
Calmodulin and Amyloid Beta as Coregulators of Critical Events during the Onset and Progression of Alzheimer’s Disease


Abstract
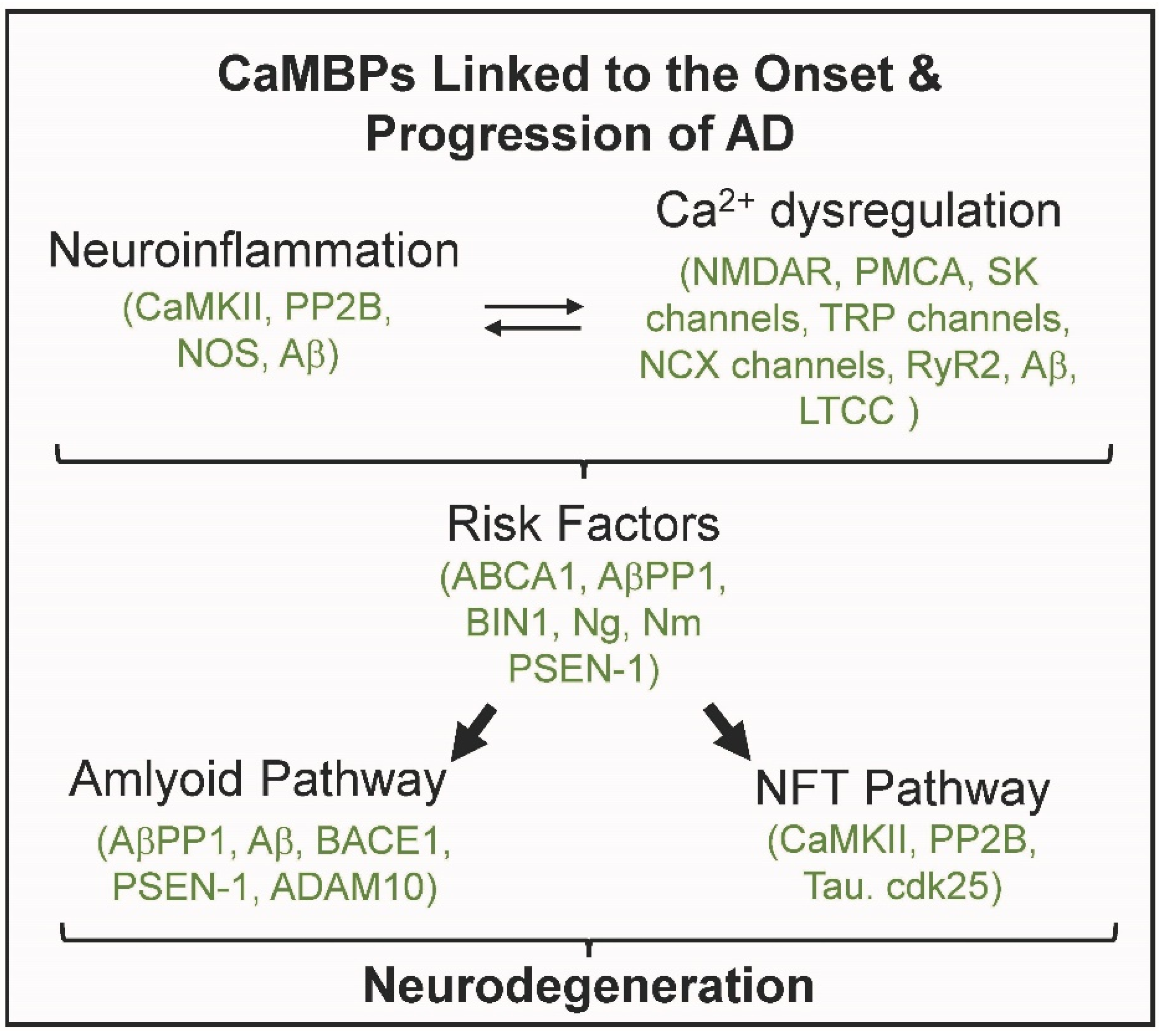
1. Calmodulin Binding Proteins and Alzheimer’s Disease
2. Aβ/CaMBP Receptors Involved in Alzheimer’s Disease
3. Aβ, CaM and Calcium Channels
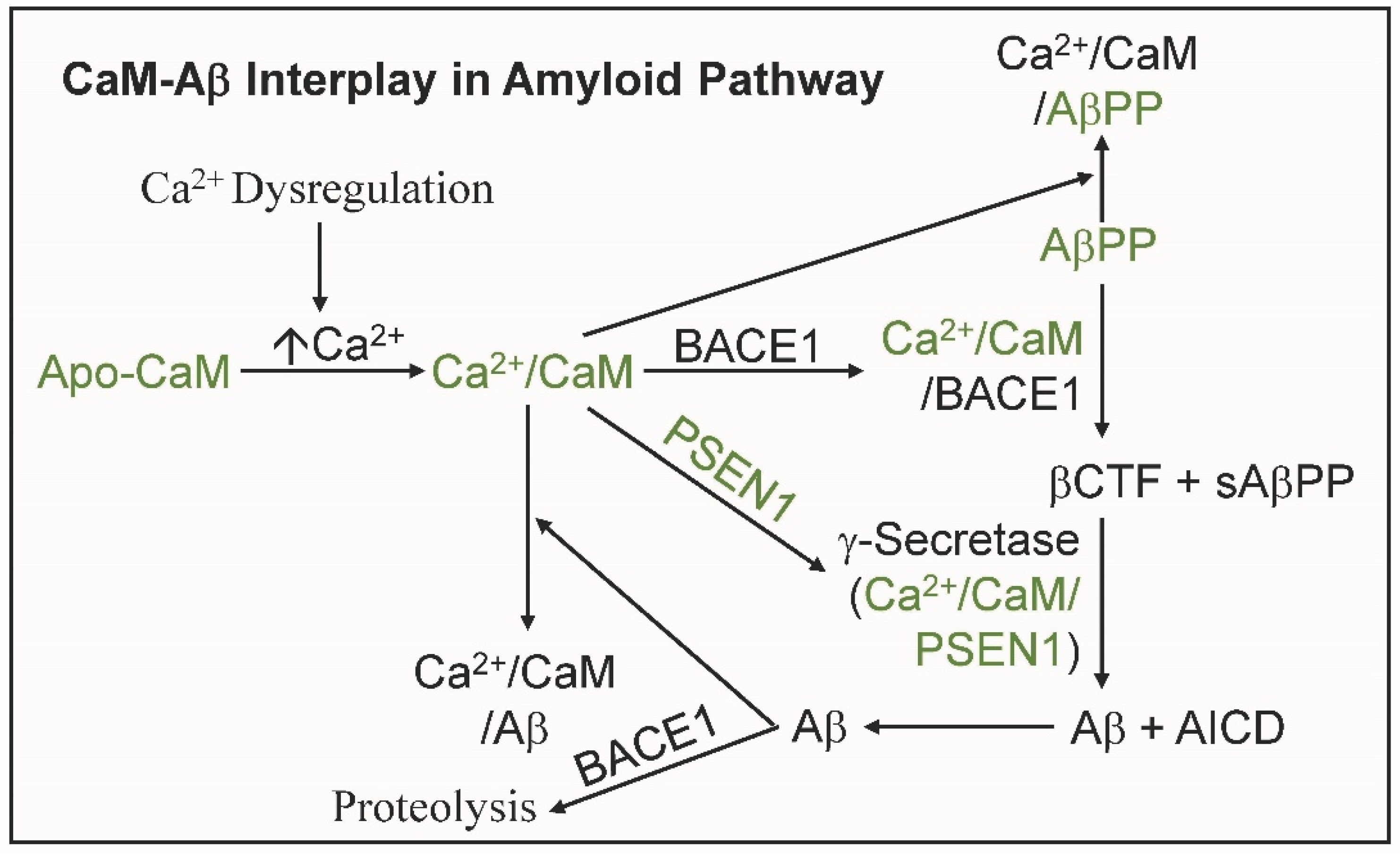
4. The Complex Interplay between CaM and Aβ in the Amyloid Pathway
5. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta |
| Aβo | amyloid beta oligomers |
| AβPP | amyloid-β precursor protein |
| AchR | acetylcholine receptor |
| AD | Alzheimer’s disease |
| APOE | apolipoprotein E |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| BACE1 | beta-secretase 1 |
| BIN1 | bridging Integrator 1 |
| CaM | calmodulin |
| CaMBD | calmodulin binding domain |
| CaMBP | calmodulin binding protein |
| CaMKII | calcium/CaM-dependent kinase II |
| PP2B | calcineurin |
| CLU | clusterin |
| CRAC | calcium release-activated calcium channels |
| CR1 | complement receptor type 1 |
| LTP | long-term potentiation |
| LTD | long-term depression |
| MARCKs | myristoylated alanine-rich, C-kinase substrate |
| mGluR5 | metabotropic glutamate receptor 5 |
| NFTs | neurofibrillary tangles |
| Ng | neurogranin |
| NMDAR | N-methyl-D-aspartate receptor |
| PMCA | plasma membrane calcium ATPase |
| pTau | phosphorylated Tau |
| RyRs | ryanodine receptors |
| TREM2 | triggering receptor expressed on myeloid cells 2 |
References
- Karen, E.; DeStrooper, B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nature 2022, 21, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Towards theories of brain aging. In Handbook of Studies on Psychiatry and Old Age; Kay, D.S., Burrows, G.W., Eds.; Elsevier Science Publishers, B.V.: Amsterdam, The Netherlands, 1984; pp. 7–30. [Google Scholar]
- Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann. N. Y. Acad. Sci. 1994, 747, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’adamio, L.; Grassi, C.; Devanand, D.; Honig, L.S.; et al. Role of amyloid-β and tau proteins in Alzheimer’s disease: Confuting the amyloid cascade. J. Alzheimer’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Solà, C.; Barrón, S.; Tusell, J.M.; Serratosa, J. The Ca2+/calmodulin system in neuronal hyperexcitability. Int. J. Bochem. Cell Biol. 2001, 33, 439–455. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Myre, M.A. Calmodulin-binding domains in Alzheimer’s disease proteins: Extending the calcium hypothesis. Biochem. Biophys. Res. Commun. 2004, 230, 1051–1054. [Google Scholar] [CrossRef]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease: A Review. J. Alzheimer’s Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef]
