
The Therapeutic Potential of Pyroptosis in Melanoma



Abstract
1. Introduction
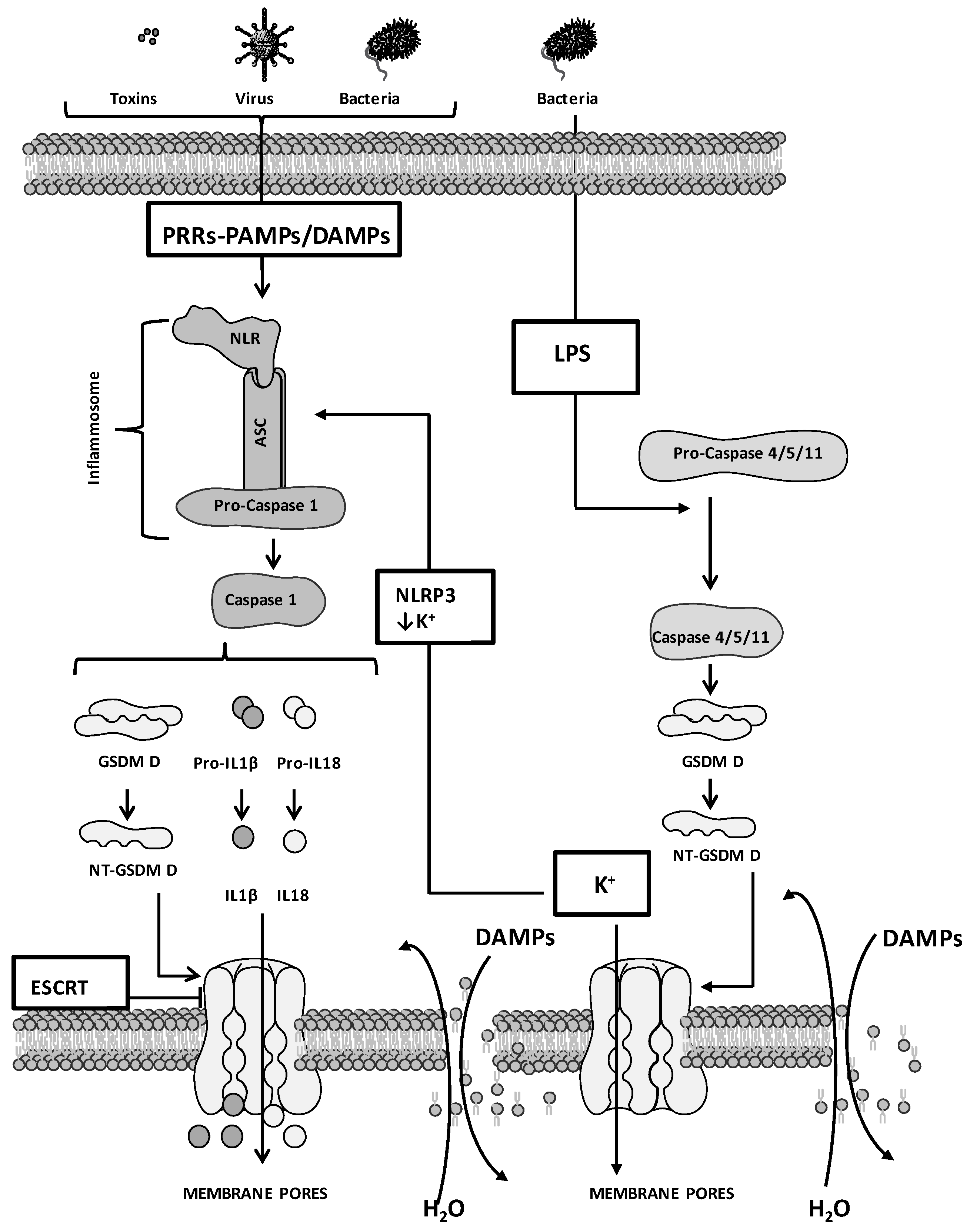
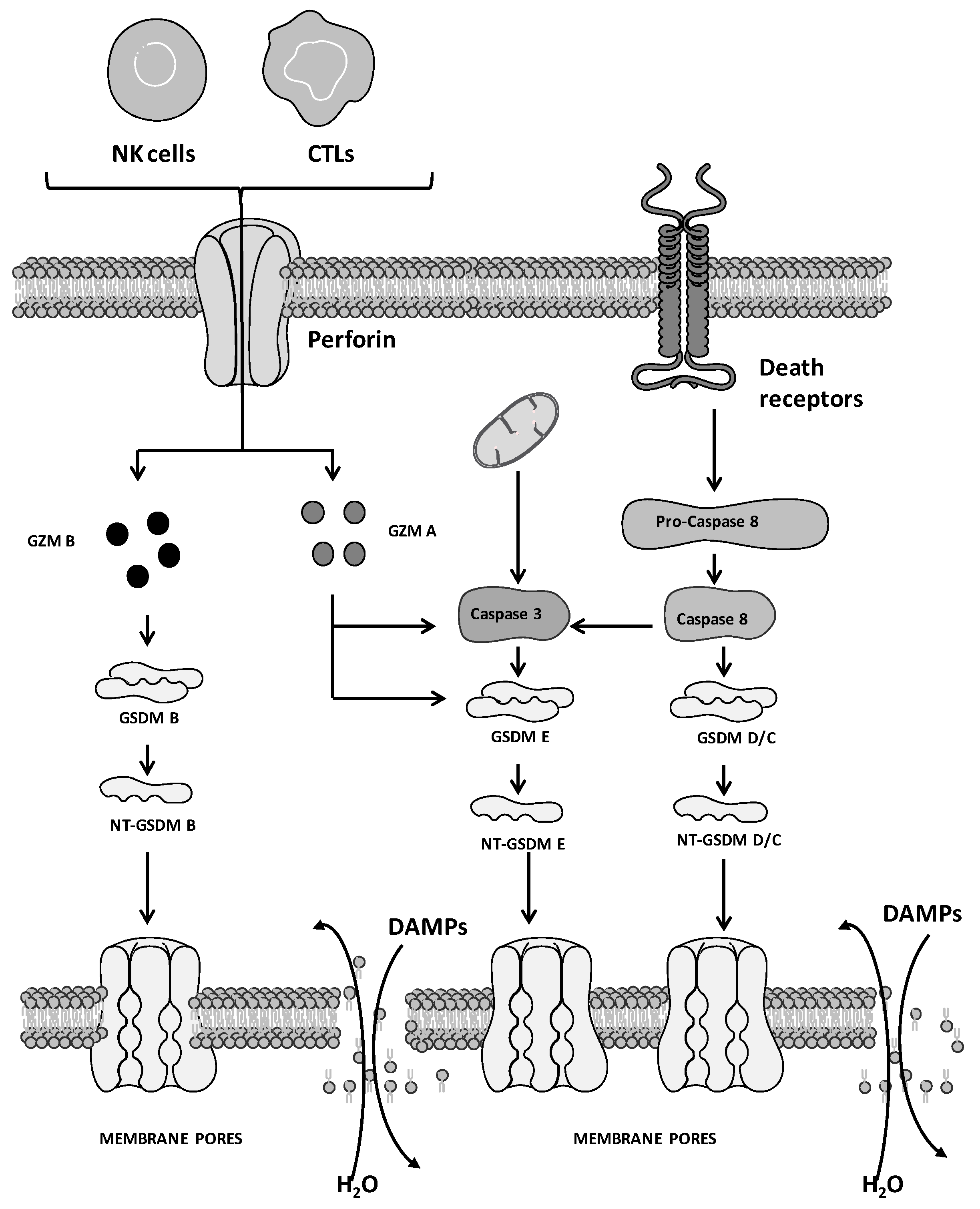
2. Molecular Mechanisms of Pyroptosis: General Overview
3. Pyroptosis and Cancer
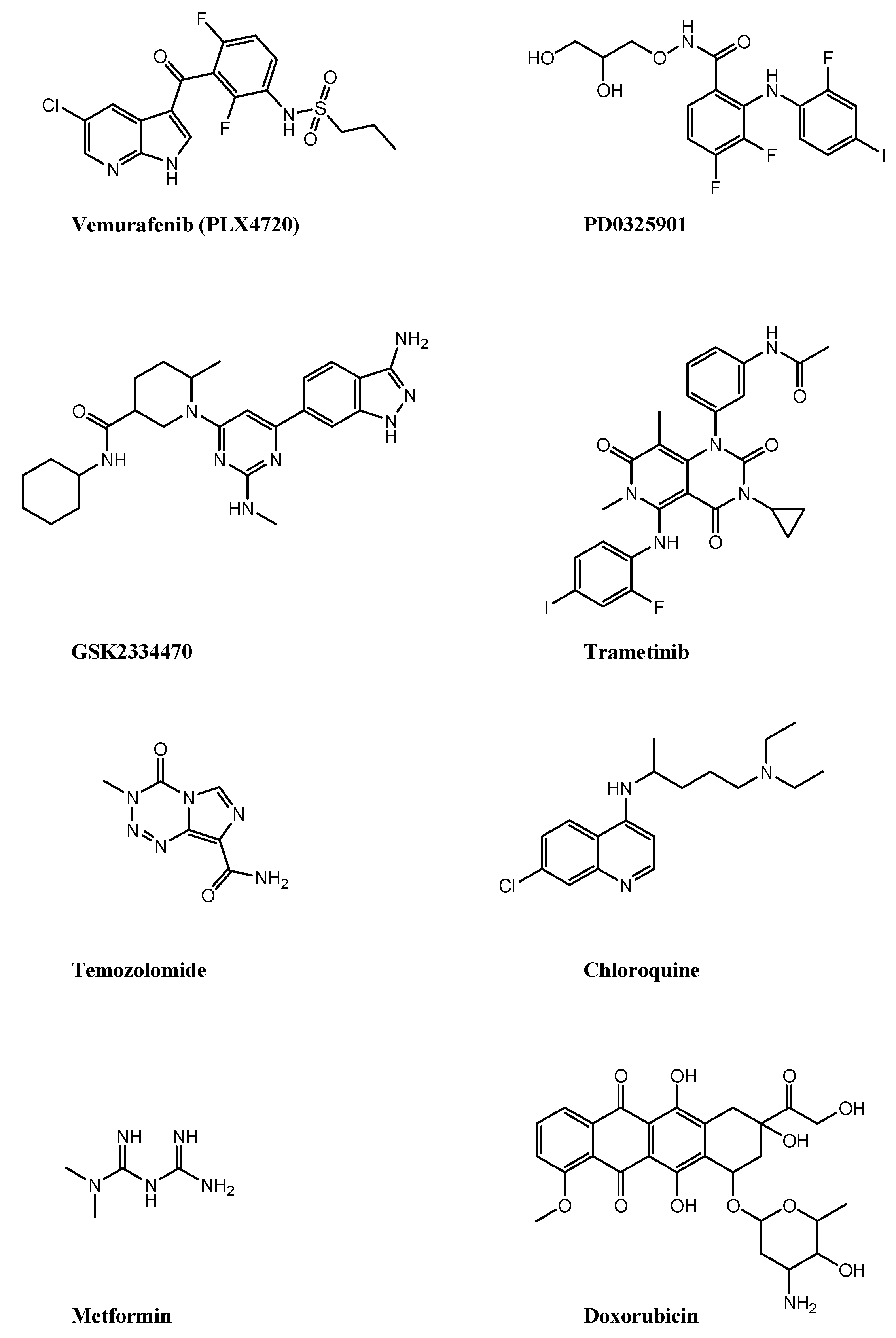
3.1. Drug Combinations That Induce Pyroptosis in Melanoma
3.2. Pyroptosis-Associated Gene Signatures in Melanoma
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Choi, J.J.; Reich, C.F.; Pisetsky, D.S. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand. J. Immunol. 2004, 60, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host. Microb. 2018, 24, 97–108.e4. [Google Scholar] [CrossRef] [PubMed]
- Canli, Ö.; Alankuş, Y.B.; Grootjans, S.; Vegi, N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016, 127, 139–148. [Google Scholar] [CrossRef]
- Haberzettl, P.; Hill, B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox. Biol. 2013, 1, 56–64. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, N.; Jiang, N. Autophagy provides a conceptual therapeutic framework for bone metastasis from prostate cancer. Cell Death Dis. 2021, 12, 909. [Google Scholar] [CrossRef]
- Zaffaroni, N.; Beretta, G.L. Nanoparticles for Ferroptosis Therapy in Cancer. Pharmaceutics 2021, 13, 1785. [Google Scholar] [CrossRef]
- Zaffaroni, N.; Beretta, G.L. Ferroptosis inducers for prostate cancer therapy. Curr. Med. Chem. 2022, 29, 4185–4201. [Google Scholar] [CrossRef]
- Beretta, G.L.; Zaffaroni, N. Necroptosis and Prostate Cancer: Molecular Mechanisms and Therapeutic Potential. Cells 2022, 11, 1221. [Google Scholar] [CrossRef]
- Goodall, M.L.; Cramer, S.D.; Thorburn, A. Autophagy RIPs into cell death. Cell Cycle 2016, 15, 3014–3015. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Lee, E.; Banoth, B.; Malireddi, R.K.S.; Samir, P.; Tuladhar, S.; Mummareddy, H.; Burton, A.R.; Vogel, P.; et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight 2020, 5, e136720. [Google Scholar] [CrossRef]
- Place, D.E.; Lee, S.; Kanneganti, T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021, 59, 42–49. [Google Scholar] [CrossRef]
- Heidaryan, F.; Bamehr, H.; Babaabasi, B.; Emamvirdizadeh, A.; Mohammadzadeh, N.; Khalili, A. The Trend of ripk1/ripk3 and mlkl Mediated Necroptosis Pathway in Patients with Different Stages of Prostate Cancer as Promising Progression Biomarkers. Clin. Lab. 2020, 66. [Google Scholar] [CrossRef]
