

Proof-of-Concept Method to Study Uncharacterized Methyltransferases Using PRDM15



Abstract
1. Introduction
2. Results
2.1. PRDM15 Dependency, Expression, and Associations
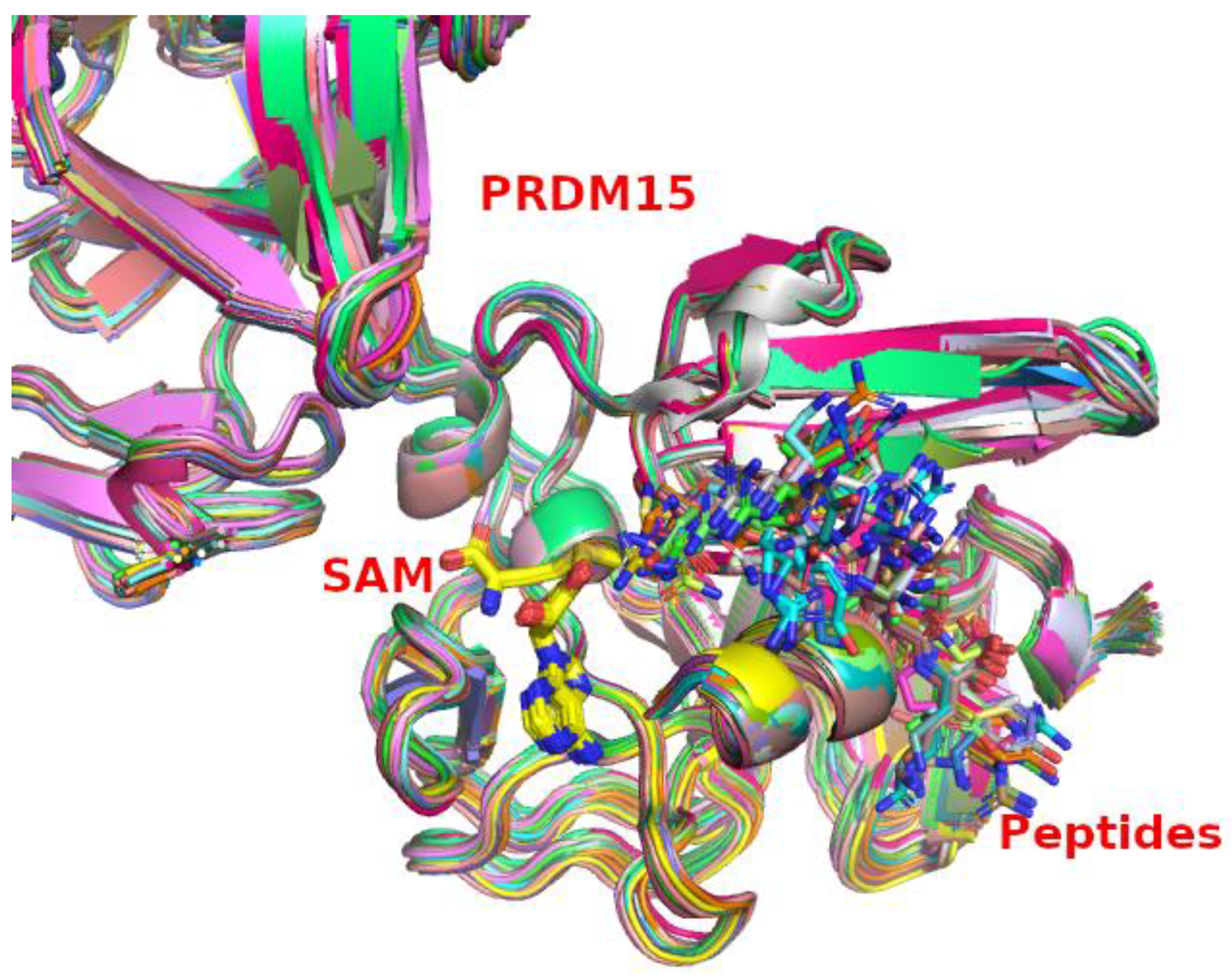
2.2. PRDM15 Native Substrates beyond Histone-Based Peptides
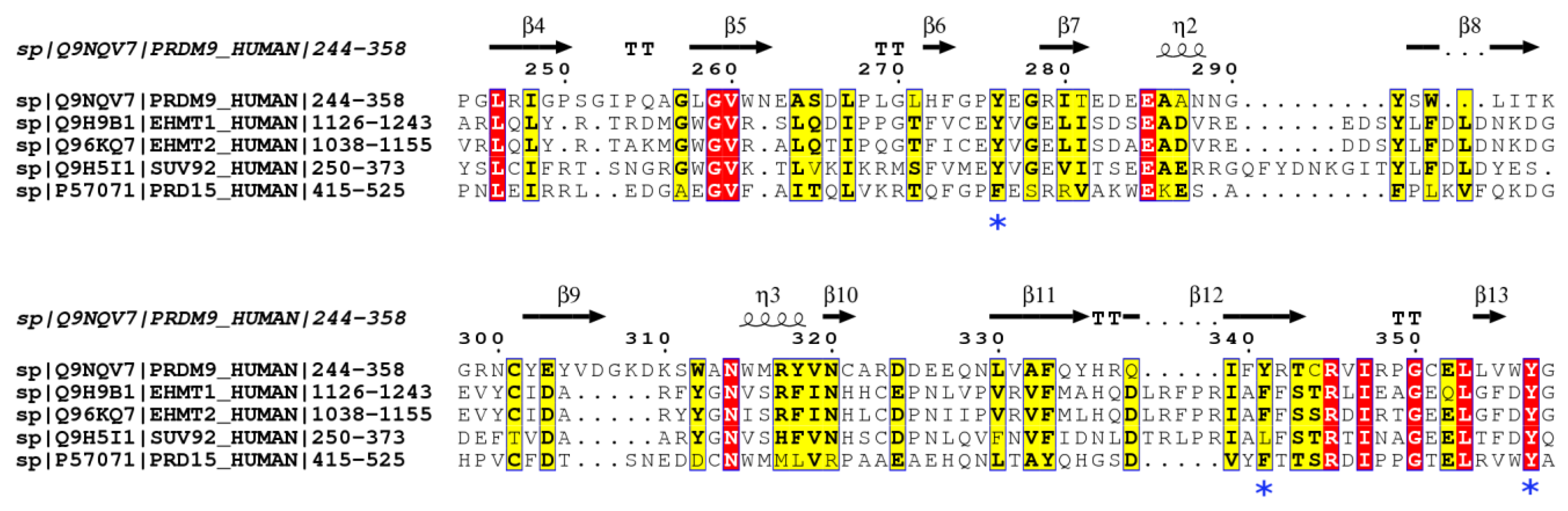
2.3. PRDM15 Potential Methylation Profile
3. Discussion
3.1. PRDM15 as a Potential Drug Target
3.2. Outlook
4. Materials and Methods
4.1. Homology Modeling of PRDM15
4.2. Theoretical Approach for the Search of Endogenous Substrate(s) of PRDM15
4.3. Binding Energy Calculations
4.4. Statistics Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Latchman, D.S. Transcription factors: An overview. Int. J. Exp. Pathol. 1993, 74, 417–422. [Google Scholar] [CrossRef]
- Karin, M. Too many transcription factors: Positive and negative interactions. New Biol. 1990, 2, 126–131. [Google Scholar] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Q.; Shyr, Y. Dysregulated transcription across diverse cancer types reveals the importance of RNA-binding protein in carcinogenesis. BMC Genom. 2015, 16, S5. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Libermann, T.A.; Zerbini, L.F. Targeting transcription factors for cancer gene therapy. Curr. Gene Ther. 2006, 6, 17–33. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Posternak, V.; Cole, M.D. Strategically targeting MYC in cancer. F1000Research 2016, 5, 408. [Google Scholar] [CrossRef]
- Mzoughi, S.; Tan, Y.X.; Low, D.; Guccione, E. The role of PRDMs in cancer: One family, two sides. Curr. Opin. Genet. Dev. 2016, 36, 83–91. [Google Scholar] [CrossRef]
- Tullio, F.D.; Schwarz, M.; Zorgati, H.; Mzoughi, S.; Guccione, E. The duality of PRDM proteins: Epigenetic and structural perspectives. FEBS J. 2022, 289, 1256–1275. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Li, H. Cancer drug discovery targeting histone methyltransferases: An update. Curr. Med. Chem. 2015, 22, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Jung, M. New lysine methyltransferase drug targets in cancer. Nat. Biotechnol. 2012, 30, 622–623. [Google Scholar] [CrossRef]
