |
| SERPINE1 | Serpin family E member 1 | 1.87 ± 0.95 | 1.58 ± 0.04 |
| ST3GAL1 | ST3 beta-galactoside alpha-2,3-sialyltransferase 1 | 1.53 ± 0.27 | 1.68 ± 0.27 |
| VLDLR | Very low density lipoprotein receptor | −1.53 ± 0.21 | 1.98 ± 0.15 |
| Glucose metabolic process | |||
| ENO4 | Enolase family member 4 | −1.9 ± 1.02 | −2.40 ± 0.40 |
| Collagen biosynthesis | |||
| COL5A3 | Collagen type V alpha 3 chain | 1.58 ± 0.11 | 1.74 ± 0.12 |
| Transmembrane transport | |||
| ATP6V1H | ATPase H+ transporting V1 subunit H | −1.51 ± 0.26 | −1.44 ± 0.24 |
| ATP9A | ATPase phospholipid transporting 9A | 2.52 ± 0.24 | 1.6 ± 0.59 |
| ODC | Ornithine decarboxylase | 1.47 ± 0.24 | 2.13 ± 0.54 |
| P2Y12R | Purinergic receptor P2Y12 | −1.71 ± 0.61 | −1.61 ± 0.35 |
| SLC11A2 | Solute carrier family 11, member 2 | 1.68 ± 0.33 | 1.97 ± 0.4 |
| SLC1A1 | Solute carrier family 1 member 1 | 1.53 ± 0.23 | 1.72 ± 0.31 |
| SLC30A10 | Solute carrier family 30, member 10 | 2.08 ± 0.68 | 2.21 ± 0.45 |
| TMEM9 | Transmembrane protein 9 | −1.56 ± 0.12 | −1.45 ± 0.35 |
| Signal transduction | |||
| ARHGAP1 | Rho GTPase activating protein 1 | 1.62 ± 0.12 | 1.7 ± 0.17 |
| CLDN11 | Claudin 11 | −1.72 ± 0.49 | −1.94 ± 0.24 |
| Transcriptional regulation | |||
| RNPC3 | RNA-binding region (RNP1, RRM) containing 3 | 1.57 ± 0.3 | 1.59 ± 0.35 |
| ZNF12 | Zinc finger protein 12 | −1.62 ± 0.34 | −1.87 ± 0.41 |
| ZNF280D | Zinc finger protein 280D | 1.61 ± 0.21 | 1.88 ± 0.4 |
| Gene Symbol | Gene Description | Fold Change (Average) ± SD | |
|---|---|---|---|
| LD_Kidney | HD_Kidney | ||
| Immune and inflammatory response | |||
| C1QA | Complement C1q A chain | 2.33 ± 1.62 | 1.51 ± 0.2 |
| C1QC | Complement C1q C chain | 2.31 ± 1.61 | 1.64 ± 0.25 |
| CCL28 | Chemokine (C-C motif) ligand 28 | 1.52 ± 0.21 | 1.58 ± 0.12 |
| CFB | Complement Factor B | 6.55 ± 2.45 | 2.82 ± 0.34 |
| CFH | Complement Factor H | 2.86 ± 2.25 | 1.91 ± 0.38 |
| CXXC1 | CXXC finger protein 1 | −1.59 ± 0.17 | −1.55 ± 0.06 |
| HSP90B1 | Heat shock protein 90kDa beta, member 1 | −1.6 ± 0.4 | −1.64 ± 0.12 |
| IFITM2 | Interferon induced transmembrane protein 2 | 1.66 ± 0.43 | 1.57 ± 0.15 |
| IL10RB | Interleukin 10 receptor, beta | 1.51 ± 0.28 | 1.93 ± 0.11 |
| IRG6 (RSAD2) | Inflammatory response protein 6 | −1.99 ± 0.17 | −2.75 ± 0.59 |
| KIF3A | Kinesin family member 3A | −1.56 ± 0.1 | −1.63 ± 0.11 |
| KNG1 | Kininogen 1 | −1.96 ± 1 | −2.13 ± 0.14 |
| PTGR1 | Prostaglandin reductase 1 | 3.4 ± 2.61 | 3.13 ± 1.91 |
| SAA2 | Serum amyloid A2 | 2.12 ± 1.37 | 1.54 ± 0.13 |
| USP18 | Ubiquitin specific peptidase 18 | −1.52 ± 0.08 | −1.68 ± 0.47 |
| VCAM1 | Vascular cell adhesion molecule 1 | 1.87 ± 1.1 | 1.57 ± 0.63 |
| Response to stress | |||
| AK3L1 | Adenylate kinase 3-like 1 | −1.58 ± 0.15 | −1.79 ± 0.1 |
| CHD2 | Chromodomain helicase DNA binding protein 2 | −1.53 ± 0.22 | −2.13 ± 0.28 |
| COL3A1 | Collagen, type III, alpha 1 | 1.57 ± 0.88 | 1.51 ± 0.17 |
| GSTA1 | Glutathione S-transferase alpha 1 | 3.23 ± 1.40 | 4.6 ± 1.27 |
| LRP11 | Low density lipoprotein receptor-related protein 11 | 1.55 ± 0.11 | 1.56 ± 0.32 |
| RAD50 | RAD50 homolog | −1.58 ± 0.03 | −1.79 ± 0.33 |
| TCEA1 | Transcription elongation factor A (SII), 1 | −1.61 ± 0.21 | −1.71 ± 0.1 |
| UBXN4 | UBX domain protein 4 | −1.52 ± 0.31 | −1.92 ± 0.24 |
| UPF3B | UPF3 regulator of nonsense transcripts Homolog B | −1.73 ± 0.3 | −1.51 ± 0.15 |
| ZCCHC11 | Zinc finger, CCHC domain containing 11 | −1.56 ± 0.19 | −1.55 ± 0.09 |
| Cell death and apoptosis | |||
| CRYAB | Crystallin, alpha B | 2.02 ± 0.43 | 2.1 ± 0.63 |
| DAB2 | Dab, mitogen-responsive Phosphoprotein, homolog 2 | −1.63 ± 0.08 | −1.62 ± 0.21 |
| DSG2 | Desmoglein 2 | 1.55 ± 0.3 | 2.12 ± 0.62 |
| IGFBP3 | Insulin-like growth factor binding protein 3 | −1.83 ± 0.98 | −2.47 ± 0.7 |
| PDPK1 | 3-phosphoinositide dependent protein kinase 1 | −1.55 ± 0.06 | −1.74 ± 0.06 |
| PPTC7 | PTC7 protein phosphatase homolog | −1.49 ± 0.51 | −2.31 ± 0.77 |
| SCAF11 | SR-related CTD-associated factor 11 | −1.67 ± 0.36 | −2.1 ± 0.49 |
| UCP2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 1.59 ± 0.22 | 1.78 ± 0.36 |
| Cell cycle process | |||
| SEPT7 | Septin 7 | −1.52 ± 0.21 | −1.63 ± 0.21 |
| CDC5L | Cell division cycle 5-like | −1.53 ± 0.12 | −1.77 ± 0.18 |
| PRIM2 | Primase, DNA, polypeptide 2 | −1.64 ± 0.22 | −1.55 ± 0.12 |
| Circadian rhythmic process | |||
| CCND2 | Cyclin D2 | −1.56 ± 0.18 | −1.7 ± 0.06 |
| CRY1 | Cryptochrome circadian clock 1 | 1.62 ± 0.32 | 1.82 ± 0.53 |
