


Shaping the Future of Immunotherapy Targets and Biomarkers in Melanoma and Non-Melanoma Cutaneous Cancers


Abstract
1. Introduction
2. State of Play of Immunotherapy in Melanoma and Non-Melanoma Skin Cancers (NMSC)
2.1. Melanoma
2.2. Non-Melanoma Skin Cancers Overview
3. Biomarkers of Immunotherapy Response/Resistance in Melanoma
4. Biomarkers of Disease Activity/Treatment Monitoring in Melanoma
5. Molecular Biomarkers in Non-Melanoma Skin Cancers
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Korn, E.L.; Liu, P.Y.; Lee, S.J.; Chapman, J.A.W.; Niedzwiecki, D.; Suman, V.J.; Moon, J.; Sondak, V.K.; Atkins, M.B.; Eisenhauer, E.A.; et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J. Clin. Oncol. 2008, 26, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies-update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Kumar, Y. Cellular and molecular mechanisms in cancer immune escape: A comprehensive review. Expert Rev. Clin. Immunol. 2014, 10, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Kirchhof, M.G.; Madrenas, J. A molecular perspective of CTLA-4 function. Annu. Rev. Immunol. 2006, 24, 65–97. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Wang, C.; Thudium, K.B.; Han, M.; Wang, X.T.; Huang, H.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A. Is It Safe to Stop Anti–PD-1 Immunotherapy in Patients with Metastatic Melanoma Who Achieve a Complete Response? J. Clin. Oncol. 2020, 38, 1645–1647. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Hamid, O.; Daud, A.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients with Metastatic Melanoma. J. Clin. Oncol. 2018, 36, 1668–1674. [Google Scholar] [CrossRef]
- Jansen, Y.J.L.; Rozeman, E.A.; Mason, R.; Goldinger, S.M.; Geukes Foppen, M.H.; Hoejberg, L.; Schmidt, H.; van Thienen, J.V.; Haanen, J.; Tiainen, L.; et al. Discontinuation of anti-PD-1 antibody therapy in the absence of disease progression or treatment limiting toxicity: Clinical outcomes in advanced melanoma. Ann. Oncol. 2019, 30, 1154–1161. [Google Scholar] [CrossRef]
- Betof Warner, A.; Palmer, J.S.; Shoushtari, A.N.; Goldman, D.A.; Panageas, K.S.; Hayes, S.A.; Bajwa, R.; Momtaz, P.; Callahan, M.K.; Wolchok, J.D.; et al. Long-Term Outcomes and Responses to Retreatment in Patients with Melanoma Treated with PD-1 Blockade. J. Clin. Oncol. 2020, 38, 1655–1663. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Duration of Anti-PD-1 Therapy in Metastatic Melanoma (STOP-GAP) 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT02821013 (accessed on 13 November 2022).
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Forsyth, P.A.; Hodi, F.S.; Algazi, A.P.; Hamid, O.; Lao, C.D.; Moschos, S.J.; Atkins, M.B.; Lewis, K.; Postow, M.A.; et al. Long-term outcomes of patients with active melanoma brain metastases treated with combination nivolumab plus ipilimumab (CheckMate 204): Final results of an open-label, multicentre, phase 2 study. Lancet Oncol. 2021, 22, 1692–1704. [Google Scholar] [CrossRef]
- Long, G.V.; Atkinson, V.; Lo, S.; Guminski, A.D.; Sandhu, S.K.; Brown, M.P.; Gonzalez, M.; Scolyer, R.A.; Emmett, L.; McArthur, G.A.; et al. Five-year overall survival from the anti-PD1 brain collaboration (ABC Study): Randomized phase 2 study of nivolumab (nivo) or nivo+ipilimumab (ipi) in patients (pts) with melanoma brain metastases (mets). J. Clin. Oncol. 2021, 39, 9508. [Google Scholar] [CrossRef]
- Di Giacomo, A.M.; Chiarion-Sileni, V.; Del Vecchio, M.; Ferrucci, P.F.; Guida, M.; Quaglino, P.; Guidoboni, M.; Marchetti, P.; Cutaia, O.; Amato, G.; et al. Primary Analysis and 4-Year Follow-Up of the Phase III NIBIT-M2 Trial in Melanoma Patients with Brain Metastases. Clin. Cancer Res. 2021, 27, 4737–4745. [Google Scholar] [CrossRef] [PubMed]
- Margolin, K.; Ernstoff, M.S.; Hamid, O.; Lawrence, D.; McDermott, D.; Puzanov, I.; Wolchok, J.D.; Clark, J.I.; Sznol, M.; Logan, T.F.; et al. Ipilimumab in patients with melanoma and brain metastases: An open-label, phase 2 trial. Lancet Oncol. 2012, 13, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.; Møller, S.; Donia, M.; Persson, G.F.; Svane, I.M.; Ellebaek, E. Real-world data on melanoma brain metastases and survival outcome. Melanoma Res. 2022, 32, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Carron, R.; Gaudy-Marqueste, C.; Amatore, F.; Padovani, L.; Malissen, N.; Balossier, A.; Loundou, A.; Bonnet, N.; Muracciole, X.; Régis, J.-M.; et al. Stereotactic radiosurgery combined with anti-PD1 for the management of melanoma brain metastases: A retrospective study of safety and efficacy. Eur. J. Cancer 2020, 135, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Gatterbauer, B.; Hirschmann, D.; Eberherr, N.; Untersteiner, H.; Cho, A.; Shaltout, A.; Göbl, P.; Fitschek, F.; Dorfer, C.; Wolfsberger, S.; et al. Toxicity and efficacy of Gamma Knife radiosurgery for brain metastases in melanoma patients treated with immunotherapy or targeted therapy—A retrospective cohort study. Cancer Med. 2020, 9, 4026–4036. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion -Sileni, V.; Lewis, K.D.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-term survival in advanced melanoma for patients treated with nivolumab plus ipilimumab in CheckMate 067. J. Clin. Oncol. 2022, 40, 9522. [Google Scholar] [CrossRef]
- Lebbé, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination with Ipilimumab in Patients with Advanced Melanoma: Results from the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol. 2019, 37, 867–875. [Google Scholar] [CrossRef]
- Lebbe, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Butler, M.O.; Eigentler, T.; Menzies, A.M.; Smylie, M.; et al. Two dosing regimens of nivolumab (NIVO) plus ipilimumab (IPI) for advanced (adv) melanoma: Three-year results of CheckMate 511. J. Clin. Oncol. 2021, 39, 9516. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Dummer, R.; Long, G.V.; Robert, C.; Tawbi, H.A.; Flaherty, K.T.; Ascierto, P.A.; Nathan, P.D.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; et al. Randomized Phase III Trial Evaluating Spartalizumab Plus Dabrafenib and Trametinib for BRAF V600–Mutant Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2022, 40, 1428–1438. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF V600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Di Giacomo, A.M.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Stephens, R.; Svane, I.M.; et al. KEYNOTE-022 part 3: A randomized, double-blind, phase 2 study of pembrolizumab, dabrafenib, and trametinib in BRAF mutant melanoma. J. Immunother.Immunother. Cancer 2020, 8, e001806. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.-G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination Dabrafenib and Trametinib Versus Combination Nivolumab and Ipilimumab for Patients with Advanced BRAF-Mutant Melanoma: The DREAMseq Trial—ECOG-ACRIN EA6134. J. Clin. Oncol. 2022, 41, JCO-22. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Mandalà, M.; Ferrucci, P.F.; Guidoboni, M.; Rutkowski, P.; Ferraresi, V.; Arance, A.; Guida, M.; Maiello, E.; Gogas, H.; et al. Sequencing of Ipilimumab Plus Nivolumab and Encorafenib Plus Binimetinib for Untreated BRAF-Mutated Metastatic Melanoma (SECOMBIT): A Randomized, Three-Arm, Open-Label Phase II Trial. J. Clin. Oncol. 2022, 41, Jco2102961. [Google Scholar] [CrossRef]
- Schadendorf, D.; Gogas, H.; Kandolf Sekulovic, L.; Meier, F.E.; Eigentler, T.; Simon, J.-C.; Terheyden, P.A.M.; Gesierich, A.H.; Herbst, R.A.; Kähler, K.C.; et al. Efficacy and safety of sequencing with vemurafenib (V) plus cobimetinib (C) followed by atezolizumab (Atezo) in patients (pts) with advanced BRAFV600-positive melanoma: Interim analysis of the ImmunoCobiVem study. J. Clin. Oncol. 2022, 40, 9548. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. Jama 1994, 271, 907–913. [Google Scholar] [CrossRef]