- Copeland, R.A. Protein methyltransferase inhibitors as precision cancer therapeutics: A decade of discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170080. [Google Scholar] [CrossRef] [PubMed]
- Morera, L.; Lübbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016, 8, 57. [Google Scholar] [CrossRef]
- Shen, L.; Chen, Q.; Yang, C.; Wu, Y.; Yuan, H.; Chen, S.; Ou, S.; Jiang, Y.; Huang, T.; Ke, L.; et al. Role of PRDM1 in Tumor Immunity and Drug Response: A Pan-Cancer Analysis. Front. Pharmacol. 2020, 11, 593195. [Google Scholar] [CrossRef]
- Fu, S.-H.; Yeh, L.-T.; Chu, C.-C.; Yen, B.L.-J.; Sytwu, H.-K. New insights into Blimp-1 in T lymphocytes: A divergent regulator of cell destiny and effector function. J. Biomed. Sci. 2017, 24, 49. [Google Scholar] [CrossRef]
- Shu, X.-S.; Geng, H.; Li, L.; Ying, J.; Ma, C.; Wang, Y.; Poon, F.F.; Wang, X.; Ying, Y.; Yeo, W.; et al. The Epigenetic Modifier PRDM5 Functions as a Tumor Suppressor through Modulating WNT/β-Catenin Signaling and Is Frequently Silenced in Multiple Tumors. PLoS ONE 2011, 6, e27346. [Google Scholar] [CrossRef]
- Seki, Y. PRDM14 Is a Unique Epigenetic Regulator Stabilizing Transcriptional Networks for Pluripotency. Front. Cell Dev. Biol. 2018, 6, 12. [Google Scholar] [CrossRef]
- Mzoughi, S.; Fong, J.Y.; Papadopoli, D.; Koh, C.M.; Hulea, L.; Pigini, P.; Di Tullio, F.; Andreacchio, G.; Hoppe, M.M.; Wollmann, H.; et al. PRDM15 is a key regulator of metabolism critical to sustain B-cell lymphomagenesis. Nat. Commun. 2020, 11, 3520. [Google Scholar] [CrossRef] [PubMed]
- Mzoughi, S.; Zhang, J.; Hequet, D.; Teo, S.X.; Fang, H.; Xing, Q.; Bezzi, M.; Seah, M.K.Y.; Ong, S.; Shin, E.M.; et al. PRDM15 safeguards naive pluripotency by transcriptionally regulating WNT and MAPK-ERK signaling. Nat. Genet. 2017, 49, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Mzoughi, S.; Di Tullio, F.; Low, D.H.P.; Motofeanu, C.