- O’Day, D.H. Calmodulin binding proteins and Alzheimer’s disease: Biomarkers, regulatory enzymes and receptors that are regulated by calmodulin. Int. J. Mol. Sci. 2020, 21, 7344. [Google Scholar] [CrossRef]
- Corbacho, I.; Berrocal, M.; Török, K.; Mata, A.M.; Gutierrez-Merino, C. High affinity binding of amyloid β-peptide to calmodulin: Structural and functional implications. Biochem. Biophys. Res. Commun. 2017, 486, 992–997. [Google Scholar] [CrossRef]
- Canobbio, I.; Catricalà, S.; Balduini, C.; Torti, M. Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim. Biophys. Acta Bioenergy 2011, 1813, 500–506. [Google Scholar] [CrossRef]
- Fogel, H.; Frere, S.; Segev, O.; Bharill, S.; Shapira, I.; Gazit, N.; O’Malley, T.; Slomowitz, E.; Berdichevsky, Y.; Walsh, D.M.; et al. APP homodimers transduce an amyloid-β mediated increase in release probability at excitatory synapses. Cell Rep. 2014, 17, 1560–1576. [Google Scholar] [CrossRef]
- Minakami, R.; Jinnai, N.; Sugiyama, H. Phosphorylation and calmodulin binding of the metatropic glutamate receptor subtype 5 (mGluR5) are antagonistic in vitro. J. Biol. Chem. 1997, 272, 20291–20298. [Google Scholar] [CrossRef]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef]
- Ehlers, M.D.; Zhang, S.; Bernhardt, J.P.; Huganir, R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 1996, 84, 745–755. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug Memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Shull, G.E.; Greeb, J. The plasma membrane calcium/calmodulin-dependent calcium ATPase (PMCA). J. Biol. Chem. 1988, 263, 8646–8657. [Google Scholar] [CrossRef]
- Berrocal, M.; Corbacho, I.; Sepulveda, M.R.; Gutierrez-Merino, C.; Mata, A.M. Phospholipids and calmodulin modulate the inhibition of PMCA activity by tau. Biochim. Biophys. Acta. 2017, 1864, 1028–1035. [Google Scholar] [CrossRef]
- Michno, K.; Knight, D.; Campussano, J.M.; van de Hoef, D.; Boulianne, G.L. Intracellular calcium deficits in Drosophila cholinergic neurons expressing wild type or FAD-mutant presenilin. PLoS ONE 2009, 4, e6904. [Google Scholar] [CrossRef]