- Liu, W.; Jin, W.; Zhu, S.; Chen, Y.; Liu, B. Targeting regulated cell death (RCD) with small-molecule compounds in cancer therapy: A revisited review of apoptosis, autophagy-dependent cell death and necroptosis. Drug Discov. Today 2022, 27, 612–625. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Wang, F.; Liu, W.; Ning, J.; Wang, J.; Lang, Y.; Jin, X.; Zhu, K.; Wang, X.; Li, X.; Yang, F.; et al. Simvastatin Suppresses Proliferation and Migration in Non-small Cell Lung Cancer via Pyroptosis. Int. J. Biol. Sci. 2018, 14, 406–417. [Google Scholar] [CrossRef]
- Teng, J.F.; Mei, Q.B.; Zhou, X.G.; Tang, Y.; Xiong, R.; Qiu, W.Q.; Pan, R.; Law, B.Y.; Wong, V.K.; Yu, C.L.; et al. Polyphyllin VI Induces Caspase-1-Mediated Pyroptosis via the Induction of ROS/NF-κB/NLRP3/GSDMD Signal Axis in Non-Small Cell Lung Cancer. Cancers 2020, 12, 193. [Google Scholar] [CrossRef]
- Chu, Q.; Jiang, Y.; Zhang, W.; Xu, C.; Du, W.; Tuguzbaeva, G.; Qin, Y.; Li, A.; Zhang, L.; Sun, G.; et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget 2016, 7, 84658–84665. [Google Scholar] [CrossRef]
- Hage, C.; Hoves, S.; Strauss, L.; Bissinger, S.; Prinz, Y.; Pöschinger, T.; Kiessling, F.; Ries, C.H. Sorafenib Induces Pyroptosis in Macrophages and Triggers Natural Killer Cell-Mediated Cytotoxicity Against Hepatocellular Carcinoma. Hepatology 2019, 70, 1280–1297. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, B.; Sun, J.; Na, H.; Chen, Z.; Zhu, Z.; Yan, L.; Ren, S.; Zuo, Y. DAC can restore expression of NALP1 to suppress tumor growth in colon cancer. Cell Death Dis. 2015, 6, e1602. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, B.; Li, D.; Wang, G.; Han, X.; Sun, X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 2018, 495, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef]
- Cai, W.; Nguyen, M.Q.; Wilski, N.A.; Purwin, T.J.; Vernon, M.; Tiago, M.; Aplin, A.E. A Genome-Wide Screen Identifies PDPK1 as a Target to Enhance the Efficacy of MEK1/2 Inhibitors in NRAS Mutant Melanoma. Cancer Res. 2022, 82, 2625–2639. [Google Scholar] [CrossRef]
- Ahmed, F.; Tseng, H.Y.; Ahn, A.; Gunatilake, D.; Alavi, S.; Eccles, M.; Rizos, H.; Gallagher, S.J.; Tiffen, J.C.; Hersey, P.; et al. Repurposing Melanoma Chemotherapy to Activate Inflammasomes in the Treatment of BRAF/MAPK Inhibitor Resistant Melanoma. J. Investig. Dermatol. 2022, 142, 1444–1455.e10. [Google Scholar] [CrossRef]