| NR1D2/REVERBB | Nuclear receptor subfamily 1, group D, member 2 | −1.99 ± 0.56 | −1.99 ± 0.77 |
| PER2 | Period circadian clock 2 | −1.56 ± 0.32 | −1.85 ± 0.29 |
| Lipid metabolism | |||
| FADS6 | Fatty acid desaturase domain family, member 6 | 1.56 ± 0.31 | 1.87 ± 0.29 |
| KRT8 | keratin 8 | 1.51 ± 0.28 | 1.93 ± 0.11 |
| LDLR | Low density lipoprotein receptor | 1.52 ± 0.2 | 1.51 ± 0.1 |
| SIRT1 | Sirtuin 1 | −1.51 ± 0.21 | −1.52 ± 0.38 |
| Protein metabolism | |||
| ARSJ | Arylsulfatase family, member J | 1.53 ± 0.02 | 1.75 ± 0.29 |
| RBM3 | RNA binding motif (RNP1, RRM) protein 3 | −1.52 ± 0.27 | −2.56 ± 0.55 |
| TET2 | Tet methylcytosine dioxygenase 2 | −1.67 ± 0.04 | −1.83 ± 0.18 |
| TTL | Tubulin tyrosine ligase | 1.84 ± 0.19 | 1.89 ± 0.36 |
| Collagen biosynthesis | |||
| COL16A1 | Collagen Type XVI alpha 1 | 1.55 ± 0.35 | 1.94 ± 0.12 |
| COL21A1 | Collagen, type XXI, alpha 1 | −1.51 ± 0.37 | −2.09 ± 0.41 |
| Signal transduction | |||
| ARHGAP29 | Rho GTPase activating protein 29 | −1.81 ± 0.17 | −1.94 ± 0.28 |
| GPNMB | Glycoprotein (transmembrane) nmb | 2.48 ± 0.39 | 2.47 ± 0.85 |
| S100A6 | S100 Calcium Binding Protein A6 | 2.49 ± 1.83 | 1.74 ± 0.29 |
| Transmembrane and ion-channel transport | |||
| FXYD4 | FXYD Domain Containing Ion Transport Regulator 4 | 1.61 ± 0.34 | 1.62 ± 0.12 |
| GAPVD1 | GTPase activating protein and VPS9 domains 1 | −1.55 ± 0.05 | −1.89 ± 0.3 |
| SLC25A25 | Solute carrier family 25 (mitochondrial; phosphate carrier), member 25 | 1.68 ± 0.27 | 2.19 ± 0.57 |
| SLC4A4 | Solute carrier family 4 (sodium bicarbonate cotransporter), member 4 | −2.34 ± 0.16 | −2.33 ± 0.42 |
| TMEM87A | Transmembrane protein 87A | −1.54 ± 0.1 | −1.53 ± 0.14 |
| Transcriptional regulation | |||
| DDX6 | DEAD box helicase 6 | −1.52 ± 0.21 | −1.76 ± 0.12 |
| GTF2IRD2 | GTF2I repeat domain containing 2 | −1.5 ± 0.41 | −1.77 ± 0.29 |
| HNRNPR | Heterogeneous nuclear ribonucleoprotein R | −1.55 ± 0.41 | −1.82 ± 0.14 |
| METTL12 | Methyltransferase like 12 | 1.51 ± 0.21 | 1.61 ± 0.22 |
| NOL8 | Nucleolar protein 8 | −1.54 ± 0.08 | −1.65 ± 0.14 |
| ZNF12 | Zinc finger protein 12 | −1.81 ± 0.25 | −1.94 ± 0.21 |
| Vascular smooth muscle contraction | |||
| CALD1 | Caldesmon 1 | −1.94 ± 0.4 | −2.55 ± 0.37 |
| GO ID. | Biological Process | Low-Dose | High-Dose | ||||
|---|---|---|---|---|---|---|---|
| No of Genes (% Genes Associated with Terms) | p-Value | Adjusted p-Value | No of Genes (% Genes Associated with Terms) | p-Value | Adjusted p-Value | ||
| GO:0006955 | Immune response | 13 (0.59%) | 0.0298 | 0.00150 | 42 (1.90%) | 9.47 × 10−4 | 0.0153 |
| GO:0006954 | Inflammatory response | 9 (1.17%) | 0.0010 | 0.04570 | 13(1.69%) | 0.0101 | 0.0428 |
| GO:0006950 | Response to stress | 41 (1.06%) | 9.30 × 10−11 | 0.02630 | 91 (2.36%) | 4.58 × 10−4 | 0.0129 |
| GO:0033554 | Cellular response to stress | 17 (0.90%) | 1.16 × 10−7 | 0.00000 | 44 (2.34%) | 0.0015 | 0.0241 |
| GO:0001666 | Response to hypoxia | 9 (2.98%) | 1.51 × 10−4 | 0.00000 | 14 (4.64%) | 0.0094 | 0.0502 |
| GO:0055114 | Oxidation-reduction process | 10 (0.91%) | 0.0115 | 0.00110 | 37 (3.39%) | 5.05 × 10−4 | 0.0136 |
| GO:0006979 | Response to oxidative stress | 4 (1.14%) | 1.12 × 10−4 | 0.02510 | 14 (3.28%) | 6.07 × 10−4 | 0.0144 |
| GO:0071345 | Cellular response to cytokine stimulus | 9 (0.88%) | 0.0012 | 0.00901 | 25 (2.46%) | 7.62 × 10−5 | 0.0033 |
| GO:0019221 | Cytokine-mediated signaling pathway | No enrichment | 18 (2.50%) | 0.0099 | 0.0496 | ||
| GO:0060333 | IFNG-mediated signaling pathway | No enrichment | 7 (7.78%) | 0.0325 | 0.0116 | ||
| GO:0002694 | Regulation of leukocyte activation | 7 (1.25%) | 0.0030 | 0.00500 | 19 (3.40%) | 0.0123 | 0.0497 |
| GO:0050900 | Leukocyte migration | 4 (0.84%) | 0.0015 | 0.00970 | 10 (2.09%) | 0.0339 | 0.1196 |
| GO:0000165 | MAPK cascade | 5 (0.56%) | 0.0013 | 0.00580 | 10 (1.12%) | 0.0466 | 0.1400 |
| GO:0007623 | Circadian rhythm | 6 (3.08%) | 0.0028 | 0.04420 | 12 (6.15%) | 0.0021 | 0.0292 |
| GO:0007049 | Cell cycle | 6 (0.34%) | 4.49 × 10−4 | 0.04190 | 22 (1.25%) | 0.0330 | 0.1173 |
| GO:0008219 | Cell death and apoptosis | 20 (0.92%) | 0.0085 | 0.00250 | 49 (2.25%) | 0.0054 | 0.0440 |
| GO:0097190 | Apoptotic signaling pathway | 7 (1.20%) | 7.45 × 10−7 | 0.02030 | 14 (2.41%) | 0.0365 | 0.0143 |
| GO:0006952 | Defense response | 14 (0.85%) | 2.93 × 10−6 | 0.00001 | 46 (2.79%) | 1.81 × 10−4 | 0.0053 |
| GO:0001525 | Angiogenesis | 8 (1.69%) | 0.0342 | 0.00010 | 14 (2.97%) | 0.0058 | 0.0459 |
| GO:0006805 | Xenobiotic metabolic process | 4 (3.22%) | 0.0011 | 0.01740 | 9 (7.26%) | 0.0157 | 0.0438 |