- Alva, A.; Daniels, G.A.; Wong, M.K.; Kaufman, H.L.; Morse, M.A.; McDermott, D.F.; Clark, J.I.; Agarwala, S.S.; Miletello, G.; Logan, T.F.; et al. Contemporary experience with high-dose interleukin-2 therapy and impact on survival in patients with metastatic melanoma and metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2016, 65, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Ding, F.; Saul, M.; Sander, C.; Tarhini, A.A.; Kirkwood, J.M.; Tawbi, H.A. High-dose interleukin-2 (HD IL-2) for advanced melanoma: A single center experience from the University of Pittsburgh Cancer Institute. J. Immunother. Cancer 2017, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Kunkel, L.; Sznol, M.; Rosenberg, S.A. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: Long-term survival update. Cancer J. Sci. Am. 2000, 6 (Suppl. S1), S11–S14. [Google Scholar] [PubMed]
- Buchbinder, E.I.; Gunturi, A.; Perritt, J.; Dutcher, J.; Aung, S.; Kaufman, H.L.; Ernstoff, M.S.; Miletello, G.P.; Curti, B.D.; Daniels, G.A.; et al. A retrospective analysis of High-Dose Interleukin-2 (HD IL-2) following Ipilimumab in metastatic melanoma. J. Immunother. Cancer 2016, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandalà, M.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; Meshcheryakov, A.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma (EORTC 1325-MG/KEYNOTE-054): Distant metastasis-free survival results from a double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 643–654. [Google Scholar] [CrossRef]
- Luke, J.J.; Rutkowski, P.; Queirolo, P.; Del Vecchio, M.; Mackiewicz, J.; Chiarion-Sileni, V.; de la Cruz Merino, L.; Khattak, M.A.; Schadendorf, D.; Long, G.V.; et al. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial. Lancet 2022, 399, 1718–1729. [Google Scholar] [CrossRef]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef]
- Blank, C.U.; Reijers, I.L.M.; Saw, R.P.M.; Versluis, J.M.; Pennington, T.; Kapiteijn, E.; Van Der Veldt, A.A.M.; Suijkerbuijk, K.; Hospers, G.; van Houdt, W.J.; et al. Survival data of PRADO: A phase 2 study of personalized response-driven surgery and adjuvant therapy after neoadjuvant ipilimumab (IPI) and nivolumab (NIVO) in resectable stage III melanoma. J. Clin. Oncol. 2022, 40, 9501. [Google Scholar] [CrossRef]
- Rozeman, E.A.; Hoefsmit, E.P.; Reijers, I.L.M.; Saw, R.P.M.; Versluis, J.M.; Krijgsman, O.; Dimitriadis, P.; Sikorska, K.; van de Wiel, B.A.; Eriksson, H.; et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat. Med. 2021, 27, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Othus, M.; Prieto, V.; Lowe, M.; Buchbinder, E.; Chen, Y.; Hyngstrom, J.; Lao, C.D.; Truong, T.; Chandra, S.; et al. LBA6—Neoadjvuant versus adjuvant pembrolizumab for resected stage III-IV melanoma (SWOG S1801). Ann. Oncol. 2022, 33 (Suppl. S7), S808–S869. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Chesney, J.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. 1037O MASTERKEY-265: A phase III, randomized, placebo (Pbo)-controlled study of talimogene laherparepvec (T) plus pembrolizumab (P) for unresectable stage IIIB–IVM1c melanoma (MEL). Ann. Oncol. 2021, 32, S868–S869. [Google Scholar] [CrossRef]
- Rohaan, M.W.; Stahlie, E.H.A.; Franke, V.; Zijlker, L.P.; Wilgenhof, S.; van der Noort, V.; van Akkooi, A.C.J.; Haanen, J.B.A.G. Neoadjuvant nivolumab + T-VEC combination therapy for resectable early stage or metastatic (IIIB-IVM1a) melanoma with injectable disease: Study protocol of the NIVEC trial. BMC Cancer 2022, 22, 851. [Google Scholar] [CrossRef] [PubMed]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Workman, C.J.; Cauley, L.S.; Kim, I.-J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A.A. Lymphocyte Activation Gene-3 (CD223) Regulates the Size of the Expanding T Cell Population Following Antigen Activation In Vivo. J. Immunol. 2004, 172, 5450. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell-cell contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-h.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, Z.; Mahesh, S.P.; Pantanelli, S.; Hwang, F.S.; Siu, W.O.; Nussenblatt, R.B. Glucocorticoid-induced Tumor Necrosis Factor Receptor Negatively Regulates Activation of Human Primary Natural Killer (NK) Cells by Blocking Proliferative Signals and Increasing NK Cell Apoptosis. J. Biol. Chem. 2008, 283, 8202–8210. [Google Scholar] [CrossRef] [PubMed]
- Schoenhals, J.E.; Cushman, T.R.; Barsoumian, H.B.; Li, A.; Cadena, A.P.; Niknam, S.; Younes, A.I.; Caetano, M.d.S.; Cortez, M.A.; Welsh, J.W. Anti-glucocorticoid-induced Tumor Necrosis Factor–Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front. Immunol. 2018, 9, 2170. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Murphy, J.T.; Wolchok, J.D. Modulation of GITR for cancer immunotherapy. Curr. Opin. Immunol. 2012, 24, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, D.L.; Watts, T.H. Cell-specific and context-dependent effects of GITR in cancer, autoimmunity, and infection. Cytokine Growth Factor Rev. 2014, 25, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef]
- Narumi, K.; Miyakawa, R.; Shibasaki, C.; Henmi, M.; Mizoguchi, Y.; Ueda, R.; Hashimoto, H.; Hiraoka, N.; Yoshida, T.; Aoki, K. Local Administration of GITR Agonistic Antibody Induces a Stronger Antitumor Immunity than Systemic Delivery. Sci. Rep. 2019, 9, 5562. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Autio, K.; Golan, T.; Dobrenkov, K.; Chartash, E.; Chen, Q.; Wnek, R.; Long, G.V. Phase I Study of MK-4166, an Anti-human Glucocorticoid-Induced TNF Receptor Antibody, Alone or with Pembrolizumab in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 1904–1911. [Google Scholar] [CrossRef]
- Arance, A.; de la Cruz-Merino, L.; Petrella, T.M.; Jamal, R.; Ny, L.; Carneiro, A.; Berrocal, A.; Márquez-Rodas, I.; Spreafico, A.; Atkinson, V.; et al. Phase II LEAP-004 Study of Lenvatinib Plus Pembrolizumab for Melanoma with Confirmed Progression on a Programmed Cell Death Protein-1 or Programmed Death Ligand 1 Inhibitor Given as Monotherapy or in Combination. J. Clin. Oncol. 2022, 41, 75–85. [Google Scholar] [CrossRef]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K.; Minoshima, Y.; Iwata, M.; Funahashi, Y. Antitumor Activity of Lenvatinib (E7080): An Angiogenesis Inhibitor That Targets Multiple Receptor Tyrosine Kinases in Preclinical Human Thyroid Cancer Models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsui, J.; Matsushima, T.; Obaishi, H.; Miyazaki, K.; Nakamura, K.; Tohyama, O.; Semba, T.; Yamaguchi, A.; Hoshi, S.S.; et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc. Cell 2014, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Kamiyama, H.; Ichikawa, K.; Fukushima, S.; Ozawa, Y.; Yamaguchi, S.; Goda, S.; Kimura, T.; Kodama, K.; Matsuki, M.; et al. Inhibition of FGFR Reactivates IFNγ Signaling in Tumor Cells to Enhance the Combined Antitumor Activity of Lenvatinib with Anti-PD-1 Antibodies. Cancer Res. 2022, 82, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Chen, L.; Lin, Z.; Liu, L.; Shao, W.; Zhang, R.; Lin, J.; Zhang, J.; Zhu, W.; Jia, H.; et al. Lenvatinib Targets FGF Receptor 4 to Enhance Antitumor Immune Response of Anti-Programmed Cell Death-1 in HCC. Hepatology 2021, 74, 2544–2560. [Google Scholar] [CrossRef] [PubMed]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye 2017, 31, 241–257. [Google Scholar] [CrossRef]