- Song, M.; Xia, W.; Tao, Z.; Zhu, B.; Zhang, W.; Liu, C.; Chen, S. Self-assembled polymeric nanocarrier-mediated co-delivery of metformin and doxorubicin for melanoma therapy. Drug Deliv. 2021, 28, 594–606. [Google Scholar] [CrossRef]
- Yang, F.; Bettadapura, S.N.; Smeltzer, M.S.; Zhu, H.; Wang, S. Pyroptosis and pyroptosis-inducing cancer drugs. Acta Pharmacol. Sin 2022, 43, 2462–2473. [Google Scholar] [CrossRef]
- Vernon, M.; Wilski, N.A.; Kotas, D.; Cai, W.; Pomante, D.; Tiago, M.; Alnemri, E.S.; Aplin, A.E. Raptinal induces Gasdermin E-dependent pyroptosis in naive and therapy-resistant melanoma. Mol. Cancer Res. 2022, 20, 1811–1821. [Google Scholar] [CrossRef]
- De Backer, J.; Maric, D.; Zuhra, K.; Bogaerts, A.; Szabo, C.; Vanden Berghe, W.; Hoogewijs, D. Cytoglobin Silencing Promotes Melanoma Malignancy but Sensitizes for Ferroptosis and Pyroptosis Therapy Response. Antioxidants 2022, 11, 1548. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, J.Y.; Liu, X.S.; Chen, H.Z.; Ai, Y.L.; Cheng, K.; Sun, R.Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Kandolf Sekulovic, L.; Guo, J.; Agarwala, S.; Hauschild, A.; McArthur, G.; Cinat, G.; Wainstein, A.; Caglevic, C.; Lorigan, P.; Gogas, H.; et al. Access to innovative medicines for metastatic melanoma worldwide: Melanoma World Society and European Association of Dermato-oncology survey in 34 countries. Eur. J. Cancer. 2018, 104, 201–209. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A. Melanoma Staging: American JoInt. Committee on Cancer (AJCC) 8th Edition and Beyond. Ann. Surg. Oncol. 2018, 25, 2105–2110. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef]
- Stamatakos, S.; Beretta, G.L.; Vergani, E.; Dugo, M.; Corno, C.; Corna, E.; Tinelli, S.; Frigerio, S.; Ciusani, E.; Rodolfo, M.; et al. Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations. Cancers 2021, 13, 2284. [Google Scholar] [CrossRef]
- Vergani, E.; Beretta, G.L.; Aloisi, M.; Costantino, M.; Corno, C.; Frigerio, S.; Tinelli, S.; Dugo, M.; Accattatis, F.M.; Granata, A.; et al. Targeting of the Lipid Metabolism Impairs Resistance to BRAF Kinase Inhibitor in Melanoma. Front. Cell Dev. Biol. 2022, 10, 927118. [Google Scholar] [CrossRef]