- Ohki, Y.; Shimada, N.; Tominaga, A.; Osawa, S.; Higo, T.; Yokoshima, S.; Fukuyama, T.; Tomita, T.; Iwatsubo, T. Binding of longer Aβ to transmembrane domain 1 of presenilin 1 impacts on Aβ42 generation. Mol. Neurodegen. 2014, 9, 7. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Ribe, E.; Al-Shawi, R.; Malik, B.; Hooper, C.; Fernandes, C.; Dobson, R.; Nolan, P.; Lourdusamy, A.; Furney, S.; et al. Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry 2014, 19, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-b blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. beta-Amyloid (1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Wang, D.; Govindaiah, G.; Liu, R.; De Arcangelis, V.; Cox, C.L.; Xiang, Y.K. Binding of amyloid β peptide to β2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J. 2010, 24, 3511–3521. [Google Scholar] [CrossRef]
- Myre, M.A.; Tesco, G.; Tanzi, R.E.; Wasco, W. Calmodulin binding to APP and the APLPs. In Molecular Mechanisms of Neurodegeneration, Proceedings of the Joint Biochemical Society/Neuroscience Ireland Focused Meeting, Dublin, Ireland, 13–16 March 2005; University College Dublin: Dublin, Ireland, 2005. [Google Scholar]
- Chavez, S.E.; O’Day, D.H. Calmodulin binds to and regulates the activity of beta-secretase (BACE1). Curr. Res. Alzheimer’s Dis. 2007, 1, 37–47. [Google Scholar]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Ghosh, A.; Geise, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef]
- Zhao, D.; Watson, J.B.; Xie, C.W. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J. Neurophysiol. 2004, 92, 2853–2858. [Google Scholar] [CrossRef]
- Padilla, R.; Maccioni, R.; Avila, J. Calmodulin binds to a tubulin binding site of the microtubule-associated protein tau. Mol. Cell. Biochem. 1990, 97, 35–41. [Google Scholar] [CrossRef]
- Huber, R.J.; Catalano, A.; O’Day, D.H. Cyclin dependent kinase 5 is a calmodulin-binding protein that associates with puromycin-sensitive amino peptidase in the nucleus of Dictyostelium. Biochim. Biophys. Acta 2013, 1833, 11–20. [Google Scholar] [CrossRef]
- Wang, H.; Muiznieks, L.D.; Ghosh, P.; Williams, D.; Solarski, M.; Fang, A.; Ruiz-Riquelme, A.; Pomès, R.; Watts, J.C.; Chakrabartty, A.; et al. Somatostatin binds to the human amyloid β peptide and favors the formation of distinct oligomers. eLife 2017, 6, e28401. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta. Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. The relevance of amyloid β-calmodulin complexation in neurons and brain degeneration in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 4976. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef]