- Chattopadhyay, C.; Kim, D.W.; Gombos, D.S.; Oba, J.; Qin, Y.; Williams, M.D.; Esmaeli, B.; Grimm, E.A.; Wargo, J.A.; Woodman, S.E.; et al. Uveal melanoma: From diagnosis to treatment and the science in between. Cancer 2016, 122, 2299–2312. [Google Scholar] [CrossRef]
- Mahendraraj, K.; Lau, C.S.; Lee, I.; Chamberlain, R.S. Trends in incidence, survival, and management of uveal melanoma: A population-based study of 7,516 patients from the Surveillance, Epidemiology, and End Results database (1973–2012). Clin. Ophthalmol. 2016, 10, 2113–2119. [Google Scholar] [CrossRef]
- Shields, C.L.; Kaliki, S.; Furuta, M.; Fulco, E.; Alarcon, C.; Shields, J.A. American Joint Committee on Cancer classification of posterior uveal melanoma (tumor size category) predicts prognosis in 7731 patients. Ophthalmology 2013, 120, 2066–2071. [Google Scholar] [CrossRef]
- Shields, C.L.; Kaliki, S.; Furuta, M.; Fulco, E.; Alarcon, C.; Shields, J.A. American Joint Committee on Cancer Classification of Uveal Melanoma (Anatomic Stage) Predicts Prognosis in 7731 Patients: The 2013 Zimmerman Lecture. Ophthalmology 2015, 122, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Rowcroft, A.; Loveday, B.P.T.; Thomson, B.N.J.; Banting, S.; Knowles, B. Systematic review of liver directed therapy for uveal melanoma hepatic metastases. HPB 2020, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Pelster, M.S.; Gruschkus, S.K.; Bassett, R.; Gombos, D.S.; Shephard, M.; Posada, L.; Glover, M.S.; Simien, R.; Diab, A.; Hwu, P.; et al. Nivolumab and Ipilimumab in Metastatic Uveal Melanoma: Results from a Single-Arm Phase II Study. J. Clin. Oncol. 2021, 39, 599–607. [Google Scholar] [CrossRef]
- Piulats, J.M.; Espinosa, E.; de la Cruz Merino, L.; Varela, M.; Alonso Carrion, L.; Martin-Algarra, S.; Lopez Castro, R.; Curiel, T.; Rodriguez-Abreu, D.; Redrado, M.; et al. Nivolumab Plus Ipilimumab for Treatment-Naive Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J. Clin. Oncol. 2021, 39, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.G.; Navrazhina, K.; Ding, F.; Bhatia, R.; Tsai, K.; Abbate, K.; Durden, B.; Eroglu, Z.; Bhatia, S.; Park, S.; et al. Ipilimumab plus nivolumab for patients with metastatic uveal melanoma: A multicenter, retrospective study. J. Immunother. Cancer 2020, 8, e000331. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Amaral, T.; Kahler, K.C.; Heinzerling, L.; Hassel, J.C.; Meissner, M.; Kreuzberg, N.; Loquai, C.; Reinhardt, L.; Utikal, J.; et al. Combined immune checkpoint blockade for metastatic uveal melanoma: A retrospective, multi-center study. J. Immunother. Cancer 2019, 7, 299. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Heinzerling, L.; Kahler, K.C.; Forschner, A.; Kirchberger, M.C.; Loquai, C.; Meissner, M.; Meier, F.; Terheyden, P.; Schell, B.; et al. Prognostic factors and outcomes in metastatic uveal melanoma treated with programmed cell death-1 or combined PD-1/cytotoxic T-lymphocyte antigen-4 inhibition. Eur. J. Cancer 2017, 82, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, K.; Takahashi, A.; Mori, T.; Tsutsumida, A.; Suzuki, S.; Motoi, N.; Jinnai, S.; Kage, Y.; Mizuta, H.; Muto, Y.; et al. Nivolumab for patients with metastatic uveal melanoma previously untreated with ipilimumab: A single-institution retrospective study. Melanoma Res. 2020, 30, 76–84. [Google Scholar] [CrossRef]
- Klemen, N.D.; Wang, M.; Rubinstein, J.C.; Olino, K.; Clune, J.; Ariyan, S.; Cha, C.; Weiss, S.A.; Kluger, H.M.; Sznol, M. Survival after checkpoint inhibitors for metastatic acral, mucosal and uveal melanoma. J. Immunother. Cancer 2020, 8, e000341b. [Google Scholar] [CrossRef]
- Taylor, A.W.; Hsu, S.; Ng, T.F. The Role of Retinal Pigment Epithelial Cells in Regulation of Macrophages/Microglial Cells in Retinal Immunobiology. Front. Immunol. 2021, 12, 724601. [Google Scholar] [CrossRef]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [PubMed]
- Vergara, I.A.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Genetic drivers of non-cutaneous melanomas: Challenges and opportunities in a heterogeneous landscape. Exp. Derm. 2022, 31, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, M.; Amaro, A.; Solari, N.; Gangemi, R.; Croce, E.; Tanda, E.T.; Spagnolo, F.; Filaci, G.; Pfeffer, U.; Croce, M. How to Make Immunotherapy an Effective Therapeutic Choice for Uveal Melanoma. Cancers 2021, 13, 2043. [Google Scholar] [CrossRef] [PubMed]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Invest. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Arguello, D.; Johnston, C.; Gatalica, Z.; Terai, M.; Weight, R.M.; Orloff, M.; Mastrangelo, M.J.; Sato, T. PD-L1 expression in tumor metastasis is different between uveal melanoma and cutaneous melanoma. Immunotherapy 2017, 9, 1323–1330. [Google Scholar] [CrossRef]
- Wessely, A.; Steeb, T.; Erdmann, M.; Heinzerling, L.; Vera, J.; Schlaak, M.; Berking, C.; Heppt, M.V. The Role of Immune Checkpoint Blockade in Uveal Melanoma. Int. J. Mol. Sci. 2020, 21, 879. [Google Scholar] [CrossRef]
- de Vos, L.; Carrillo Cano, T.M.; Zarbl, R.; Klumper, N.; Ralser, D.J.; Franzen, A.; Herr, E.; Gabrielpillai, J.; Vogt, T.J.; Dietrich, J.; et al. CTLA4, PD-1, PD-L1, PD-L2, TIM-3, TIGIT, and LAG3 DNA Methylation Is Associated with BAP1 -Aberrancy, Transcriptional Activity, and Overall Survival in Uveal Melanoma. J. Immunother. 2022, 45, 324–334. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsen, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.R.; Mehnert, J.M. Mucosal Melanoma: Epidemiology, Biology and Treatment. Cancer Treat. Res. 2016, 167, 295–320. [Google Scholar] [CrossRef]
- Postow, M.A.; Hamid, O.; Carvajal, R.D. Mucosal melanoma: Pathogenesis, clinical behavior, and management. Curr. Oncol. Rep. 2012, 14, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Nassar, K.W.; Tan, A.C. The mutational landscape of mucosal melanoma. Semin. Cancer Biol. 2020, 61, 139–148. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H. PD-L1 expression in 117 sinonasal mucosal melanomas and its association with clinical outcome. Ann. Diagn Pathol 2022, 60, 151976. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Qi, Z.; Zhang, L.; Guo, J.; Si, L. Immunotherapy in Acral and Mucosal Melanoma: Current Status and Future Directions. Front. Immunol. 2021, 12, 680407. [Google Scholar] [CrossRef] [PubMed]
- Moya-Plana, A.; Herrera Gomez, R.G.; Rossoni, C.; Dercle, L.; Ammari, S.; Girault, I.; Roy, S.; Scoazec, J.Y.; Vagner, S.; Janot, F.; et al. Evaluation of the efficacy of immunotherapy for non-resectable mucosal melanoma. Cancer Immunol. Immunother. 2019, 68, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Shoushtari, A.N.; Wagstaff, J.; Ascierto, P.A.; Butler, M.O.; Lao, C.D.; Marquez-Rodas, I.; Chiarion-Sileni, V.; Dummer, R.; Ferrucci, P.F.; Lorigan, P.; et al. CheckMate 067: Long-term outcomes in patients with mucosal melanoma. J. Clin. Oncol. 2020, 38, 10019. [Google Scholar] [CrossRef]
- Simonetti, O.; Lucarini, G.; Rubini, C.; Lazzarini, R.; Di Primio, R.; Offidani, A. Clinical and prognostic significance of survivin, AKT and VEGF in primary mucosal oral melanoma. Anticancer. Res. 2015, 35, 2113–2120. [Google Scholar]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; Lian, B.; Wang, X.; et al. Axitinib in Combination with Toripalimab, a Humanized Immunoglobulin G4 Monoclonal Antibody Against Programmed Cell Death-1, in Patients with Metastatic Mucosal Melanoma: An Open-Label Phase IB Trial. J. Clin. Oncol. 2019, 37, 2987–2999. [Google Scholar] [CrossRef]
- Li, S.; Wu, X.; Yan, X.; Zhou, L.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Mao, L.; Lian, B.; et al. Toripalimab plus axitinib in patients with metastatic mucosal melanoma: 3-year survival update and biomarker analysis. J. Immunother. Cancer 2022, 10, e004036. [Google Scholar] [CrossRef]