- Friedlander, A.M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986, 261, 7123–7126. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Gong, Y.N.; Guy, C.; Crawford, J.C.; Green, D.R. Biological events and molecular signalling following MLKL activation during necroptosis. Cell Cycle 2017, 16, 1748–1760. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Martin, S.J. Cell death and inflammation: The case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef]
- Bozza, M.T.; Jeney, V. Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front. Immunol. 2020, 11, 1323. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Paquette, N.; Conlon, J.; Sweet, C.; Rus, F.; Wilson, L.; Pereira, A.; Rosadini, C.V.; Goutagny, N.; Weber, A.N.; Lane, W.S.; et al. Serine/threonine acetylation of TGFβ-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 12710–12715. [Google Scholar] [CrossRef]
- Ruano-Gallego, D.; Sanchez-Garrido, J.; Kozik, Z.; Núñez-Berrueco, E.; Cepeda-Molero, M.; Mullineaux-Sanders, C.; Naemi-Baghshomali Clark, J.; Slater, S.L.; Wagner, N.; Glegola-Madejska, I.; et al. Type III secretion system effectors form robust and flexible intracellular virulence networks. Science 2021, 371, eabc9531. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V. Bacterial Outer Membrane 457 Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Chen, X.; He, W.T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Fox, D.; Man, S.M. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef] [PubMed]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mo, F.; Wang, Y.; Li, W.; Chen, Y.; Liu, J.; Chen-Mayfield, T.J.; Hu, Q. Enhancing Gasdermin-induced tumor pyroptosis through preventing ESCRT-dependent cell membrane repair augments antitumor immune response. Nat. Commun. 2022, 13, 6321. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–6671. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Brough, D. Potassium efflux fires the canon: Potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur. J. Immunol. 2015, 45, 2758–2761. [Google Scholar] [CrossRef]
- Baker, P.J.; Boucher, D.; Bierschenk, D.; Tebartz, C.; Whitney, P.G.; D’Silva, D.B.; Tanzer, M.C.; Monteleone, M.; Robertson, A.A.; Cooper, M.A.; et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 2015, 45, 2918–2926. [Google Scholar] [CrossRef]