- Liu, S.; Park, S.; Allington, G. Targeting apolipoprotein E/amyloid β binding by peptoid CPO_Aβ17-21P ameliorates Alzheimer’s disease related pathology and cognitive decline. Sci. Rep. 2017, 7, 8009. [Google Scholar] [CrossRef]
- Babaei, P. NMDA and AMPA receptors dysregulation in Alzheimer’s disease. Eur. J. Pharmacol. 2021, 908, 174310. [Google Scholar] [CrossRef]
- Iacobucci, G.J.; Popescu, G.K. Resident calmodulin primes NMDA receptors for Ca2+-dependent inactivation. Biophys. J. 2017, 113, 2236–2248. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza, M.; Fernandez-Villalobos, G.; Stein, I.S.; Kasumova, G.; Zhang, P.; Bayer, K.U.; Otmakhov, N.; Hell, J.W.; Lisman, J. Role of the CaMKII/NMDA receptor complex I the maintenance of synaptic strength. J. Neurosci. 2011, 31, 9170–9178. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, W.; Yan, Z. β-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin dependent protein kinase II synaptic distribution. J. Biol. Chem. 2009, 284, 10639–10649. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.W.; Klyubin, I.; Anwyl, R.; Rowan, M.J. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20504–20509. [Google Scholar] [CrossRef] [PubMed]
- Malinow, R. New developments on the role of NMDA receptors in Alzheimer’s disease. Curr. Opin. Neurobiol. 2012, 22, 559–563. [Google Scholar] [CrossRef]
- Giliberto, L.; Borghi, R.; Piccini, A.; Mangerini, R.; Sorbi, S.; Cirmena, G.; Garuti, A.; Ghetti, B.; Tagliavini, F.; Mughal, M.R.; et al. Mutant Presenilin 1 Increases the Expression and Activity of BACE1. J. Biol. Chem. 2009, 284, 9027–9038. [Google Scholar] [CrossRef]
- Dear, A.J.; Michaels, T.C.T.; Meisl, G.; Klenerman, D.; Wu, S.; Perrett, S.; Linse, S.; Dobson, C.M.; Knowles, T.P.J. Kinetic diversity of amyloid oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 12087–12094. [Google Scholar] [CrossRef]
- Lee, E.B.; Zhang, B.; Liu, K.; Greenbaum, E.A.; Doms, R.W.; Trojanowski, J.Q.; Lee, V.M.-Y. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J. Cell Biol. 2005, 168, 291–302. [Google Scholar] [CrossRef]