| GO:0006629 | Lipid metabolic process | No enrichment | 44 (3.12%) | 2.23 × 10−4 | 0.0083 | ||
| GO:0006631 | Fatty acid metabolic process | 6 (1.66%) | 0.0118 | 0.00460 | 14 (3.87%) | 0.0284 | 0.1085 |
| GO:0006006 | Glucose metabolic process | 4 (1.98%) | 0.0083 | 0.02540 | 9 (4.46%) | 2.58 × 10−4 | 0.0069 |
| GO:0034762 | Transmembrane transport regulation | 12 (2.30%) | 0.03964 | 0.04260 | 18 (3.45%) | 0.0105 | 0.0717 |
| hsa0415 | PI3K-AKT signaling pathway | 5 (1.41%) | 0.0141 | 0.03076 | 11 (3.11%) | 0.0056 | 0.0003 |
| hsa03320 | PPAR signaling pathway | No enrichment | 7 (9.46%) | 0.0099 | 0.0129 | ||
| GO ID | Biological Process | Low-Dose | High-Dose | ||||
|---|---|---|---|---|---|---|---|
| No of Genes (% Genes Associated with Terms) | p-Value | Adjusted p-Value | No of Genes (% Genes Associated with Terms) | p-Value | Adjusted p-Value | ||
| GO:0007623 | Circadian rhythm | 7 (3.60%) | 0.0016 | 0.0439 | 13 (6.67%) | 3.97 × 10−5 | 0.0028 |
| GO:0006955 | Immune response | 11 (0.54%) | 0.0227 | 0.0902 | 27 (1.28%) | 0.0021 | 0.0417 |
| GO:0045087 | Innate immune response | No enrichment | 17 (1.86%) | 4.73 × 10−2 | 0.0194 | ||
| GO:0006954 | Inflammatory response | 9 (1.17%) | 0.0022 | 0.0261 | 17 (2.21%) | 6.59 × 10−6 | 8.14 × 10−4 |
| GO:0006950 | Response to stress | 35 (0.91%) | 3.09 × 10−6 | 3.09 × 10−4 | 69 (1.79%) | 5.79 × 10−8 | 2.68 × 10−5 |
| GO:0001666 | Response to hypoxia | 16 (5.30%) | 1.47 × 10−9 | 9.49 × 10−7 | No enrichment | ||
| GO:0055114 | Oxidation-reduction process | 13 (1.19%) | 0.0457 | 0.1269 | 22 (2.01%) | 0.0322 | 0.0462 |
| GO:0000302 | Response to reactive oxygen species | 8 (3.79%) | 2.76 × 10−4 | 0.0065 | 7 (3.32%) | 0.0372 | 0.1728 |
| GO:0006956 | Complement activation | 5 (2.65%) | 9.47 × 10−5 | 0.0032 | 8 (4.23%) | 1.67 × 10−7 | 4.69 × 10−5 |
| GO:0071345 | Cellular response to cytokine stimulus | 8 (0.79%) | 1.10 × 10−4 | 0.0034 | 15 (1.48%) | 0.0250 | 0.0204 |
| GO:0019221 | Cytokine-mediated signaling pathway | No enrichment | 11 (1.53%) | 0.0117 | 0.0998 | ||
| GO:0050900 | Leukocyte migration | No enrichment | 7 (1.46%) | 0.0258 | 0.0446 | ||
| GO:0032496 | Response to lipopolysaccharide | No enrichment | 11 (3.45%) | 0.0273 | 0.0478 | ||
| GO:0008219 | Cell death and apoptosis | 13 (0.60%) | 0.0307 | 0.0437 | 27 (1.24%) | 0.0135 | 0.0148 |
| GO:0009611 | Response to wounding | No enrichment | 17 (2.68%) | 1.06 × 10−4 | 0.0054 | ||
| GO:0006952 | Defense response | 23 (1.39%) | 3.13 × 10−4 | 0.0071 | 36 (2.18%) | 5.73 × 10−6 | 7.59 × 10−4 |
| GO:0006631 | Fatty acid metabolic process | No enrichment | 9 (2.49%) | 0.0298 | 0.0453 | ||
| hsa0415 | PI3K-AKT signaling pathway | 5 (1.41%) | 0.0096 | 3.08 × 10−6 | 5 (1.41%) | 0.0366 | 0.0389 |
| Gene Symbol | Gene Description | Fold Change ± SD | |||
|---|---|---|---|---|---|
| LD_Liver | HD_Liver | LD_Kidney | HD_Kidney | ||
| Circadian rhythm | |||||
| ARNTL/BMAL1 | Aryl hydrocarbon receptor nuclear translocator like | 2.07 ± 0.46 * | 2.69 ± 0.1 * | 1.48 ± 0.35 | 1.9 ± 0.24 * |
| BHLHE41 | Basic helix-loop-helix family member e41 | −1.52 ± 0.29 * | −2.41 ± 0.32 * | −1.1 ± 0.2 | −1.51 ± 0.09 |
| CRY1 | Cryptochrome 1 | 4.13 ± 1.23 * | 4.28 ± 0.51 * | 1.62 ± 0.32 * | 1.82 ± 0.53 * |
| DBP | D-box binding PAR bZIP transcription factor | −1.83 ± 0.35 * | −2.41 ± 0.65 * | −1.38 ± 0.33 | −1.55 ± 0.08 * |
| NPAS2 | Neuronal PAS domain protein 2 | 1.3 ± 0.2 | 2.46 ± 0.51 * | 1.15 ± 0.21 | 1.47 ± 0.18 |
| NR1D2/REVERBbeta | Nuclear receptor subfamily 1 group D member 2 | −2.79 ± 0.77 * | −5.25 ± 1.01 * | −1.99 ± 0.56 * | −1.99 ± 0.77 * |
| PER2 | Period circadian clock 2 | −1.38 ± 0.69 | −2.6 ± 1.36 * | −1.56 ± 0.32 * | −1.85 ± 0.29 * |
| RORC | RAR related orphan receptor C | 1.76 ± 0.26 * | 1.41 ± 0.08 | −1.02 ± 0.11 | 1.01 ± 0.05 |
| Immune and inflammatory response | |||||
| BIRC3 | Baculoviral IAP repeat containing 3 | 1.52 ± 0.26 * | 1.64 ± 0.23 * | 1.03 ± 0.11 | 1.16 ± 0.09 |
| CD27 | CD27 molecule | −1.41 ± 0.15 | −1.53 ± 0.15 * | 1.23 ± 0.09 | 1.04 ± 0.03 |
| CSF2RB | Colony stimulating factor 2 receptor beta | 1.51 ± 0.92 * | 1.51 ± 0.11 * | 1.25 ± 0.53 | 1.07 ± 0.17 |
| DDX58 | DExD/H-box helicase 58 | −1.06 ± 0.17 | −1.57 ± 0.07 * | −1.13 ± 0.19 | −1.27 ± 0.16 |
| IL5 | Interleukin 5 | −1.1 ± 0.25 | −1.52 ± 0.15 * | −1.04 ± 0.05 | −1.04 ± 0.07 |
| LYZ | Lysozyme | 2.78 ± 1.93 * | 5.89 ± 1.04 * | 5.31 ± 5.17 | 3.27 ± 0.93 * |
| MAPKAPK3 | Mitogen-activated protein kinase-activated protein kinase 3 | 1.16 ± 0.23 | 1.55 ± 0.11 * | 1.08 ± 0.16 | 1.08 ± 0.07 |