- Rühl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Muñoz-Planillo, R.; Liu, Q.; Núñez, G. Caspase-11 requires the Pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 2017, 24, 507–514.e4. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef]
- Demarco, B.; Grayczyk, J.P. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 2020, 6, eabc3465. [Google Scholar] [CrossRef]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, S.; Zhang, Y.; Li, P.; Wang, K. The multifaceted roles of pyroptotic cell death pathways in cancer. Cancers 2019, 11, 1313. [Google Scholar] [CrossRef]
- Minton, K. Pyroptosis heats tumour immunity. Nat. Rev. Drug Discov. 2020, 19, 309. [Google Scholar] [CrossRef]
- Rosenbaum, S.R.; Wilski, N.A.; Aplin, A.E. Fueling the fire: Inflammatory forms of cell death and implications for cancer immunotherapy. Cancer Discov. 2021, 11, 266–281. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Silke, J. The intersection of cell death and inflammasome activation. Cell Mol. Life Sci. 2016, 73, 2349–2367. [Google Scholar] [CrossRef]
- Zhivaki, D.; Kagan, J.C. NLRP3 inflammasomes that induce antitumor immunity. Trends Immunol. 2021, 42, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef]
- Zhivaki, D.; Borriello, F.; Chow, O.A.; Doran, B.; Fleming, I.; Theisen, D.J.; Pallis, P.; Shalek, A.K.; Sokol, C.L.; Zanoni, I.; et al. Inflammasomes within Hyperactive Murine Dendritic Cells Stimulate Long-Lived T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2020, 33, 108381. [Google Scholar] [CrossRef]
- Fassi, E.M.A.; Sgrignani, J.; D’Agostino, G.; Cecchinato, V.; Garofalo, M.; Grazioso, G.; Uguccioni, M.; Cavalli, A. Oxidation State Dependent Conformational Changes of HMGB1 Regulate the Formation of the CXCL12/HMGB1 Heterocomplex. Comput. Struct. Biotechnol. J. 2019, 17, 886–894. [Google Scholar] [CrossRef]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundbäck, P.; Long, W.; Valdes-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef]
- Nyström, S.; Antoine, D.J.; Lundbäck, P.; Lock, J.G.; Nita, A.F.; Högstrand, K.; Grandien, A.; Erlandsson-Harris, H.; Andersson, U.; Applequist, S.E. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 2013, 32, 86–99. [Google Scholar] [CrossRef]
- Kazama, H.; Ricci, J.E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility Group Box-1 protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef]