- Maloney, B.; Lahiri, D.K. The Alzheimer’s amyloid β-peptide (Aβ) binds a specific DNA Aβ-interacting domain (AβID) in the APP, BACE1 and APOE promoters in a sequence specific manner: Characterizing a new regulatory motif. Gene 2011, 488, 1–12. [Google Scholar] [CrossRef]
- Sturchio, A.; Dwivedi, A.K.; Malm, T.; Wood, M.J.; Cilia, R.; Sharma, J.S.; Hill, E.J.; Schneider, L.S.; Graff-Radford, N.R.; Mori, H.; et al. High Soluble Amyloid-β42 Predicts Normal Cognition in Amyloid-Positive Individuals with Alzheimer’s Disease-Causing Mutations. J. Alzheimer’s Dis. 2022, 90, 343–348. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| A. DIRECT REGULATION | |||
|---|---|---|---|
| 1. Validated CaMBPs | |||
| Aβ Receptor | Example Function | CaMBP Reference | Aβ Receptor Reference |
| Aβ | oligos/fibrils/plaques | [11] | Not applicable |
| AβPP1 | source of Aβ | [12] | [13] |
| mGluR | Ca2+ homeostasis | [14] | [15] |
| NMDAR | Ca2+ homeostasis | [16] | [17] |
| PMCA | Ca2+ homeostasis | [18] | [19] |
| PSEN-1 | γ-secretase subunit | [20] | [21] |
| 2. Presumptive CaMBPs | |||
| Aβ Receptor | Example Function | CaMBP Reference | Aβ Receptor Reference |
| APOE 2-4 | risk factor | [9] | [22] |
| CLU/ApoJ | risk factor | [9] | [23] |
| PICALM | risk factor | [9] | [24] |
| TREM2 | risk factor | [10] | [25] |
| B. INDIRECT REGULATION | |||
| Aβ Receptor | Example Function | CaMBP Reference | Aβ Receptor Reference |
| α7nAChR | Ca2+ homeostasis | Regulated by CaMKII | [26] |
| AMPAR | Ca2+ homeostasis | Regulated by PP2B | [27] |
| β2AR | adrenergic function | Regulated by CaMKII | [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Day, D.H. Calmodulin and Amyloid Beta as Coregulators of Critical Events during the Onset and Progression of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 1393. https://doi.org/10.3390/ijms24021393
O’Day DH. Calmodulin and Amyloid Beta as Coregulators of Critical Events during the Onset and Progression of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(2):1393. https://doi.org/10.3390/ijms24021393
Chicago/Turabian StyleO’Day, Danton H. 2023. "Calmodulin and Amyloid Beta as Coregulators of Critical Events during the Onset and Progression of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 2: 1393. https://doi.org/10.3390/ijms24021393
APA StyleO’Day, D. H. (2023). Calmodulin and Amyloid Beta as Coregulators of Critical Events during the Onset and Progression of Alzheimer’s Disease. International Journal of Molecular Sciences, 24(2), 1393. https://doi.org/10.3390/ijms24021393