- Mao, L.; Fang, M.; Chen, Y.; Wei, X.; Cao, J.; Lin, J.; Zhang, P.; Chen, L.; Cao, X.; Chen, Y.; et al. Atezolizumab Plus Bevacizumab in Patients with Unresectable or Metastatic Mucosal Melanoma: A Multicenter, Open-label, Single-arm Phase 2 Study. Clin. Cancer Res. 2022, 28, 4642–4648. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Cancer, C.; Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Kacew, A.J.; Dharaneeswaran, H.; Starrett, G.J.; Thakuria, M.; LeBoeuf, N.R.; Silk, A.W.; DeCaprio, J.A.; Hanna, G.J. Predictors of immunotherapy benefit in Merkel cell carcinoma. Oncotarget 2020, 11, 4401–4410. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Rischin, D.; Khushalani, N.I.; Schmults, C.D.; Guminski, A.; Chang, A.L.S.; Lewis, K.D.; Lim, A.M.; Hernandez-Aya, L.; Hughes, B.G.M.; Schadendorf, D.; et al. Integrated analysis of a phase 2 study of cemiplimab in advanced cutaneous squamous cell carcinoma: Extended follow-up of outcomes and quality of life analysis. J. Immunother. Cancer 2021, 9, e002757. [Google Scholar] [CrossRef]
- Hughes, B.G.M.; Munoz-Couselo, E.; Mortier, L.; Bratland, A.; Gutzmer, R.; Roshdy, O.; Gonzalez Mendoza, R.; Schachter, J.; Arance, A.; Grange, F.; et al. Pembrolizumab for locally advanced and recurrent/metastatic cutaneous squamous cell carcinoma (KEYNOTE-629 study): An open-label, nonrandomized, multicenter, phase II trial. Ann. Oncol. 2021, 32, 1276–1285. [Google Scholar] [CrossRef]
- Ramella Munhoz, R.; De Camargo, V.P.; Nader Marta, G.; Queiroz, M.M.; Cury-Martins, J.; Nardo, M.; Chaul-Barbosa, C.; Ricci, H.; De Mattos, M.R.; De Menezes, T.A.F.; et al. 1064P Final results of CA209-9JC: A phase II study of first-line nivolumab in patients with advanced cutaneous squamous cell carcinoma. Ann. Oncol. 2021, 32, S885. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Lebbe, C.; Mortier, L.; Brohl, A.S.; Fazio, N.; Grob, J.J.; Prinzi, N.; Hanna, G.J.; Hassel, J.C.; Kiecker, F.; et al. First-line avelumab in a cohort of 116 patients with metastatic Merkel cell carcinoma (JAVELIN Merkel 200): Primary and biomarker analyses of a phase II study. J. Immunother. Cancer 2021, 9, e002646. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients with Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Stratigos, A.J.; Sekulic, A.; Peris, K.; Bechter, O.; Prey, S.; Kaatz, M.; Lewis, K.D.; Basset-Seguin, N.; Chang, A.L.S.; Dalle, S.; et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: An open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Schmults, C.D.; Blitzblau, R.; Aasi, S.Z.; Alam, M.; Andersen, J.S.; Baumann, B.C.; Bordeaux, J.; Chen, P.L.; Chin, R.; Contreras, C.M.; et al. NCCN Guidelines(R) Insights: Squamous Cell Skin Cancer, Version 1.2022. J. Natl. Compr. Canc. Netw. 2021, 19, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Gross, N.D.; Miller, D.M.; Khushalani, N.I.; Divi, V.; Ruiz, E.S.; Lipson, E.J.; Meier, F.; Su, Y.B.; Swiecicki, P.L.; Atlas, J.; et al. Neoadjuvant Cemiplimab for Stage II to IV Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2022, 387, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.L.; Healy, M.A.; Nghiem, P.; Sober, A.J.; Johnson, T.M.; Bichakjian, C.K.; Wong, S.L. Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann. Surg. Oncol. 2016, 23, 3564–3571. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbe, C.; Lewis, K.D.; et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: Long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J. Immunother. Cancer 2020, 8, e000674. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Three-year survival, correlates and salvage therapies in patients receiving first-line pembrolizumab for advanced Merkel cell carcinoma. J. Immunother. Cancer 2021, 9, e002478. [Google Scholar] [CrossRef] [PubMed]
- Wadhera, A.; Fazio, M.; Bricca, G.; Stanton, O. Metastatic basal cell carcinoma: A case report and literature review. How accurate is our incidence data? Derm. Online J. 2006, 12, 7. [Google Scholar] [CrossRef]
- Bichakjian, C.K.; Olencki, T.; Aasi, S.Z.; Alam, M.; Andersen, J.S.; Berg, D.; Bowen, G.M.; Cheney, R.T.; Daniels, G.A.; Glass, L.F.; et al. Basal Cell Skin Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2016, 14, 574–597. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weissgraeber, S.; Han, C.T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Piro, G.; Carbone, C.; Carbognin, L.; Pilotto, S.; Ciccarese, C.; Iacovelli, R.; Milella, M.; Bria, E.; Tortora, G. Revising PTEN in the Era of Immunotherapy: New Perspectives for an Old Story. Cancers 2019, 11, 1525. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Richards, J.A.; Gupta, R.; Aung, P.P.; Emley, A.; Kluger, Y.; Dogra, S.K.; Mahalingam, M.; Wajapeyee, N. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 2014, 33, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Olbryt, M.; Rajczykowski, M.; Widlak, W. Biological Factors behind Melanoma Response to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4071. [Google Scholar] [CrossRef]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schüller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. OncoImmunology 2018, 7, e1462430. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.-L.; Reuben, A.; Spencer Christine, N.; Prieto Peter, A.; Miller John, P.; Gopalakrishnan, V.; Wang, F.; Cooper Zachary, A.; Reddy Sangeetha, M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef]
- Spiliopoulou, P.; Yang, S.Y.C.; Bruce, J.P.; Wang, B.X.; Berman, H.K.; Pugh, T.J.; Siu, L.L. All is not lost: Learning from 9p21 loss in cancer. Trends Immunol. 2022, 43, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Yang, G.; Hao, D.; Lu, Y.; Thein, K.; Simpson, B.S.; Chen, J.; Sun, R.; Alhalabi, O.; Wang, R.; et al. 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat. Commun. 2021, 12, 5606. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Uno, H.; Wooten Eric, C.; Elledge Stephen, J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Mitra, A.; Andrews, M.C.; Roh, W.; De Macedo, M.P.; Hudgens, C.W.; Carapeto, F.; Singh, S.; Reuben, A.; Wang, F.; Mao, X.; et al. Spatially resolved analyses link genomic and immune diversity and reveal unfavorable neutrophil activation in melanoma. Nat. Commun. 2020, 11, 1839. [Google Scholar] [CrossRef]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef]
- Ribas, A.; Robert, C.; Hodi, F.S.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.-J.; Weber, J.S.; Zarour, H.M.; Kefford, R.; Loboda, A.; et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J. Clin. Oncol. 2015, 33, 3001. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Rossi, N.; Lee, K.A.; Bermudez, M.V.; Visconti, A.; Thomas, A.M.; Bolte, L.A.; Björk, J.R.; de Ruijter, L.K.; Newton-Bishop, J.; Harland, M.; et al. Circulating inflammatory proteins associate with response to immune checkpoint inhibition therapy in patients with advanced melanoma. eBioMedicine 2022, 83, 104235. [Google Scholar] [CrossRef]
- Lebrun, J.J. The Dual Role of TGFbeta in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Invest. 2016, 126, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.H. CD20+ B Cells: The Other Tumor-Infiltrating Lymphocytes. J. Immunol. 2010, 185, 4977–4982. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Wu, H.; Li, L.; Luo, Y.; Li, X.; Huang, G. Regulatory T cells in the immunotherapy of melanoma. Tumour. Biol. 2016, 37, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Tobin, R.P.; Jordan, K.R.; Kapoor, P.; Spongberg, E.; Davis, D.; Vorwald, V.M.; Couts, K.L.; Gao, D.; Smith, D.E.; Borgers, J.S.W.; et al. IL-6 and IL-8 Are Linked with Myeloid-Derived Suppressor Cell Accumulation and Correlate with Poor Clinical Outcomes in Melanoma Patients. Front. Oncol. 2019, 9, 1223. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614. [Google Scholar] [CrossRef]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef]