- Li, A.A.; Zhang, Y.; Tong, W.L.; Chen, J.W.; Huang, S.H.; Liu, J.M.; Liu, Z.L. Identification of a Novel Pyroptosis-Related Gene Signature Indicative of Disease Prognosis and Treatment Response in Skin Cutaneous Melanoma. Int. J. Gen. Med. 2022, 15, 6145–6163. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, Y.; Niu, Z.; Xing, J.; Yang, Z.; Yin, X.; Guo, L.; Zhang, Q.; Qiu, H.; Han, Y. A Novel Pyroptotic and Inflammatory Gene Signature Predicts the Prognosis of Cutaneous Melanoma and the Effect of Anticancer Therapies. Front. Med. 2022, 9, 841568. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Shi, L.Y.; Zhu, Z.T.; Wang, Q.J. A new pyroptosis model can predict the immunotherapy response and immune microenvironment characteristics and prognosis of patients with cutaneous melanoma based on TCGA and GEO databases. Ann. Transl. Med. 2022, 10, 353. [Google Scholar] [CrossRef]
- Niu, Z.; Xu, Y.; Li, Y.; Chen, Y.; Han, Y. Construction and validation of a novel pyroptosis-related signature to predict prognosis in patients with cutaneous melanoma. Math. Biosci. Eng. 2022, 19, 688–706. [Google Scholar] [CrossRef]
- Meng, J.; Huang, X.; Qiu, Y.; Zheng, X.; Huang, J.; Wen, Z.; Yao, J. Pyroptosis-related gene mediated modification patterns and immune cell infiltration landscapes in cutaneous melanoma to aid immunotherapy. Aging 2021, 13, 24379–24401. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, L.; Jin, C.; Xu, J.; Zhang, X.; Yao, Y. A novel pyroptosis-associated gene signature for immune status and prognosis of cutaneous melanoma. PeerJ 2021, 9, e12304. [Google Scholar] [CrossRef]
- Ju, A.; Tang, J.; Chen, S.; Fu, Y.; Luo, Y. Pyroptosis-Related Gene Signatures Can Robustly Diagnose Skin Cutaneous Melanoma and Predict the Prognosis. Front. Oncol. 2021, 11, 709077. [Google Scholar] [CrossRef]
- Lou, X.; Li, K.; Qian, B.; Li, Y.; Zhang, D.; Cui, W. Pyroptosis correlates with tumor immunity and prognosis. Commun. Biol. 2022, 5, 917. [Google Scholar] [CrossRef]
- Shi, Z.; Gu, J.; Yao, Y.; Wu, Z. Identification of a predictive gene signature related to pyroptosis for the prognosis of cutaneous melanoma. Medicine 2022, 101, e30564. [Google Scholar] [CrossRef]