| PDE4B | Phosphodiesterase 4B | 1.32 ± 0.41 | 1.59 ± 0.07 * | −1.1 ± 0.08 | 1.31 ± 0.22 |
| Drug metabolic process | |||||
| CYP1A1 | Cytochrome P450 family 1 subfamily A member 1 | 4.86 ± 3.71 | 2.66 ± 2.83 | 17.05 ± 25.15 | 1.75 ± 0.72 * |
| CYP2C19 | Cytochrome P450 family 2 subfamily C member 19 | −1.59 ± 0.2 * | −1.51 ± 0.28 * | −1.14 ± 0.15 | 1.04 ± 0.03 |
| CYP2C42 (CYP2C9) | Cytochrome P450 family 2 subfamily C member 9 | −1.67 ± 1.11 | −2.05 ± 0.27 * | 1.01 ± 0.01 | 1.54 ± 1.01 |
| CYP3A29 (CYP3A4) | Cytochrome P450 family 3 subfamily A member 4 | −1.24 ± 0.12 | −1.57 ± 0.28 * | −1.27 ± 0.14 | −1.62 ± 0.67 |
| Cellular response to stress | |||||
| ANGPTL4 | Angiopoietin like 4 | −1.58 ± 0.85 * | −1.54 ± 0.29 * | −1.32 ± 0.13 | −1.01 ± 0.04 |
| CALU | Calumenin | 1.18 ± 0.19 | 1.55 ± 0.25 * | 1.01 ± 0.15 | 1.02 ± 0.09 |
| HSP90AA1 | Heat shock protein 90 alpha family class A member 1 | 1.58 ± 0.2 * | 2.01 ± 0.3 * | −1.25 ± 0.17 | −1.15 ± 0.24 |
| MAOB | Monoamine oxidase B | −1.32 ± 0.3 | −1.86 ± 0.22 * | −1.04 ± 0.1 | 1.05 ± 0.07 |
| MAT1A | Methionine adenosyltransferase 1A | −1.06 ± 0.24 | −1.53 ± 0.22 * | −1.14 ± 0.19 | −1.04 ± 0.02 |
| Oxidation-reduction process | |||||
| ALOX5AP | Arachidonate 5-lipoxygenase activating protein | 1.33 ± 0.36 | 1.87 ± 0.31 * | 1.8 ± 1.24 | 1.41 ± 0.26 |
| APOA2 | Apolipoprotein A2 | −1.31 ± 0.13 | −1.57 ± 0.07 * | −1.03 ± 0.1 | −1.07 ± 0.05 |
| CREG1 | Cellular repressor of E1A stimulated genes 1 | −1.11 ± 0.1 | −1.57 ± 0.17 * | 1.07 ± 0.15 | 1.05 ± 0.04 |
| DAO | D-amino acid oxidase | 1 ± 0.99 | −2 ± 0.93 * | −1.09 ± 0.19 | −1.36 ± 0.06 |
| GRHPR | Glyoxylate and hydroxypyruvate reductase | −1.26 ± 0.46 | −1.61 ± 0.18 * | −1.13 ± 0.05 | −1.1 ± 0.08 |
| PAM | Peptidylglycine alpha-amidating monooxygenase | −1.18 ± 0.19 | −1.5 ± 0.29 * | 1.19 ± 0.16 | −1.07 ± 0.19 |
| Interferon-gamma-mediated signaling pathway | |||||
| B2M | Beta-2-microglobulin | −1.21 ± 0.25 | −1.53 ± 0.34 * | −1.03 ± 0.07 | −1.03 ± 0.03 |
| MT2A | Metallothionein 2A | 1.26 ± 0.38 | 1.65 ± 0.37 * | 1.85 ± 0.79 * | 1.85 ± 1.55 |
| SP100 | SP100 nuclear antigen | 1.08 ± 0.15 | 1.74 ± 0.57 * | −1.09 ± 0.12 | 1.31 ± 0.18 |
| Cell cycle and arrest | |||||
| CDKN1C | Cyclin dependent kinase inhibitor 1C | 1.8 ± 0.69 * | 2.74 ± 1.11 * | 1.43 ± 0.35 | 1.2 ± 0.08 |
| GADD45G | growth arrest and DNA damage inducible gamma | 2.09 ± 1.65 * | 1.75 ± 0.29 * | 1.29 ± 0.03 | 1.42 ± 0.71 |
| GAS1 | Growth arrest specific 1 | 1.09 ± 0.22 | −1.51 ± 0.32 * | −1.24 ± 0.08 | −1.35 ± 0.23 |
| TOP2A | Topoisomerase (DNA) II alpha | −1.62 ± 0.49 * | −1.77 ± 0.33 * | 1.79 ± 1.14 | −1.02 ± 0.09 |
| Response to glucocorticoid stimulus and metabolism | |||||
| LMO3 | LIM domain only 3 | −1.1 ± 0.09 | −1.6 ± 0.38 * | −1.18 ± 0.1 | −1.22 ± 0.18 |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 | 1.57 ± 0.08 * | 1.22 ± 0.06 | −1.15 ± 0.12 | −1.21 ± 0.28 |
| SERPINA6 | Serpin family A member 6 | −1.6 ± 0.57 * | −1.54 ± 0.33 * | −1.15 ± 0.05 | −1.13 ± 0.11 |
| STC1 | Stanniocalcin 1 | 1.54 ± 0.3 * | 1.58 ± 0.35 * | −1.15 ± 0.16 | 1.02 ± 0.02 |
| TAT | Tyrosine aminotransferase | 1.11 ± 0.35 | −2.15 ± 0.45 * | −1.13 ± 0.05 | −1.13 ± 0.19 |
| Lipid metabolism | |||||
| AFP | Alpha fetoprotein | −1.57 ± 0.36 * | −1.33 ± 0.13 | 1.11 ± 0.29 | 2.12 ± 1.01 * |
| LPIN1 | Lipin 1 | −1.09 ± 0.12 | −1.55 ± 0.35 * | −1.19 ± 0.06 | −1.08 ± 0.07 |
| Aminoacid synthesis | |||||
| PHGDH | Phosphoglycerate dehydrogenase | −1.42 ± 0.56 | −2.49 ± 1.05 * | −1.03 ± 0.05 | −1.31 ± 0.19 |
| Carbohydrate metabolic process | |||||
| XYLB | Xylulokinase | −1.25 ± 0.38 | −1.52 ± 0.35 * | −1.52 ± 0.27 * | −1.60 ± 0.27 * |
| Transmembrane transport | |||||
| ABCA8 | ATP binding cassette subfamily A member 8 | 1.05 ± 0.22 | −1.55 ± 1.03 * | −1.44 ± 0.34 | −1.47 ± 0.31 |
| ATP1A2 | ATPase Na+/K+ transporting subunit alpha 2 | −1.34 ± 0.54 | −1.69 ± 0.26 * | 1.22 ± 0.19 | 1.35 ± 0.18 |
| ATP1B1 | ATPase Na+/K+ transporting subunit beta 1 | 1.21 ± 0.35 | 1.51 ± 0.37 * | 1.02 ± 0.02 | 1.02 ± 0.03 |
| ATP9A | ATPase phospholipid transporting 9A | 2.35 ± 0.35 * | 1.6 ± 0.22 * | 1.3 ± 0.11 | 1.3 ± 0.29 |
| Extracellular matrix organization | |||||
| ANXA2 | Annexin A2 | −1.42 ± 0.66 | −1.78 ± 0.27 * | 1.46 ± 0.62 | 1.09 ± 0.11 |
| FMOD | Fibromodulin | −1.2 ± 0.23 | −1.52 ± 0.39 * | −1.07 ± 0.07 | −1.18 ± 0.12 |
| ITGA8 | Integrin subunit alpha 8 | 1.24 ± 0.27 | 1.51 ± 0.03 * | −1 ± 0.12 | 1.24 ± 0.39 |