-M.; Ong, S.L.M.; Wollmann, H.; Wun, C.M.; Kruszka, P.; Muenke, M.; Hildebrandt, F.; et al. PRDM15 loss of function links NOTCH and WNT/PCP signaling to patterning defects in holoprosencephaly. Sci. Adv. 2020, 6, eaax9852. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Kaldis, P. Cascading proton transfers are a hallmark of the catalytic mechanism of SAM-dependent methyltransferases. FEBS Lett. 2020, 594, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Perna, S.K.; Huye, L.E.; Savoldo, B. Management of patients with non-Hodgkin’s lymphoma: Focus on adoptive T-cell therapy. Immunotargets Ther. 2015, 4, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Agapito, G.; Cannataro, M. Using BioPAX-Parser (BiP) to Annotate Lists of Biological Entities with Pathway Data. In Advances in Conceptual Modeling; Grossmann, G., Ram, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 92–101. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef]
- Petrossian, T.C.; Clarke, S.G. Uncovering the Human Methyltransferasome. Mol. Cell. Proteom. 2011, 10, M110.000976. [Google Scholar] [CrossRef] [PubMed]
- Bon, C.; Si, Y.; Arimondo, P.B. Chapter 4—Targeting DOT1L for mixed-lineage rearranged leukemia. In Histone Modifications in Therapy; Castelo-Branco, P., Jeronimo, C., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 81–99. [Google Scholar] [CrossRef]
- Vougiouklakis, T.; Bernard, B.J.; Nigam, N.; Burkitt, K.; Nakamura, Y.; Saloura, V. Clinicopathologic significance of protein lysine methyltransferases in cancer. Clin. Epigenet. 2020, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Min, J.; Lunin, V.V.; Antoshenko, T.; Dombrovski, L.; Zeng, H.; Allali-Hassani, A.; Campagna-Slater, V.; Vedadi, M.; Arrowsmith, C.; et al. Structural Biology of Human H3K9 Methyltransferases. PLoS ONE 2010, 5, e8570. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta 2009, 1789, 45–57. [Google Scholar] [CrossRef]
- Qian, C.; Zhou, M.-M. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell. Mol. Life Sci. 2006, 63, 2755–2763. [Google Scholar] [CrossRef]