- Wu, G.; Chen, B.; Jiang, J.; Chen, Y.; Chen, Y.; Wang, H. Identification of a pyroptosis-based model for predicting clinical outcomes from immunotherapy in patients with metastatic melanoma. Cancer Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, Z.; Gao, L.; Zeng, J.; Xiang, Y.; Zhou, L.; Tong, X.; Wang, X.Q.; Lu, J. Increased IRF9-STAT2 Signaling Leads to Adaptive Resistance toward Targeted Therapy in Melanoma by Restraining GSDME-Dependent Pyroptosis. J. Investig. Dermatol. 2022, 142, 2476–2487. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, H.; Chen, L.; Cao, Y.; Hu, Y.; Zhu, Z.; Wang, M.; Shi, J. A Novel Pyroptosis-Related lncRNA Signature for Predicting the Prognosis of Skin Cutaneous Melanoma. Int. J. Gen. Med. 2021, 14, 6517–6527. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, G.; He, Y.W.; Chen, R.; Wu, Z.Y. Identification of a pyroptosis-associated long non-coding RNA signature for predicting the immune status and prognosis in skin cutaneous melanoma. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 5597–5609. [Google Scholar] [CrossRef]
- Zhong, J.; Wang, Z.; Hounye, A.H.; Liu, J.; Zhang, J.; Qi, M.; Hou, M. A novel pyroptosis-related LncRNA signature predicts prognosis and indicates tumor immune microenvironment in skin cutaneous melanoma. Life Sci. 2022, 307, 120832. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Combination | Cell Lines | In Vivo Studies | Pyroptosis Markers |
|---|---|---|---|
| PLX4720/PD0325901 | Mouse D4M3.A and YUMM1.7 Human A375 and TJUMEL57 | yes | GSDM E HMGB1 |
| GSK2334470/trametinib | Human WM1361A, WM1633, SK-MEL-30 and SK-MEL-173 | yes | GSDM E HMGB1 |
| Temozolomide/chloroquine | Human primary culture melanoma cell lines | yes | GSDM E GSDM D |
| Metformin/doxorubicin | Human A375 and SK-MEL-28 | yes | GSDM D |
| Gene Signature | Cell Line Validation | Drug Sensitivity Prediction | References |
|---|---|---|---|
| GSDM A, GSDM C, IL18, NLRP6, AIM2 | Healthy HaCaT and melanoma A375, HS294T and M14 | Paclitaxel, docetaxel, cisplatin, sorafenib, PD0325901 | [92] |
| TLR1, CCL8, EMP3, IFNGR2, CCL25, IL15, RTP4, NLRP6 | Healthy HaCaT and PIG1 and melanoma A375, SK-MEL-28 | Afatinib, sorafenib, refametinib, docetaxel, rapamycin, cisplatin | [93] |
| GSDM C, GSDM D, NLRP6, IL18, AIM2, PRKACA | No | Anti PD1, anti CTLA4 | [94] |
| GSDM A, GSDM C, AIM2, NOD2 | No | Anti PD1, anti CTLA4 | [95] |
| AIM2, IL1B, NLRC4, NLRP3, NLRP6, NLRP7, TNF, ELANE, GSDM A, GSDM B, GSDM C, NLRP1 | No | Anti PD1, anti CTLA4 | [96] |