| SPP1 | Secreted phosphoprotein 1 | −1.53 ± 0.13 * | −1.17 ± 0.67 | 3 ± 3.56 | 1.38 ± 0.28 |
| Signal transduction | |||||
| NR1I3 (CAR) | Nuclear receptor subfamily 1 group I member 3 | 1.51 ± 0.34 * | 1.77 ± 0.1 * | 1.07 ± 0.09 | −1.01 ± 0.08 |
| PLEK | Pleckstrin | 1.21 ± 0.69 | 1.51 ± 0.12 * | 1.62 ± 0.66 * | 1.56 ± 0.17 * |
| REEP5 | Receptor accessory protein 5 | −1 ± 0.29 | 1.79 ± 0.42 * | 1.04 ± 0.14 | 1.02 ± 0.1 |
| Protein complex assembly and cell adhesion | |||||
| CLDN14 | Claudin 14 | −1.34 ± 0.57 | −1.69 ± 0.32 * | 1.13 ± 0.07 | 1.44 ± 0.82 |
| CNTN4 | Contactin 4 | 1.09 ± 0.35 | −1.95 ± 0.52 * | 1.05 ± 0.01 | 1.37 ± 0.25 |
| MOGS | Mannosyl-oligosaccharide glucosidase | 1.25 ± 0.28 | 1.68 ± 0.28 * | 1.25 ± 0.12 | 1.25 ± 0.26 |
| SERPINI1 | Serpin family I member 1 | −1.52 ± 0.18 * | −1.01 ± 0.09 | −1.22 ± 0.08 | −1.02 ± 0.04 |
| Transcriptional regulation | |||||
| RPL32 | Ribosomal protein L32 | 1.32 ± 0.22 | 1.56 ± 0.35 * | 1.03 ± 0.11 | 1 ± 0.01 |
| YIPF1 | Yip1 domain family member 1 | −1.08 ± 0.22 | −1.51 ± 0.17 * | −1.03 ± 0.04 | −1.27 ± 0.03 |
| Probeset ID | Gene Symbol | Description | Liver | Kidney | ||
|---|---|---|---|---|---|---|
| LD | HD | LD | HD | |||
| Fold Change (Average) ± SD | ||||||
| Lipogenesis | ||||||
| Ssc.4891.1.A1_at | AGPAT9 (GPAT3) | Glycerol-3-phosphate acyltransferase 3 | 1.14 ± 0.39 | 2.99 ± 1.11 * | 1.16 ± 0.29 | 1.08 ± 0.02 |
| Ssc.12241.1.A1_at | ANXA2 | Annexin A2 | −1.42 ± 0.66 | −1.78 ± 0.27 * | 1.46 ± 0.62 | 1.09 ± 0.11 |
| Ssc.3703.1.S1_at | APOA2 | Apolipoprotein A2 | −1.31 ± 0.13 | −1.57 ± 0.07 * | −1.03 ± 0.1 | −1.07 ± 0.05 |
| Ssc.14503.1.S1_at | APOA4 | Apolipoprotein A4 | 1.16 ± 4.9 | 1.56 ± 0.29 * | 1.01 ± 0.08 | 1.07 ± 0.16 |
| Ssc.1039.1.S1_at | APOC3 | Apolipoprotein C3 | −1.01 ± 0.01 | −1.03 ± 0.04 | −3.97 ± 1.42 * | 1.01 ± 0.11 |
| Ssc.5848.1.S1_at | B4GALT5 | Beta-1,4-galactosyltransferase 5 | 1.98 ± 1.28 | 1.51 ± 0.35 * | 1.58 ± 0.76 * | 1.15 ± 0.14 |
| Ssc.2224.1.S1_at | CYP2C19 | Cytochrome P450 family 2 subfamily C member 19 | −1.59 ± 0.2 * | −1.51 ± 0.28 * | 1.11 ± 0.15 | −1.04 ± 0.11 |
| Ssc.19298.2.S1_at | DHCR24 | 24-dehydrocholesterol reductase | −1.01 ± 0.39 | 1.01 ± 0.12 | 1.36 ± 0.2 | 1.56 ± 0.03 * |
| Ssc.30467.1.A1_at | ELOVL2 | ELOVL fatty acid elongase 2 | 2.66 ± 2.9 | 3.53 ± 0.59 * | 1.04 ± 0.03 | −1.18 ± 0.08 |
| Ssc.1595.1.S1_a_at | FADS1 | Fatty acid desaturase 1 | −1.64 ± 3.48 | −1.91 ± 0.79 * | 1 ± 0.1 | 1.55 ± 0.06 * |
| Ssc.18175.1.A1_at | FASN | Fatty acid synthase | −1.61 ± 0.22 * | −2.12 ± 0.54 * | 1.03 ± 0.04 | 1.14 ± 0.11 |
| Ssc.4417.1.A1_at | GALNT2 | Polypeptide N-acetylgalactosaminyltransferase 2 | 1.55 ± 0.77 * | 1.45 ± 0.1 | 1.01 ± 0.02 | −1.14 ± 0.05 |
| Ssc.11103.1.S1_at | MDH1 | Malate dehydrogenase 1 | 1.04 ± 0.06 | 1.54 ± 0.07 * | 1.01 ± 0.03 | 1.09 ± 0.09 |
| Ssc.3345.2.S1_at | MVK | Mevalonate kinase | −1.53 ± 1.07 | −2.36 ± 0.93 * | 1.05 ± 0.07 | −1.08 ± 0.07 |
| Ssc.9781.1.S1_at | SERPINE1 | Serpin family E member 1 | 1.87 ± 0.95 * | 1.58 ± 0.04 * | 3.32 ± 3.93 | 1.41 ± 0.53 |
| Ssc.4552.1.S1_at | VLDLR | Very low density lipoprotein receptor | −1.53 ± 0.21 * | 1.98 ± 0.15 * | 1.08 ± 0.06 | 1.08 ± 0.02 |
| Fatty acid oxidation/mitochondrial stress | ||||||
| Ssc.2176.1.A1_at | ACSL3 | Acyl-CoA synthetase long-chain family member 3 | −1.52 ± 0.83 * | −1.66 ± 0.58 * | 1.03 ± 0.08 | 1.07 ± 0.06 |
| Ssc.18933.1.A1_at | ADH4 | Alcohol dehydrogenase 4 | −1.72 ± 0.86 * | −2.93 ± 1.27 * | 1.01 ± 0.08 | −1.06 ± 0.11 |
| Ssc.17183.1.S1_at | ATP5D | ATP synthase subunit delta, mitochondrial-like | 1.28 ± 0.06 | 1.56 ± 0.36 * | 1.08 ± 0.13 | 1.1 ± 0.04 |
| Ssc.26021.1.S1_at | FOXA1 | Forkhead box A1 | 1.6 ± 0.55 * | 1.79 ± 0.37 * | 1.12 ± 0.17 | 1.01 ± 0.05 |
| Lipid transport | ||||||
| Ssc.13622.1.S1_at | AQP4 | Aquaporin 4 | −1.56 ± 0.24 * | −1.14 ± 0.15 | 1 ± 0.02 | 1.09 ± 0.17 |
| Ssc.604.1.S1_at | FABP1 | Fatty acid binding protein 1, liver | −1.12 ± 0.56 | 1.01 ± 0.11 | 1.59 ± 0.18 * | 1.32 ± 0.29 |
| Ssc.6605.1.S1_at | HDLBP | High density lipoprotein binding protein | 1.35 ± 0.18 | 1.64 ± 0.28 * | −1.18 ± 0.18 | 1.03 ± 0.08 |
| Ssc.21926.1.S1_at | LDLR | Low density lipoprotein receptor | −1.11 ± 0.51 | −1.13 ± 0.13 | 1.52 ± 0.2 * | 1.59 ± 0.1 * |
| Ssc.29043.1.S1_at | NR5A2 | Nuclear receptor subfamily 5, group A, member 2 | −1.3 ± 0.64 | −1.57 ± 0.46 * | 1.1 ± 0.1 | −1.31 ± 0.3 |
| LD growth/ER stress | ||||||
| Ssc.29181.1.A1_s_at | ATP2A2 (SERCA2) | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 | 1.44 ± 0.31 | 2.04 ± 1.08 * | 1.01 ± 0.04 | 1.11 ± 0.14 |