- Vanhersecke, L.; Brunet, M.; Guegan, J.P.; Rey, C.; Bougouin, A.; Cousin, S.; Moulec, S.L.; Besse, B.; Loriot, Y.; Larroquette, M.; et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2021, 2, 794–802. [Google Scholar] [CrossRef]
- Lauss, M.; Donia, M.; Svane, I.M.; Jonsson, G. B Cells and Tertiary Lymphoid Structures: Friends or Foes in Cancer Immunotherapy? Clin. Cancer Res. 2022, 28, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Jian, C.Z.; Lin, L.I.; Low, G.S.; Ou, P.Y.; Hsu, C.; Ou, D.L. Potential Role of CXCL13/CXCR5 Signaling in Immune Checkpoint Inhibitor Treatment in Cancer. Cancers 2022, 14, 294. [Google Scholar] [CrossRef] [PubMed]
- Sucker, A.; Zhao, F.; Real, B.; Heeke, C.; Bielefeld, N.; Maßen, S.; Horn, S.; Moll, I.; Maltaner, R.; Horn, P.A.; et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin. Cancer Res. 2014, 20, 6593–6604. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Wang, Z.; Arienti, F.; Rivoltini, L.; Parmiani, G.; Ferrone, S. beta2-Microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J. Clin. Investig. 1998, 101, 2720–2729. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Hogan, S.A.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.F.; Nguyen-Kim, T.D.L.; et al. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2019, 7, 77–85. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef]
- Han, J.; Duan, J.; Bai, H.; Wang, Y.; Wan, R.; Wang, X.; Chen, S.; Tian, Y.; Wang, D.; Fei, K.; et al. TCR Repertoire Diversity of Peripheral PD-1(+)CD8(+) T Cells Predicts Clinical Outcomes after Immunotherapy in Patients with Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2020, 8, 146–154. [Google Scholar] [CrossRef]
- Poran, A.; Scherer, J.; Bushway, M.E.; Besada, R.; Balogh, K.N.; Wanamaker, A.; Williams, R.G.; Prabhakara, J.; Ott, P.A.; Hu-Lieskovan, S.; et al. Combined TCR Repertoire Profiles and Blood Cell Phenotypes Predict Melanoma Patient Response to Personalized Neoantigen Therapy plus Anti-PD-1. Cell Rep. Med. 2020, 1, 100141. [Google Scholar] [CrossRef]
- Valpione, S.; Mundra, P.A.; Galvani, E.; Campana, L.G.; Lorigan, P.; De Rosa, F.; Gupta, A.; Weightman, J.; Mills, S.; Dhomen, N.; et al. The T cell receptor repertoire of tumor infiltrating T cells is predictive and prognostic for cancer survival. Nat. Commun. 2021, 12, 4098. [Google Scholar] [CrossRef] [PubMed]
- Au, L.; Hatipoglu, E.; Robert de Massy, M.; Litchfield, K.; Beattie, G.; Rowan, A.; Schnidrig, D.; Thompson, R.; Byrne, F.; Horswell, S.; et al. Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma. Cancer Cell 2021, 39, 1497–1518. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Li, J.; Chang, L.; Zhao, C.; Jia, R.; Tan, Z.; Liu, R.; Zhang, Y.; Li, Y.; Yin, G.; et al. Peripheral blood T-cell receptor repertoire as a predictor of clinical outcomes in gastrointestinal cancer patients treated with PD-1 inhibitor. Clin. Transl. Oncol. 2021, 23, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Valpione, S.; Galvani, E.; Tweedy, J.; Mundra, P.A.; Banyard, A.; Middlehurst, P.; Barry, J.; Mills, S.; Salih, Z.; Weightman, J.; et al. Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat. Cancer 2020, 1, 210–221. [Google Scholar] [CrossRef]
- He, Y.; Liu, T.; Dai, S.; Xu, Z.; Wang, L.; Luo, F. Tumor-Associated Extracellular Matrix: How to Be a Potential Aide to Anti-tumor Immunotherapy? Front. Cell Dev. Biol. 2021, 9, 739161. [Google Scholar] [CrossRef]
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Chen, L.; Wang, J.; Byers, L.A.; Wei, Y.; Chapman, H.A.; Yamauchi, M.; Behrens, C.; et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat. Commun. 2020, 11, 4520. [Google Scholar] [CrossRef]
- Lebbink, R.J.; Raynal, N.; de Ruiter, T.; Bihan, D.G.; Farndale, R.W.; Meyaard, L. Identification of multiple potent binding sites for human leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol. 2009, 28, 202–210. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef]
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 108. [Google Scholar] [CrossRef]
- Xavier, J.B.; Young, V.B.; Skufca, J.; Ginty, F.; Testerman, T.; Pearson, A.T.; Macklin, P.; Mitchell, A.; Shmulevich, I.; Xie, L.; et al. The Cancer Microbiome: Distinguishing Direct and Indirect Effects Requires a Systemic View. Trends Cancer 2020, 6, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.C.; Duong, C.P.M.; Gopalakrishnan, V.; Iebba, V.; Chen, W.S.; Derosa, L.; Khan, M.A.W.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Fidelle, M.; Iebba, V.; Alla, L.; Pasolli, E.; Segata, N.; Desnoyer, A.; Pietrantonio, F.; Ferrere, G.; et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. Eur. Urol. 2020, 78, 195–206. [Google Scholar] [CrossRef]
- Jiang, S.; Geng, S.; Chen, Q.; Zhang, C.; Cheng, M.; Yu, Y.; Zhang, S.; Shi, N.; Dong, M. Effects of Concomitant Antibiotics Use on Immune Checkpoint Inhibitor Efficacy in Cancer Patients. Front. Oncol. 2022, 12, 823705. [Google Scholar] [CrossRef]
- Yang, M.; Wang, Y.; Yuan, M.; Tao, M.; Kong, C.; Li, H.; Tong, J.; Zhu, H.; Yan, X. Antibiotic administration shortly before or after immunotherapy initiation is correlated with poor prognosis in solid cancer patients: An up-to-date systematic review and meta-analysis. Int. Immunopharmacol. 2020, 88, 106876. [Google Scholar] [CrossRef]
- Hopkins, A.M.; Kichenadasse, G.; Karapetis, C.S.; Rowland, A.; Sorich, M.J. Concomitant Antibiotic Use and Survival in Urothelial Carcinoma Treated with Atezolizumab. Eur. Urol. 2020, 78, 540–543. [Google Scholar] [CrossRef]
- Elkrief, A.; El Raichani, L.; Richard, C.; Messaoudene, M.; Belkaid, W.; Malo, J.; Belanger, K.; Miller, W.; Jamal, R.; Letarte, N.; et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1568812. [Google Scholar] [CrossRef]
- He, Y.; Fu, L.; Li, Y.; Wang, W.; Gong, M.; Zhang, J.; Dong, X.; Huang, J.; Wang, Q.; Mackay, C.R.; et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8(+) T cell immunity. Cell Metab. 2021, 33, 988–1000. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, X.; Wang, M.; He, Z.; Li, H.; Wang, J.; Li, Q. Gut microbiota influence immunotherapy responses: Mechanisms and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.C.; Araya, R.E.; Huang, A.; Chen, Q.; Di Modica, M.; Rodrigues, R.R.; Lopes, A.; Johnson, S.B.; Schwarz, B.; Bohrnsen, E.; et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 2021, 184, 5338–5356.e21. [Google Scholar] [CrossRef]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- Mohan, V.; Das, A.; Sagi, I. Emerging roles of ECM remodeling processes in cancer. Semin. Cancer Biol. 2020, 62, 192–200. [Google Scholar] [CrossRef]
- Henstra, C.; van Praagh, J.; Olinga, P.; Nagelkerke, A. The gastrointestinal microbiota in colorectal cancer cell migration and invasion. Clin. Exp. Metastasis. 2021, 38, 495–510. [Google Scholar] [CrossRef]
- Fluckiger, A.; Daillere, R.; Sassi, M.; Sixt, B.S.; Liu, P.; Loos, F.; Richard, C.; Rabu, C.; Alou, M.T.; Goubet, A.G.; et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 2020, 369, 936–942. [Google Scholar] [CrossRef]