- Guo, H.-B.; Guo, H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proc. Natl. Acad. Sci. USA 2007, 104, 8797–8802. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.-F.; Dirk, L.M.A.; Brunzelle, J.S.; Houtz, R.L.; Trievel, R.C. Structural origins for the product specificity of SET domain protein methyltransferases. Proc. Natl. Acad. Sci. USA 2008, 105, 20659–20664. [Google Scholar] [CrossRef] [PubMed]
- Giallourakis, C.C.; Benita, Y.; Molinie, B.; Cao, Z.; Despo, O.; Pratt, H.E.; Zukerberg, L.R.; Daly, M.J.; Rioux, J.D.; Xavier, R.J. Genome-wide analysis of immune system genes by expressed sequence Tag profiling. J. Immunol. 2013, 190, 5578–5587. [Google Scholar] [CrossRef]
- Lichtarge, O.; Bourne, H.R.; Cohen, F.E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996, 257, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Warshel, A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 1992, 97, 3100–3107. [Google Scholar] [CrossRef]
- King, G.; Warshel, A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 1989, 91, 3647–3661. [Google Scholar] [CrossRef]
- Schutz, C.N.; Warshel, A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins Struct. Funct. Bioinform. 2001, 44, 400–417. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Peptides | Binding Energy | ID | Peptides | Binding Energy |
|---|---|---|---|---|---|
| 1 | AALKKAL | 1.52 | 33 | KAAKKAG | 1.04 |
| 2 | AGAKKAV | 1.83 | 34 | KAAKPKA | 1.41 |
| 3 | AGVKKVA | −0.44 | 35 | KAAKPKV | 0.27 |
| 4 | AKAKKPA | 0.41 | 36 | KATKKAA | 2.64 |
| 5 | AKPKAAK | 0.7 | 37 | KDGKKRK | 0.06 |
| 6 | AKPKKAT | 0.48 | 38 | KPKKAAK | −0.46 |
| 7 | ALKKALA | 0.44 | 39 | KSPKKAK | 1.75 |
| 8 | APKKGSK | 0.03 | 40 | KTPKKAK | 2.1 |
| 9 | ARAKAKT | 0.74 | 41 | LATKAAR | 0.39 |
| 10 | ARTKQTA | −1.42 | 42 | LGLKSLV | 1.73 |
| 11 | ATPKKAK | 2.09 | 43 | LIRKLPF | 2.63 |
| 12 | ATPKKSA | −1.3 | 44 | LITKAVA | 0.19 |
| 13 | AVTKAQK | 0.33 | 45 | LLRKGNY | 0.44 |
| 14 | DVEKNNS | 1.62 | 46 | PAEKAPV | −0.75 |