| Ssc.3106.1.S1_at | CALR | Calreticulin | 1.35 ± 0.25 | 2.42 ± 1.21 * | 1.32 ± 0.14 | 1.79 ± 0.37 * |
| Ssc.12842.1.S1_at | CAV1 | Caveolin 1 | −2.05 ± 0.39 * | −1.74 ± 0.38 * | 1.2 ± 0.27 | −1.2 ± 0.08 |
| Ssc.250.1.S1_at | CBR1 | Carbonyl reductase 1 | −1.97 ± 0.69 * | −1.6 ± 1.08 | −1.49 ± 0.64 | −1.09 ± 0.08 |
| Ssc.6784.1.S1_at | LIPE | Lipase E | 1.04 ± 0.09 | −1.23 ± 0.31 | 1.14 ± 0.13 | 1.57 ± 0.29 * |
| Ssc.6323.1.S1_at | PLIN2 | Perilipin 2 | 3.01 ± 1.39 * | 4.7 ± 1.15 * | 1.06 ± 0.12 | 1.32 ± 0.28 |
| Ssc.17391.2.S1_at | RAB35 | RAB35, member RAS oncogene family | −1.2 ± 0.38 | −1.57 ± 0.38 * | 1.08 ± 0.07 | −1.26 ± 0.27 |
| Ssc.29036.1.S1_at | TUBA4A | Tubulin, alpha 4a | 1.59 ± 0.61 * | 1.62 ± 0.43 * | 1.2 ± 0.14 | 1.26 ± 0.03 |
| Oxidative stress/signalling events | ||||||
| Ssc.22673.1.S1_at | AKR1C1 | Aldo-keto reductase family 1, member C1 | −1.16 ± 0.03 | −1.81 ± 0.31 * | −1.3 ± 0.45 | −1.7 ± 0.31 * |
| Ssc.8987.1.A1_at | ARHGAP1 | Rho GTPase activating protein 1 | 1.62 ± 0.12 * | 1.7 ± 0.17 * | 1.27 ± 0.18 | 1.18 ± 0.19 |
| Ssc.27539.2.A1_a_at | ASPH | Aspartate beta-hydroxylase | 1.13 ± 0.15 | 1.57 ± 0.14 * | 1.08 ± 0.08 | 1.11 ± 0.06 |
| Ssc.22002.2.A1_at | CXCL13 | Chemokine lignad 13 | 1.11 ± 0.14 | 1.54 ± 0.16 * | −1.07 ± 0.14 | 1.26 ± 0.22 |
| Ssc.20578.1.S1_at | CXCL14 | Chemokine ligand 14 | 1.01 ± 0.05 | 1.24 ± 0.16 | 1.61 ± 0.68 * | 1.58 ± 0.4 * |
| Ssc.19692.1.S1_at | CXCL2 | Chemokine ligand 2 | 4.33 ± 1.26 * | 2.56 ± 1.05 * | 4.05 ± 5.08 | 1.76 ± 0.56 * |
| Ssc.390.1.A1_at | HIF1A | Hypoxia inducible factor 1 alpha subunit | −1.67 ± 0.04 * | −1.08 ± 0.18 | 1.16 ± 0.09 | 1.11 ± 0.11 |
| Ssc.12191.1.A1_at | HSP90AA1 | Heat shock protein 90kDa | 1.58 ± 0.2 * | 1.32 ± 0.3 | −1.25 ± 0.17 | −1.15 ± 0.24 |
| Ssc.308.1.S1_at | HSP90B1 | Heat shock protein 90 beta family member 1 | 1.26 ± 0.31 | 1.93 ± 0.8 * | −1.6 ± 0.4 * | −1.64 ± 0.12 * |
| Ssc.9056.1.A1_at | HSPA5 | Heat shock protein family A (Hsp70) member 5 | 1.66 ± 0.3 * | 1.81 ± 0.1 * | 1.16 ± 0.21 | 1.2 ± 0.04 |
| Ssc.23054.1.S1_at | JAK3 | Janus kinase 3 | 1.27 ± 0.41 | 1.52 ± 0.19 * | 1.42 ± 0.49 | 1.34 ± 0.13 |
| Ssc.18557.1.S1_at | KNG1 | Kininogen 1 | −1.2 ± 0.04 | −1.61 ± 0.15 * | −1.96 ± 1 * | −2.13 ± 0.14 * |
| Ssc.7297.1.S1_at | MAOB | Monoamine oxidase B | −1.32 ± 0.3 | −1.86 ± 0.22 * | −1.04 ± 0.1 | 1.05 ± 0.07 |
| Ssc.7478.1.A1_at | MARCH5 | Membrane associated ring-CH-type finger 5 | −1.18 ± 0.07 | −1.5 ± 0.29 * | −1.11 ± 0.06 | −1.25 ± 0.25 |
| Ssc.10955.1.S1_at | MGST3 | Microsomal glutathione S-transferase 3 | −1.21 ± 0.25 | −1.7 ± 0.36 * | 1.21 ± 0.15 | 1.11 ± 0.09 |
| Ssc.9170.1.A1_at | PRKD1 | Protein kinase D1 | −2.04 ± 1.13 * | −2.52 ± 0.42 * | 1.06 ± 0.1 | 1.14 ± 0.05 |
| Ssc.830.1.S1_at | PSME2 | Proteasome activator subunit 2 | −1.14 ± 0.15 | −1.51 ± 0.26 * | 1.14 ± 0.27 | −1.09 ± 0.07 |
| Ssc.14490.1.S1_at | PTGR1 | Prostaglandin reductase 1 | −1.27 ± 0.68 | −1.18 ± 0.22 | 3.4 ± 2.61 * | 3.13 ± 1.91 * |
| Ssc.3706.1.S2_at | SOD2 | Superoxide dismutase 2, mitochondrial | 2.22 ± 0.9 * | 1.91 ± 0.53 * | 1.09 ± 0.14 | 1.07 ± 0.05 |
| Ssc.11252.1.S1_at | SPR | Sepiapterin reductase | −1.61 ± 0.49 * | −1.67 ± 0.38 * | −1.22 ± 0.05 | −1.24 ± 0.02 |
| Ssc.14066.2.S1_at | TAT | Tyrosine aminotransferase | 1.11 ± 0.35 | −2.15 ± 0.45 * | −1.13 ± 0.05 | −1.13 ± 0.19 |
| Ssc.3753.1.S1_at | TFRC (CD71) | Transferrin receptor | 1.64 ± 0.69 * | 1.34 ± 0.35 | 1.68 ± 0.31 * | 1.44 ± 0.36 |
| Ssc.28413.1.A1_at | UBXN4 | UBX domain-protein 4 | 1.06 ± 0.04 | 1.12 ± 0.15 | −1.52 ± 0.31 * | −1.92 ± 0.24 * |
| Ssc.16350.1.S1_at | UCP2 | Uncoupling protein 2 | 1.25 ± 0.04 | 1.17 ± 0.12 | 1.59 ± 0.22 * | 1.78 ± 0.36 * |
| Ssc.3185.1.S1_at | TXNDC5 | Thioredoxin domain-containing protein 5 | 1.07 ± 0.16 | 1.53 ± 0.02 * | 1.12 ± 0.18 | 1.28 ± 0.01 |
| Lipid metabolism marker genes | ||||||
| Ssc.16377.1.A1_s_at | GSTA1 | Glutathione S-transferase alpha 1 | −1.08 ± 0.1 | −1.61 ± 1.07 | 3.23 ± 1.40 * | 4.60 ± 1.27 * |
| Ssc.5991.1.A1_at | KRT18 | Keratin 18 | 1.74 ± 0.62 * | 1.67 ± 0.25 * | 4.01 ± 5.19 | −1.06 ± 0.05 |
| Ssc.5955.1.A1_at | KRT8 | keratin 8 | 1.9 ± 0.38 * | 2.6 ± 0.28 * | 1.51 ± 0.28 * | 1.93 ± 0.11 * |
| Glucose metabolism | ||||||
| Ssc.8980.1.A1_at | ANGPTL4 | Angiopoietin-like 4 | −1.58 ± 1.15 | −1.54 ± 0.29 * | −1.32 ± 0.13 | −1.01 ± 0.04 |
| Ssc.27727.1.S1_at | PGM5 | Phosphoglucomutase 5 | −1.39 ± 0.33 | −1.79 ± 0.11 * | −1.59 ± 0.28 * | −1.48 ± 0.48 |
| Ssc.21431.3.A1_s_at | PHGDH | Phosphoglycerate dehydrogenase | −1.42 ± 0.56 | −2.49 ± 1.05 * | −1.03 ± 0.05 | −1.31 ± 0.19 |