- Bessell, C.A.; Isser, A.; Havel, J.J.; Lee, S.; Bell, D.R.; Hickey, J.W.; Chaisawangwong, W.; Glick Bieler, J.; Srivastava, R.; Kuo, F.; et al. Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI Insight. 2020, 5, 8. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Siravegna, G. How to use liquid biopsies to treat patients with cancer. ESMO Open 2021, 6, 100060. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.C.; Belloum, Y.; Deitert, B.; Sementsov, M.; Heidrich, I.; Gebhardt, C.; Keller, L.; Pantel, K. Current and Future Clinical Applications of ctDNA in Immuno-Oncology. Cancer Res. 2022, 82, 349–358. [Google Scholar] [CrossRef]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann. Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef]
- Elez, E.; Chianese, C.; Sanz-Garcia, E.; Martinelli, E.; Noguerido, A.; Mancuso, F.M.; Caratu, G.; Matito, J.; Grasselli, J.; Cardone, C.; et al. Impact of circulating tumor DNA mutant allele fraction on prognosis in RAS-mutant metastatic colorectal cancer. Mol. Oncol. 2019, 13, 1827–1835. [Google Scholar] [CrossRef]
- Cai, Z.; Chen, G.; Zeng, Y.; Dong, X.; Li, Z.; Huang, Y.; Xin, F.; Qiu, L.; Xu, H.; Zhang, W.; et al. Comprehensive Liquid Profiling of Circulating Tumor DNA and Protein Biomarkers in Long-Term Follow-Up Patients with Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 5284–5294. [Google Scholar] [CrossRef]
- Nygård, L.; Ahlborn, L.B.; Persson, G.F.; Chandrananda, D.; Langer, J.W.; Fischer, B.M.; Langer, S.W.; Gabrielaite, M.; Kjær, A.; Rosenfeld, N.; et al. Circulating cell free DNA during definitive chemo-radiotherapy in non-small cell lung cancer patients—Initial observations. PLoS ONE 2020, 15, e0231884. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Forde, P.M.; White, J.R.; Niknafs, N.; Hruban, C.; Naidoo, J.; Marrone, K.; Sivakumar, I.K.A.; Bruhm, D.C.; Rosner, S.; et al. Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Res. 2019, 79, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Burgener, J.M.; Zou, J.; Zhao, Z.; Zheng, Y.; Shen, S.Y.; Huang, S.H.; Keshavarzi, S.; Xu, W.; Liu, F.F.; Liu, G.; et al. Tumor-Naive Multimodal Profiling of Circulating Tumor DNA in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2021, 27, 4230–4244. [Google Scholar] [CrossRef]
- García-Pardo, M.; Makarem, M.; Li, J.J.N.; Kelly, D.; Leighl, N.B. Integrating circulating-free DNA (cfDNA) analysis into clinical practice: Opportunities and challenges. Br. J. Cancer 2022, 127, 592–602. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Chopra, N.; Comino-Méndez, I.; Beaney, M.; Tovey, H.; Cutts, R.J.; Swift, C.; Kriplani, D.; Afentakis, M.; Hrebien, S.; et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol. 2019, 5, 1473–1478. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Lee, J.H.; Saw, R.P.; Thompson, J.F.; Lo, S.; Spillane, A.J.; Shannon, K.F.; Stretch, J.R.; Howle, J.; Menzies, A.M.; Carlino, M.S.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. J. Eur. Soc. Med. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fisher, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Lorigan, P.; et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Genta, S.; Araujo, D.V.; Keshavarzi, S.; Muniz, T.P.; Kamil, Z.S.; Howarth, K.; Terrell, S.; Joad, A.; Ventura, R.; Covelli, A.; et al. Leveraging personalized circulating tumor DNA (ctDNA) for detection and monitoring of molecular residual disease in high-risk melanoma. J. Clin. Oncol. 2022, 40, 9579. [Google Scholar] [CrossRef]
- McEvoy, A.C.; Pereira, M.R.; Reid, A.; Pearce, R.; Cowell, L.; Al-Ogaili, Z.; Khattak, M.A.; Millward, M.; Meniawy, T.M.; Gray, E.S.; et al. Monitoring melanoma recurrence with circulating tumor DNA: A proof of concept from three case studies. Oncotarget 2019, 10, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Adam, G.; Rampasek, L.; Safikhani, Z.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Machine learning approaches to drug response prediction: Challenges and recent progress. NPJ Precis. Oncol. 2020, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Desai, K.; Tang, T.; Weber, J.S.; Dolfi, S.; Ritchings, C.; Huang, S.P.; Bolisetty, M.; Sausen, M.; Del Vecchio, M.; et al. 788O Association of pre-treatment ctDNA with disease recurrence and clinical and translational factors in patients with stage IIIB-D/IV melanoma treated with adjuvant immunotherapy (CheckMate 915). Ann. Oncol. 2022, 33, S904. [Google Scholar] [CrossRef]
- Gracie, L.; Pan, Y.; Atenafu, E.G.; Ward, D.G.; Teng, M.; Pallan, L.; Stevens, N.M.; Khoja, L. Circulating tumour DNA (ctDNA) in metastatic melanoma, a systematic review and meta-analysis. Eur. J. Cancer 2021, 158, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, P.; Buszka, K.; Zabel, M.; Nowicki, M.; Alix-Panabieres, C.; Budna-Tukan, J. Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. Int. J. Mol. Sci. 2021, 22, 9714. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Bratman, S.V.; Yang, S.Y.C.; Iafolla, M.A.J.; Liu, Z.; Hansen, A.R.; Bedard, P.L.; Lheureux, S.; Spreafico, A.; Razak, A.A.; Shchegrova, S.; et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat. Cancer 2020, 1, 873–881. [Google Scholar] [CrossRef]
- Marsavela, G.; Lee, J.; Calapre, L.; Wong, S.Q.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Abed, A.; et al. Circulating Tumor DNA Predicts Outcome from First-, but not Second-line Treatment and Identifies Melanoma Patients Who May Benefit from Combination Immunotherapy. Clin. Cancer Res. 2020, 26, 5926–5933. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients with Metastatic Melanoma Treated with Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients with Non-Small Cell Lung Cancer with Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Velcheti, V.; Mekhail, T.; Yun, C.; Shagan, S.M.; Hu, S.; Chae, Y.K.; Leal, T.A.; Dowell, J.E.; Tsai, M.L.; et al. Blood-based tumor mutational burden as a biomarker for atezolizumab in non-small cell lung cancer: The phase 2 B-F1RST trial. Nat. Med. 2022, 28, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Kuziora, M.; Quinn, K.J.; Helman, E.; Ye, J.; Liu, F.; Scheuring, U.; Peters, S.; Rizvi, N.A.; Brohawn, P.Z.; et al. A Blood-based Assay for Assessment of Tumor Mutational Burden in First-line Metastatic NSCLC Treatment: Results from the MYSTIC Study. Clin. Cancer Res. 2021, 27, 1631–1640. [Google Scholar] [CrossRef]
- Chen, X.; Fang, L.; Zhu, Y.; Bao, Z.; Wang, Q.; Liu, R.; Sun, W.; Du, H.; Lin, J.; Yu, B.; et al. Blood tumor mutation burden can predict the clinical response to immune checkpoint inhibitors in advanced non-small cell lung cancer patients. Cancer Immunol. Immunother. 2021, 70, 3513–3524. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, X.; Gao, C.; Gong, J.; Wang, X.; Gao, J.; Li, Z.; Wang, J.; Yang, B.; Wang, L.; et al. Plasma-based microsatellite instability detection strategy to guide immune checkpoint blockade treatment. J. Immunother. Cancer 2020, 8, e001297. [Google Scholar] [CrossRef]
- Georgiadis, A.; Durham, J.N.; Keefer, L.A.; Bartlett, B.R.; Zielonka, M.; Murphy, D.; White, J.R.; Lu, S.; Verner, E.L.; Ruan, F.; et al. Noninvasive Detection of Microsatellite Instability and High Tumor Mutation Burden in Cancer Patients Treated with PD-1 Blockade. Clin. Cancer Res. 2019, 25, 7024–7034. [Google Scholar] [CrossRef]
- Cabel, L.; Proudhon, C.; Romano, E.; Girard, N.; Lantz, O.; Stern, M.H.; Pierga, J.Y.; Bidard, F.C. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2018, 15, 639–650. [Google Scholar] [CrossRef]
- Snyder, A.; Morrissey, M.P.; Hellmann, M.D. Use of Circulating Tumor DNA for Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 6909–6915. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Shi, W.; Zhu, M.; Liu, Z.; Luo, N.; Zeng, Y.; He, Y. Serial ultra-deep sequencing of circulating tumor DNA reveals the clonal evolution in non-small cell lung cancer patients treated with anti-PD1 immunotherapy. Cancer Med. 2019, 8, 7669–7678. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, D.L.; Wang, F.; Yang, C.P.; Chen, X.X.; You, J.Q.; Huang, J.S.; Shao, Y.; Zhu, D.Q.; Ouyang, Y.M.; et al. The predicting role of circulating tumor DNA landscape in gastric cancer patients treated with immune checkpoint inhibitors. Mol. Cancer 2020, 19, 154. [Google Scholar] [CrossRef] [PubMed]