| 15 | EGTKAVT | 0.74 | 47 | PKAKKAG | 0.14 |
| 16 | EHAKRKT | 2.55 | 48 | PVEKSPA | 0.72 |
| 17 | ELAKHAV | 1.04 | 49 | QDFKTDL | 1.9 |
| 18 | ELNKLLG | 0.56 | 50 | RDNKKTR | 1.85 |
| 19 | EPAKSAP | −0.07 | 51 | RHRKVLR | 2.88 |
| 20 | GAAKKPK | 1.86 | 52 | RSRKESY | 1.95 |
| 21 | GAAKRKA | 1.15 | 53 | RYQKSTE | 1.48 |
| 22 | GEAKPKV | 2.49 | 54 | SAAKAVK | 0.85 |
| 23 | GGTKPKK | 2.4 | 55 | SHHKAKG | 2.77 |
| 24 | GGVKKPH | −1.66 | 56 | SPAKPKA | 1.04 |
| 25 | GGVKRIS | 1.46 | 57 | TGGKAPR | 0.71 |
| 26 | GITKPAI | 1.36 | 58 | TPRKASG | 2.31 |
| 27 | GLGKGGA | 1.09 | 59 | TVTKKVA | −0.3 |
| 28 | GRGKGGK | 0.32 | 60 | VKPKAAK | −0.35 |
| 29 | GRGKQGG | 1.3 | 61 | VKPKKAA | 1.08 |
| 30 | GSFKLNK | 0.22 | 62 | VQTKGTG | 1.84 |
| 31 | HYNKRST | 2.75 | 63 | YALKRQG | 2.84 |
| 32 | IHAKRVT | 2.36 | 64 | YVYKVLK | 3.44 |
| PRDM15 | Histone | Peptides | Binding Energy |
| H3K37 | GGVKKPH | −1.66 | |
| H3K5 | ARTKQTA | −1.42 | |
| H1.2K148 | ATPKKSA | −1.3 | |
| H1.2K17 | PAEKAPV | −0.75 | |
| H1.2K184 | KPKKAAK | −0.46 | |
| H1.5K168 | AGVKKVA | −0.44 | |
| PRDM9 | H3K4 | ARTKQTA | −2.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.-N.; Guccione, E.; Kaldis, P. Proof-of-Concept Method to Study Uncharacterized Methyltransferases Using PRDM15. Int. J. Mol. Sci. 2023, 24, 1327. https://doi.org/10.3390/ijms24021327
Zhao L-N, Guccione E, Kaldis P. Proof-of-Concept Method to Study Uncharacterized Methyltransferases Using PRDM15. International Journal of Molecular Sciences. 2023; 24(2):1327. https://doi.org/10.3390/ijms24021327
Chicago/Turabian StyleZhao, Li-Na, Ernesto Guccione, and Philipp Kaldis. 2023. "Proof-of-Concept Method to Study Uncharacterized Methyltransferases Using PRDM15" International Journal of Molecular Sciences 24, no. 2: 1327. https://doi.org/10.3390/ijms24021327
APA StyleZhao, L.-N., Guccione, E., & Kaldis, P. (2023). Proof-of-Concept Method to Study Uncharacterized Methyltransferases Using PRDM15. International Journal of Molecular Sciences, 24(2), 1327. https://doi.org/10.3390/ijms24021327