| Ssc.4717.1.S1_at | PHKB | Phosphorylase b kinase regulatory subunit beta | −1.32 ± 0.34 | −1.58 ± 0.16 * | −1.04 ± 0.1 | −1.17 ± 0.02 |
| Gene Symbol | Gene Description | Fold Change (Average) ± SD | |||
|---|---|---|---|---|---|
| LD_Liver | HD_Liver | LD_Kidney | HD_Kidney | ||
| Immune and inflammatory response | |||||
| C9 | Complement component 9 | 2.73 ± 0.77 * | 1 ± 0.05 | −1.81 ± 0.78 * | −1.23 ± 0.32 |
| CD163 | CD163 antigen | 1.49 ± 0.75 | 2.04 ± 0.42 * | 2.01 ± 1.67 * | 1.36 ± 0.45 |
| CXCL2 | Chemokine (C-X-C motif) ligand 2 | 4.33 ± 0.37 * | 2.56 ± 1.05 * | 4.05 ± 5.08 | 1.76 ± 0.56 * |
| GJA1 | Gap junction protein, alpha 1 | 1.63 ± 0.16 * | 1.32 ± 0.25 | 1.86 ± 1.5 * | −1.59 ± 0.19 * |
| IFI44L | Interferon-induced protein 44-like | −1.18 ± 0.31 | −2.03 ± 0.41 * | −1.18 ± 0.08 | −1.51 ± 0.31 * |
| IL10RB | Interleukin 10 receptor, beta | 1.9 ± 0.06 * | 2.6 ± 0.28 * | 1.51 ± 0.28 * | 1.93 ± 0.11 * |
| IRG6 (RSAD2) | Inflammatory response protein 6 | −1.82 ± 0.84 * | −3.24 ± 1.23 * | −1.99 ± 0.17 * | −2.75 ± 0.59 * |
| ISG15 | ISG15 ubiquitin-like modifier | −1.2 ± 0.15 | 1.86 ± 0.11 * | −1.32 ± 0.12 | −1.91 ± 0.24 * |
| KNG1 | Kininogen 1 | −1.2 ± 0.22 | −1.61 ± 0.15 * | −1.96 ± 1 * | −2.13 ± 0.14 * |
| LYZ | Lysozyme | 2.78 ± 1.93 * | 5.89 ± 1.04 | 5.31 ± 5.17 | 3.27 ± 0.93 * |
| S100A9 | S100 calcium binding protein A9 | 7.03 ± 5.21 | 4.13 ± 2.55 * | 6.17 ± 8.48 | 2.58 ± 0.65 * |
| TFRC | Transferrin receptor (CD71) | 1.64 ± 0.69 * | 1.34 ± 0.35 | 1.68 ± 0.31 * | 1.44 ± 0.36 |
| TNFAIP6 | Tumor necrosis factor, alpha-induced protein 6 | −1.58 ± 0.27 * | −1 ± 0.13 | −2.09 ± 0.39 * | −1.18 ± 0.28 |
| USP18 | Ubiquitin specific peptidase 18 | −1.39 ± 0.53 | −2.25 ± 0.11 * | −1.52 ± 0.08 * | −1.68 ± 0.47 * |
| VCAM1 | Vascular cell adhesion molecule 1 | −1.21 ± 0.07 | 1.85 ± 1.1 * | 1.87 ± 1.1 * | 1.57 ± 0.63 * |
| Drug metabolism | |||||
| CYB5D2 | Cytochrome B5 domain containing 2 | −1.07 ± 0.45 | 1.8 ± 0.29 * | 1.15 ± 0.37 | 1.56 ± 0.24 * |
| CYP3A29 | Cytochrome P450 3A29 | −1.24 ± 0.25 | −1.57 ± 0.28 * | −1.27 ± 0.14 | −1.62 ± 0.67 * |
| Response to stress | |||||
| IGFBP2 | Insulin-like growth factor binding protein 2 | −1.37 ± 0.29 | −1.85 ± 0.37 * | 1.52 ± 0.28 * | 1.26 ± 0.09 |
| RBM3 | RNA binding motif (RNP1, RRM) protein 3 | −1.51 ± 0.15 | −1.93 ± 0.12 * | −1.52 ± 0.27 * | −2.56 ± 0.55 * |
| Oxidation-reduction process | |||||
| ACAD10 | Acyl-CoA dehydrogenase family, member 10 | 1.23 ± 0.46 | 1.93 ± 0.35 * | 1.19 ± 0.44 | 2.14 ± 0.1 * |
| AKR1C1 | Aldo-keto reductase family 1, member C1 | −1.81 ± 0.03 * | −1.81 ± 0.31 * | −1.3 ± 0.45 | −1.7 ± 0.31 * |
| PIR | Pirin-like | −1.15 ± 0.77 | −1.53 ± 0.34 * | 2.1 ± 0.83 * | 1.25 ± 0.08 |
| Response to hypoxia | |||||
| ANGPT1 | Angiopoietin 1 | −1.15 ± 0.19 | −1.51 ± 0.39 * | −1.56 ± 0.37 * | −1.14 ± 0.22 |
| Cell death and apoptosis | |||||
| CALR | Calreticulin | 1.35 ± 0.25 | 2.42 ± 1.21 * | 1.32 ± 0.14 | 1.79 ± 0.37 * |
| EVA1A | Protein FAM176A-like | −1.54 ± 3.19 | −2.06 ± 0.14 * | −1.44 ± 0.79 | −1.82 ± 0.52 * |
| HRG | Histidine-rich glycoprotein | −1.52 ± 0.38 * | −2.26 ± 0.53 * | −1.16 ± 0.28 | −1.75 ± 0.34 * |
| HSP90B1 | Heat shock protein 90kDa beta, member 1 | 1.34 ± 0.31 | 1.93 ± 0.80 * | −1.59 ± 0.40 * | −1.64 ± 0.12 * |
| Circadian rhythmic process | |||||
| ARNTL/BMAL1 | Aryl hydrocarbon receptor nuclear translocator-like | 2.07 ± 0.46 * | 2.69 ± 0.1 * | 1.48 ± 0.35 | 1.9 ± 0.24 * |
| BHLHE41 | Basic helix-loop-helix family, member e41 | −1.52 ± 1.23 * | −2.41 ± 0.32 * | −1.1 ± 0.2 | −1.51 ± 0.09 * |
| CRY1 | Cryptochrome 1 | 4.13 ± 1.23 * | 4.28 ± 0.51 * | 1.62 ± 0.32 * | 1.82 ± 0.53 * |
| NPAS2 | Neuronal PAS domain protein 2 | 1.3 ± 0.1 | 2.46 ± 0.51 * | 1.15 ± 0.21 | 1.57 ± 0.18 * |
| NR1D2/REVERBB | Nuclear receptor subfamily 1, group D, member 2 | −2.79 ± 0.38 * | −5.25 ± 1.01 * | −1.99 ± 0.56 * | −1.99 ± 0.77 * |
| PER2 | Period homolog 2 | −1.38 ± 0.2 | −2.6 ± 1.36 * | −1.56 ± 0.32 * | −1.85 ± 0.29 * |
| Lipid metabolism | |||||
| ARSD | Arylsulfatase D | −1.05 ± 0.23 | −1.73 ± 0.11 * | −1.12 ± 0.18 | −1.73 ± 0.15 * |
| KRT8 | keratin 8 | 1.9 ± 0.38 * | 2.6 ± 0.28 * | 1.51 ± 0.28 * | 1.93 ± 0.11 * |
| PLEK | Pleckstrin | 1.21 ± 0.69 | 1.5 ± 0.12 * | 1.62 ± 0.66 * | 1.56 ± 0.17 * |
| Protein metabolism | |||||
| ANKRD28 | Ankyrin repeat domain 28 | 1.09 ± 0.14 | 1.51 ± 0.12 * | −1.13 ± 0.23 | −1.52 ± 0.07 * |
| ASRGL1 | L-asparaginase-like | −1.19 ± 0.29 | −1.53 ± 0.07 * | −1.38 ± 0.08 | −1.63 ± 0.14 * |