- Takai, E.; Omata, W.; Totoki, Y.; Nakamura, H.; Shiba, S.; Takahashi, A.; Namikawa, K.; Mori, T.; Yamazaki, N.; Shibata, T.; et al. Clonal dynamics of circulating tumor DNA during immune checkpoint blockade therapy for melanoma. Cancer Sci. 2021, 112, 4748–4757. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Derm. 2015, 151, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Didona, D.; Di Pietro, F.R.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous Squamous Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Rischin, D.; Fury, M.G.; Lowy, I.; Stankevich, E.; Han, H.; Porceddu, S. A phase III, randomized, double-blind study of adjuvant cemiplimab versus placebo post-surgery and radiation therapy (RT) in patients (pts) with high-risk cutaneous squamous cell carcinoma (CSCC). J. Clin. Oncol. 2020, 38, TPS10084. [Google Scholar] [CrossRef]
- Grob, J.J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef]
- Maubec, E.; Boubaya, M.; Petrow, P.; Beylot-Barry, M.; Basset-Seguin, N.; Deschamps, L.; Grob, J.J.; Dreno, B.; Scheer-Senyarich, I.; Bloch-Queyrat, C.; et al. Phase II Study of Pembrolizumab As First-Line, Single-Drug Therapy for Patients with Unresectable Cutaneous Squamous Cell Carcinomas. J. Clin. Oncol. 2020, 38, 3051–3061. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Amit, M.; Nagarajan, P.; Rubin, M.L.; Yuan, Y.; Bell, D.; El-Naggar, A.K.; Johnson, J.M.; Morrison, W.H.; Rosenthal, D.I.; et al. Pilot Phase II Trial of Neoadjuvant Immunotherapy in Locoregionally Advanced, Resectable Cutaneous Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2021, 27, 4557–4565. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbe, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis Prim. 2017, 3, 17077. [Google Scholar] [CrossRef]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [PubMed]
- Lyngaa, R.; Pedersen, N.W.; Schrama, D.; Thrue, C.A.; Ibrani, D.; Met, O.; Thor Straten, P.; Nghiem, P.; Becker, J.C.; Hadrup, S.R. T-cell responses to oncogenic merkel cell polyomavirus proteins distinguish patients with merkel cell carcinoma from healthy donors. Clin. Cancer Res. 2014, 20, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Bhatia, S.; Amin, A.; Kudchadkar, R.R.; Sharfman, W.H.; Lebbe, C.; Delord, J.P.; Dunn, L.A.; Shinohara, M.M.; Kulikauskas, R.; et al. Neoadjuvant Nivolumab for Patients with Resectable Merkel Cell Carcinoma in the CheckMate 358 Trial. J. Clin. Oncol. 2020, 38, 2476–2487. [Google Scholar] [CrossRef]
- Spassova, I.; Ugurel, S.; Kubat, L.; Zimmer, L.; Terheyden, P.; Mohr, A.; Bjorn Andtback, H.; Villabona, L.; Leiter, U.; Eigentler, T.; et al. Clinical and molecular characteristics associated with response to therapeutic PD-1/PD-L1 inhibition in advanced Merkel cell carcinoma. J. Immunother. Cancer 2022, 10, e003198. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Basset-Seguin, N.; Dreno, B.; Garbe, C.; Gutzmer, R.; Hauschild, A.; Krattinger, R.; Lear, J.T.; Malvehy, J.; et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: A joint expert opinion. J. Eur. Acad Derm. Venereol 2020, 34, 1944–1956. [Google Scholar] [CrossRef]



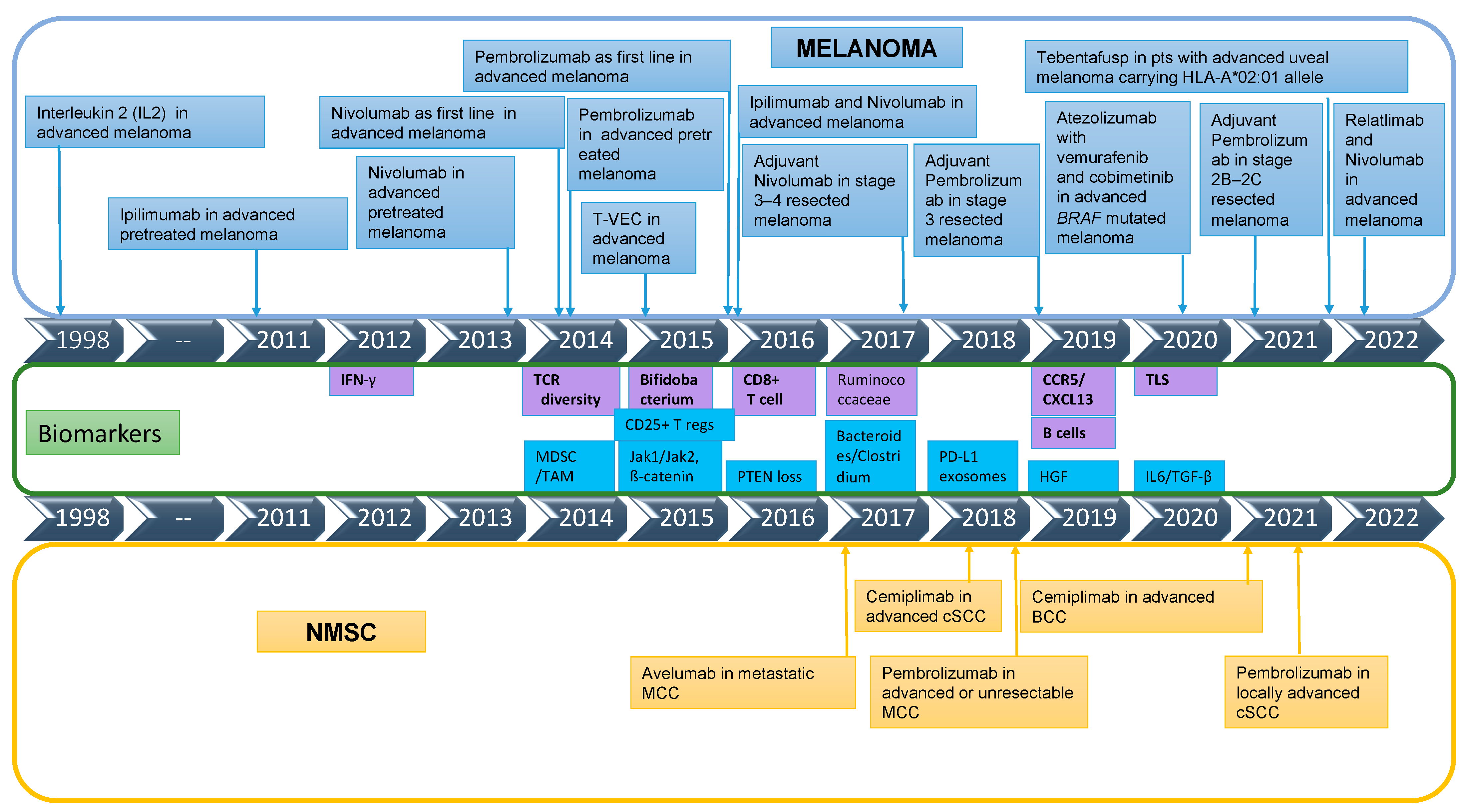{kind=link}
{kind=link}
| Type of Intervention | Targets | Agents | Number of Patients | Phase | NCT Number |
|---|---|---|---|---|---|
| Novel Immune Pathways | |||||
| Anti-PD-1 plus anti-TIGIT (IV) or anti-PD-1 + Coxsackievirus (IT) or anti-PD-1 + ILT-4 or anti-PD-1/anti-LAG-3 or anti-PD-1 + retinoid | Pembrolizumab + vibostolimab or pembrolizumab + gebasaxturev or pembrolizumab + MK-4830 or favezelimab or pembrolizumab + all-trans retinoic acid (ATRA) | 90 | 1/2 | NCT04303169 | |
| Anti-CTLA-4/anti-PD-1 +/− anti-interleukin 8 antibody | Ipilimumab/Nivolumab +/− BMS-986253 (HuMax-IL8) | 372 | 1/2 | NCT03400332 | |
| IL-2Rβ/γ-selective IL-2 variant +/− anti-PD-1 +/− TLR7/8 agonist | Pembrolizumab +/− TransCon IL-2 β/γ +/− TransCon TLR7/8 agonist | 317 * | 1/2 | NCT05081609 | |
| IL-2 superkine +/− ICI | MDNA11 +/− ICI | 100 * | 1/2 | NCT05086692 | |
| T cell engager +/− anti-PD-1 +/− anti-CTLA-4 | Tebentafusp +/− durvalumab +/− tremelimumab | 312 | 1/2 | NCT02535078 | |
| IL-12 Fc-fusion protein +/− anti-PD-1 | BMS-986415 +/− Nivolumab | 473 * | 1/2 | NCT04423029 | |
| CD40 agonist +/− anti-PD-1 | APX005M + pembrolizumab | 41 | 1/2 | NCT02706353 | |
| CXCR1/2 inhibitor + anti-PD-1 | SX-682 + pembrolizumab | 77 | 1 | NCT03161431 | |
| STING agonist + anti-PD-L1 | SB 11285 + atezolizumab | 110 * | 1 | NCT04096638 | |
| Anti-TIGIT + anti-PD-1 | AB154 + AB122 | 26 | 2 | NCT05130177 | |
| Interleukin (IL-)15 and IL-15 receptor alpha + anti-PD-1 | NIZ985 +/− spartalizumab | 110 * | 1 | NCT04261439 | |
| mAb Specific to B-and T-Lymphocyte Attenuator (BTLA) | JS004 | 156 * | 1 | NCT04773951 | |
| PD-L1/IDO peptide vaccine + anti-PD-1 | PD-L1/IDO peptide vaccine + nivolumab | 50 | 1/2 | NCT03047928 | |
| Anti-CCR8 +/− anti-PD-1 | BAY3375968 | 270 * | 1 | NCT05537740 | |
| TGFβ1 inhibitor +/− anti-PD-L1 | SRK-181 +/− anti-PD-L1 | 200 * | 1 | NCT04291079 | |
| Anti-PD-1 inhibitor/OX40 agonist | SL-279252 (PD1-Fc-OX40L) | 87 * | 1 | NCT03894618 | |
| Granulocyte Macrophage-colony stimulating factor + anti-PD-1 | Sargramostim + pembrolizumab | 30 | 2 | NCT04703426 | |
| Adoptive cell therapy | |||||
| PD-1 knock out TILs | IOV-4001 | 53 * | 1/2 | NCT05361174 | |
| Adoptive autologous cellular therapy + HDAC + DNMT inhibitors | MAGE-C2/HLA-A2 TCR T cells + azacytidine + valproic acid | 20 | 1/2 | NCT04729543 | |
| CD20 CAR transduced T cells | MB-CART20.1 | 15 | 1 | NCT03893019 | |
| Autologous alpha-type-1 polarized dendritic cells (alphaDC1)/TBVA vaccine + cytokine modulating regimen | TBVA vaccine intatolimod + IFN-alpha2b + celecoxib | 24 | 2 | NCT04093323 | |
| Autologous neoantigen-specific T-cell product | NEO-PTC-01 | 52 | 1 | NCT04625205 | |
| Oncolytic viral therapy | |||||
| Rhinovirus targeting poliovirus receptor CD155 (IT) +/− anti-PD-1 | Lerapolturev (IT) +/− anti-PD-1 | 56 | 2 | NCT04577807 | |
| Oncolytic vesicular stomatitis virus expressing human IFNβ (IT or IV) +/− anti-PD-1 +/− anti-CTLA-4 | Voyager V1 +/− cemiplimab +/− ipilimumab | 152 * | 2 | NCT04291105 | |
| Anti-PD-1 + oncolytic HSV type virus expressing anti-PD-1 antibody and IL-12 (IT) | MVR-T3011 (IT) +/− pembrolizumab | 10 | 1/2 | NCT04370587 | |
| Herpes simplex virus type 2 strain HG52 +/− anti-PD-1 | OH2 +/− nivolumab | 30 * | 1/2 | NCT04386967 | |
| Adenovirus with TMA-CD40L and 4-1BBL transgenes + anti-PD-L1 | LOAd703 + atezolizumab | 35 | 1/2 | NCT04123470 | |
| Great ape Adenoviral (GAd)/Modified Vaccinia Ankara (MVA) boosts with personalised patient neoantigens + anti-PD-1 | NOUS-PEV + pembrolizumab | 34 * | 1 | NCT04990479 | |
| Anti-PD-1 + oncolytic HSV type 1 virus (IT) | Pembrolizumab + RP1 | 300 * | 2 | NCT03767348 | |
| Anti-PD-1 plus vaccinia virus encoding transgenes for Flt3 ligand, anti-CTLA-4 antibody and IL-12 cytokine, (IT or IV) | Pembrolizumab + TBio-6517 (IV or IT) | 138 * | 1/2 | NCT04301011 | |
| Targeted treatment plus immunotherapy | |||||
| PI3K inhibitor + anti-PD-1 | Duvelisib + nivolumab | 42 | 1/2 | NCT04688658 | |
| ETBR inhibitor +/− anti-PD-1 | ENB003 +/− pembrolizumab | 130 | 1/2 | NCT04205227 | |
| Anti-VEGF + anti-PD-L1 | Bevacizumab + atezolizumab | 30 | 2 | NCT04356729 | |