| GSDM C, GZM A, AIM2, PD-L1 | No | Nelarabine, dexamethasone decadron, fluphenazine, arsenic trioxide, procarbazine, olaparib, fludarabine, simvastatin, cyclophosphamide, asparaginase | [97] |
| NLRP9, DHX9, CASP3, NLRC4, AIM2, NLRP3, IL1B, GSDM E, GSDM D | No | No | [98] |
| CASP5, NEK7, AIM2, CASP1, NLRC4, GSDM D | A375 | Anti PD1 | [99] |
| BST2, GBP5, AIM2 | No | No | [100] |
| CASP5, NLRP6, NLRP7, PYCARD | No | Anti PD1, anti CTLA4, adoptive T cell therapy | [101] |
| IRF9, STAT2 | A375 and SK-MEL-28 | Increased vemurafenib sensitivity following IRF9 and STAT2 silencing | [102] |
| AL121603.2, AC107464.2, AC245128.3, AC092171.5, AC242842.1, IRF2-DT, HLA-DQB1-AS1, AC004585.1, LINC00582 | No | Bexarotene, bryostatin, docetaxel, bortezomib, bosutinib, camptothecin | [103] |
| AC004847.1, USP30-AS1, AC082651.3, AL033384.1, AC138207.5, AC245041.1, U62317.1, AL512274.1, AC018755.4, MIR200CHG, LINC02362, LINC00861, AL683807.1, AC010503.4, AL512363.1, LINC02437, LINC01527, AL049555.1, AC245041.2, AL365361.1, AC015819.1, MIR205HG | No | Imatinib, isotretinoin, bendamustine, nilotinib, fluphenazine, nelfinavir, oxaliplatin, megestrol acetate, ifosfamide, palbociclib, etoposide, alectinib, dromostanolone propionate | [104] |
| LINC01234, ZEB1-AS1, SLFN12L, MATN1-AS1, ZNF529-AS1, HOXC-AS2, PLA2G4E-AS1, LRP4-AS1, LINC01028, TM4SF1-AS1, RNF216P1, SNHG17 | A375, SK-MEL-28, PIG1 | Diastereomer 1, buparlisib, tivozanib, pyrazole anthrone, dasatinib, rapamycin, chelerythrine, JQ1, belinostat, vincristin, methylprednisolone, hydroxyurea | [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaffaroni, N.; Beretta, G.L. The Therapeutic Potential of Pyroptosis in Melanoma. Int. J. Mol. Sci. 2023, 24, 1285. https://doi.org/10.3390/ijms24021285
Zaffaroni N, Beretta GL. The Therapeutic Potential of Pyroptosis in Melanoma. International Journal of Molecular Sciences. 2023; 24(2):1285. https://doi.org/10.3390/ijms24021285
Chicago/Turabian StyleZaffaroni, Nadia, and Giovanni L. Beretta. 2023. "The Therapeutic Potential of Pyroptosis in Melanoma" International Journal of Molecular Sciences 24, no. 2: 1285. https://doi.org/10.3390/ijms24021285
APA StyleZaffaroni, N., & Beretta, G. L. (2023). The Therapeutic Potential of Pyroptosis in Melanoma. International Journal of Molecular Sciences, 24(2), 1285. https://doi.org/10.3390/ijms24021285