| GTF2H3 | General transcription factor IIH, polypeptide 3 | 1.07 ± 0.19 | 1.82 ± 0.05 * | −1.07 ± 0.07 | 1.59 ± 0.15 * |
| IGFBP3 | Insulin-like growth factor binding protein 3 | −1.41 ± 0.04 | −1.65 ± 0.13 * | −1.83 ± 0.98 | −2.47 ± 0.7 * |
| Carbohydrate metabolism | |||||
| XYLB | Xylulose kinase-like | −1.25 ± 0.3 | −1.52 ± 0.35 * | −1.52 ± 0.27 * | −1.60 ± 0.27 * |
| Signal transduction | |||||
| GPNMB | Glycoprotein (transmembrane) nmb | −1.1 ± 0.19 | −2.01 ± 0.35 * | 2.48 ± 0.39 * | 2.47 ± 0.85 * |
| RND3 | Rho family GTPase 3 | 1.32 ± 0.15 | 1.80 ± 0.02 * | 1.27 ± 0.15 | 1.55 ± 0.24 * |
| Master Regulatory Genes | No of Genes | Score | FDR | Z-Score | Fold Change (Average) ± SD | |
|---|---|---|---|---|---|---|
| Total No. of Genes in the Network | Statistically Significant DEGs | |||||
| DCF_low dose_liver | ||||||
| Cryptochrome 1 (CRY1) | 51 | 27 | 0.27785 | 0.02 | 2.8496 | 4.13 ± 1.23 |
| Hypoxia inducible factor 1, alpha subunit (HIF1A) | 76 | 39 | 0.47052 | 0.011 | 1.90028 | −1.67 ± 0.04 |
| Nuclear receptor subfamily 3, group C, member 1 (NR3C1) | 90 | 42 | 0.58276 | 0.047 | 1.48431 | 1.57 ± 0.08 |
| DCF_high dose_liver | ||||||
| Cryptochrome 1 (CRY1) | 113 | 66 | 0.23593 | 0.024 | 3.41268 | 4.28 ± 0.51 |
| Insulin-like growth factor binding protein 2 (IGFBP2) | 196 | 111 | 0.36927 | 0.016 | 3.01892 | −1.85 ± 0.37 |
| Lipopolysaccharide-induced TNF factor (LITAF) | 199 | 111 | 0.40046 | 0.027 | 2.20201 | 1.81 ± 0.22 |
| Angiopoietin-like 4 (ANGPTL4) | 130 | 77 | 0.22103 | 0.028 | 2.10328 | −1.54 ± 0.29 |
| DCF_low dose_kidney | ||||||
| Dipeptidyl-peptidase 4 (DPP4) | 83 | 40 | 0.50686 | 0.005 | 2.69108 | −1.52 ± 0.51 |
| Angiopoietin 1 (ANGPT1) | 43 | 25 | 0.25006 | 0.011 | 2.86388 | −1.56 ± 0.37 |
| Sirtuin 1 (SIRT1) | 77 | 38 | 0.57072 | 0.021 | 1.51382 | −1.51 ± 0.21 |
| DCF_high dose_kidney | ||||||
| Matrix metallopeptidase 7 (MMP7) | 103 | 52 | 0.44009 | 0.007 | 3.18595 | 2.99 ± 0.75 |
| Complement component 1, q subcomponent, alpha polypeptide (C1QA) | 102 | 49 | 0.47405 | 0.002 | 3.33235 | 1.51 ± 0.2 |
| Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, zeta (NFKBIZ) | 114 | 56 | 0.37981 | 0.02 | 2.35022 | 1.59 ± 0.2 |
| Insulin-like growth factor binding protein 3 (IGFBP3) | 113 | 56 | 0.48865 | 0.029 | 1.92918 | −2.47 ± 0.7 |
| Antibody | Vendor | Cat no. | Lot Number | Dilution | Antigen Retrieval |
|---|---|---|---|---|---|
| Cortisol Binding Globulin (CBG) | Abcam (Cambridge, UK) | ab107368 | GR74750-20 | 1:100 | pH 6 |
| CPS1 / Hepar-1 | Santa Cruz Biotechnology, (Heidelberg, Germany) | sc-58693 | C1213 | 1:7500 | pH 6 |
| CRF1 | Abcam | ab150561 | GR139717-15 | 1:250 | pH 9 |
| CYP1A1 | Abcam | ab3568 | 1:200 | pH 6 | |
| Cryptochrome I (CRY1) | Abcam | ab3518 | GR307241-6 | 1:300 | pH 6 |
| ELOVL2 | Abcam | ab111162 | GR48113-1 | 1:100 | pH 6 |
| Glucocorticoid Receptor (GR) | Abcam | ab233165 | GR3222243-1 | 1:100 | pH 9 |
| HIF1a | Abcam | ab463 | GR252860 | 1:40 | pH 6 |
| KAT-13D (CLOCK) | Abcam | ab65033 | GR88646-1 | 1:50 | pH 6 |
| MPO | Dako (Hamburg, Germany) | REFA0398 | 20001076 | 1:500 | pH 6 |
| Per2 | Abcam | ab200388 | GR221389-8 | 1:300 | pH 6 |
| SOD-1 | Santa Cruz | sc-11407 | B0415 | 1:500 | pH 6 |
| SOD-2 | Santa Cruz | sc-30080 | J0713 | 1:250 | pH 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selvaraj, S.; Oh, J.-H.; Yoon, S.; Borlak, J. Diclofenac Disrupts the Circadian Clock and through Complex Cross-Talks Aggravates Immune-Mediated Liver Injury—A Repeated Dose Study in Minipigs for 28 Days. Int. J. Mol. Sci. 2023, 24, 1445. https://doi.org/10.3390/ijms24021445
Selvaraj S, Oh J-H, Yoon S, Borlak J. Diclofenac Disrupts the Circadian Clock and through Complex Cross-Talks Aggravates Immune-Mediated Liver Injury—A Repeated Dose Study in Minipigs for 28 Days. International Journal of Molecular Sciences. 2023; 24(2):1445. https://doi.org/10.3390/ijms24021445
Chicago/Turabian StyleSelvaraj, Saravanakumar, Jung-Hwa Oh, Seokjoo Yoon, and Jürgen Borlak. 2023. "Diclofenac Disrupts the Circadian Clock and through Complex Cross-Talks Aggravates Immune-Mediated Liver Injury—A Repeated Dose Study in Minipigs for 28 Days" International Journal of Molecular Sciences 24, no. 2: 1445. https://doi.org/10.3390/ijms24021445
APA StyleSelvaraj, S., Oh, J.-H., Yoon, S., & Borlak, J. (2023). Diclofenac Disrupts the Circadian Clock and through Complex Cross-Talks Aggravates Immune-Mediated Liver Injury—A Repeated Dose Study in Minipigs for 28 Days. International Journal of Molecular Sciences, 24(2), 1445. https://doi.org/10.3390/ijms24021445