| Integrin alpha-V/beta-8 Antagonist | PF-06940434 | 122 | 1 | NCT04152018 | |
| p38 inhibitor + anti-PD-1 or anti-CTLA-4/anti-PD-1 | ARRY-614 + nivolumab or nivolumab/ipilimumab | 144 * | 1/2 | NCT04074967 | |
| Hedgehog pathway inhibitor + anti-PD-1 | Sonidegib + Pembrolizumab | 45 * | 1 | NCT04007744 | |
| Wnt/β-catenin inhibitor +/− anti-PD-1 | E7386 +/− pembrolizumab | 108 * | 1/2 | NCT05091346 | |
| PARP inhibition + anti-PD-1 | Olaparib + pembrolizumab | 41 | 2 | NCT04633902 | |
| “Velcrin” that triggers the formation of a complex of two proteins called SLFN12 and PDE3A | BAY2666605 | 89 | 1 | NCT04809805 | |
| Heparanase inhibitor (TAMs inhibitor) + anti-PD-1 + metronomic chemotherapy | Pixatimod (PG545) + Nivolumab + cyclophosphamide | 61 | 2 | NCT05061017 | |
| Anti-CTLA-4/anti-PD-1 +/− glutamate modulator | Ipilimumab/Nivolumab +/− troriluzole | 108 | 2 | NCT04899921 | |
| MDM2 inhibitor + anti-PD-1 | APG-115 + pembrolizumab | 224 | 1/2 | NCT03611868 | |
| Multi-tyrosine kinase inhibitor + anti-PD-1 | Cabozantinib + pembrolizumab | 39 | 1/2 | NCT03957551 | |
| BTK inhibitor + anti-PD-1 | Ibrutinib + pembrolizumab | 23 | 1 | NCT03021460 | |
| ATR inhibitor +/− anti-PD-L1 | Ceralasertib +/− durvalumab | 195 | 2 | NCT05061134 | |
| Anti-PD-1 plus NLRP3 inhibitor | Pembrolizumab + Dapansutrile | 26 | 1/2 | NCT04971499 | |
| Vaccine-based treatment | |||||
| Melanoma HLA-restricted peptides vaccine +/− anti-CD27 | 6MHP +/− CDX-1127 | 30 | 1/2 | NCT03617328 | |
| DNA vaccine encoding Tyrosinase-Related Protein 2 (TRP2) and gp100 | SCIB1 DNA vaccine | 87 | 2 | NCT04079166 | |
| mRNA-encoded with tumor-associated antigens vaccine + anti-PD-1 | BNT111 + cemiplimab | 180 | 2 | NCT04526899 | |
| Antibody drug conjugates | |||||
| ADC against CD228 | SGN-CD228A | 275 * | 1 | NCT05571839 | |
| ROR2-targeted antibody drug conjugate (CAB-ROR2-ADC) +/− anti-PD-1 | BA3021 +/− pembrolizumab | 420 * | 2 | NCT03504488 | |
| Target Population | Type of Treatment | Intervention | Number of Patients | Phase | NCT Number | |
|---|---|---|---|---|---|---|
| Uveal Melanoma | Local treatment + anti-PD-1 and anti CTLA-4 | Yttrium 90 + Ipilimumab + Nivolumab | 26 | 1/2 | NCT02913417 | |
| Melphalan percutaneous hepatic perfusion + Ipilimumab + Nivolumab | 83 | 1/2 | NCT04283890 | |||
| Immunoembolization +IPI/NIVO | 14 | 2 | NCT03472586 | |||
| Ipilimumab + Nivolumab + Tumor Treating Fields | 10 | 1 | NCT05004025 | |||
| Stereotactic RT + Ipilimumab + Nivolumab | 40 | 2 | NCT05077280 | |||
| Anti-PD-1 + Anti-LAG-3 | Nivolumab + Relatlimab | 27 | 2 | NCT04552223 | ||
| Anti-PD-1 and anti CTLA-4 + TLR9 agonists | Nivolumab + Ipilimumab + intrahepatic SD-101 | 80 | 1 | NCT04935229 | ||
| Anti-PD-1 and anti CTLA-4 + anti-arginine | ADI-PEG20 + Ipilimumab + Nivolumab | 9 | 1 | NCT03922880 | ||
| Anti-CTLA-4 + cell therapy | CD8+ SLC45A2-Specific T Lymphocytes + Cyclophosphamide, Aldesleukin, and Ipilimumab | 30 | 1 | NCT03068624 | ||
| Cell therapy | Tumor Infiltrating Lymphocytes | 47 | 2 | NCT03467516 | ||
| 10 | 1 | NCT05607095 | ||||
| Dendritic cells + tumor RNA | 200 | 3 | NCT01983748 | |||
| Anti-PD-1 + epigenetic modulators | Pembrolizimab + Etinostat | 29 | 2 | NCT02697630 | ||
| Anti-PD-1 + Anti-VEGF | Pembrolizumab + Lenvatinib | 54 | 2 | NCT05282901 | ||
| 30 | 2 | NCT05308901 | ||||
| Anti-PD-1 + Anti-DDR | Pembrolizumab + Olaparib | 37 | 2 | NCT 05524935 | ||
| Mucosal Melanoma | Anti-PD-1 + local treatment | Adjuvant Pembrolizumab + hypofractionated RT | 16 | 2 | NCT04318717 | |
| Anti-PD-1 + chemotherapy + local treatment | Adjuvant Toripalimab + chemotherapy or RT | 45 | 2 | NCT04879654 | ||
| Anti-PD-1 + Anti-VEGF | Neoadjuvant Pembrolizumab + Lenvatinib | 44 | 2 | NCT05545969 | ||
| 26 | 2 | NCT04622566 | ||||
| SHR-1210 + Apatinib | 40 | 2 | NCT03986515 | |||
| Adjuvant Nivolumab +/− Cabozantinib | 99 | 2 | NCT05111574 | |||
| Nivolumab + Axitinib | 20 | 2 | NCT05384496 | |||
| Toripalib + Axitinib | 30 | 2 | NCT04180995 | |||
| 99 | 2 | NCT03941795 | ||||
| Anti-PD-1 + anti-TGFβ | Pembrolizumab + Vactosertib | 30 | 2 | NCT05436990 | ||
| Anti-PD-1 + anti CD40 + chemotherapy | YH003 + Pembrolizumab + Paclitaxel | 43 | 2 | NCT05420324 | ||
| Anti-PD-1 + chemotherapy | Nivolumab + Decitabine/Cedazuridine | 30 | 1/2 | NCT05089370 | ||
| IL-2 agonists | ALKS 4230 | 110 | 2 | NCT04830124 | ||
| Study Title | Tumor Type | Intervention | Phase | NCT Number |
|---|---|---|---|---|
| Neo-adjuvant Nivolumab or Nivolumab With Ipilimumab in Advanced Cutaneous Squamous Cell Carcinoma Prior to Surgery | cSCC | Ipilimumab Nivolumab | 2 | NCT04620200 |
| Neoadjuvant Plus Adjuvant Treatment With Cemiplimab in Cutaneous Squamous Cell Carcinoma | cSCC | Cemiplimab | 2 | NCT04632433 |
| Neoadjuvant Pembrolizumab in Cutaneous Squamous Cell Carcinoma | cSCC | Pembrolizumab | 2 | NCT05025813 |
| Atezolizumab Before Surgery for the Treatment of Regionally Metastatic Head and Neck Squamous Cell Cancer With an Unknown or Historic Primary Site | cSCC | Atezolizumab | 2 | NCT05110781 |
| Avelumab With or Without Cetuximab in Treating Patients With Advanced Skin Squamous Cell Cancer | cSCC | Avelumab Cetuximab | 2 | NCT03944941 |
| Phase II Study of Peptide Receptor Radionuclide Therapy in Combination With Immunotherapy for Patients With Merkel Cell Cancer | MCC | Pembrolizumab, Lutetium Lu 177 dotatate | 2 | NCT05583708 |
| Neoadjuvant Lenvatinib Plus Pembrolizumab in Merkel Cell Carcinoma | MCC | Pembrolizumab, Lenvatinib | 2 | NCT04869137 |
| Navtemadlin (KRT-232) With or Without Anti-PD-1/Anti-PD-L1 for the Treatment of Patients With Merkel Cell Carcinoma | MCC | KRT-232, Avelumab | 2 | NCT03787602 |
| Adjuvant Avelumab in Merkel Cell Cancer | MCC | Avelumab, placebo | 2 | NCT03271372 |
| Testing Pembrolizumab Versus Observation in Patients With Merkel Cell Carcinoma After Surgery, STAMP Study | MCC | Pembrolizumab, placebo | 3 | NCT03712605 |
| Intralesional Cemiplimab for Patients With Cutaneous Squamous Cell Carcinoma or Basal Cell Carcinoma | cSCC, BCC | Cemiplimab intralesional | 1 | NCT03889912 |
| Nivolumab Alone or Plus Relatlimab or Ipilimumab for Patients With Locally-Advanced Unresectable or Metastatic Basal Cell Carcinoma | BCC | Nivolumab, Relatlimab | 2 | NCT03521830 |
| Neoadjuvant-Adjuvant Pembrolizumab in Resectable Advanced Basal Cell Carcinoma of H&N | BCC | Pembrolizumab | 1 | NCT04323202 |
| Anti-PD1-antibody and Pulsed HHI for Advanced BCC | BCC | Cemiplimab, Sonidegib | 2 | NCT04679480 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiliopoulou, P.; Vornicova, O.; Genta, S.; Spreafico, A. Shaping the Future of Immunotherapy Targets and Biomarkers in Melanoma and Non-Melanoma Cutaneous Cancers. Int. J. Mol. Sci. 2023, 24, 1294. https://doi.org/10.3390/ijms24021294
Spiliopoulou P, Vornicova O, Genta S, Spreafico A. Shaping the Future of Immunotherapy Targets and Biomarkers in Melanoma and Non-Melanoma Cutaneous Cancers. International Journal of Molecular Sciences. 2023; 24(2):1294. https://doi.org/10.3390/ijms24021294
Chicago/Turabian StyleSpiliopoulou, Pavlina, Olga Vornicova, Sofia Genta, and Anna Spreafico. 2023. "Shaping the Future of Immunotherapy Targets and Biomarkers in Melanoma and Non-Melanoma Cutaneous Cancers" International Journal of Molecular Sciences 24, no. 2: 1294. https://doi.org/10.3390/ijms24021294
APA StyleSpiliopoulou, P., Vornicova, O., Genta, S., & Spreafico, A. (2023). Shaping the Future of Immunotherapy Targets and Biomarkers in Melanoma and Non-Melanoma Cutaneous Cancers. International Journal of Molecular Sciences, 24(2), 1294. https://doi.org/10.3390/ijms24021294