
Platelet–Neutrophil Crosstalk in Thrombosis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
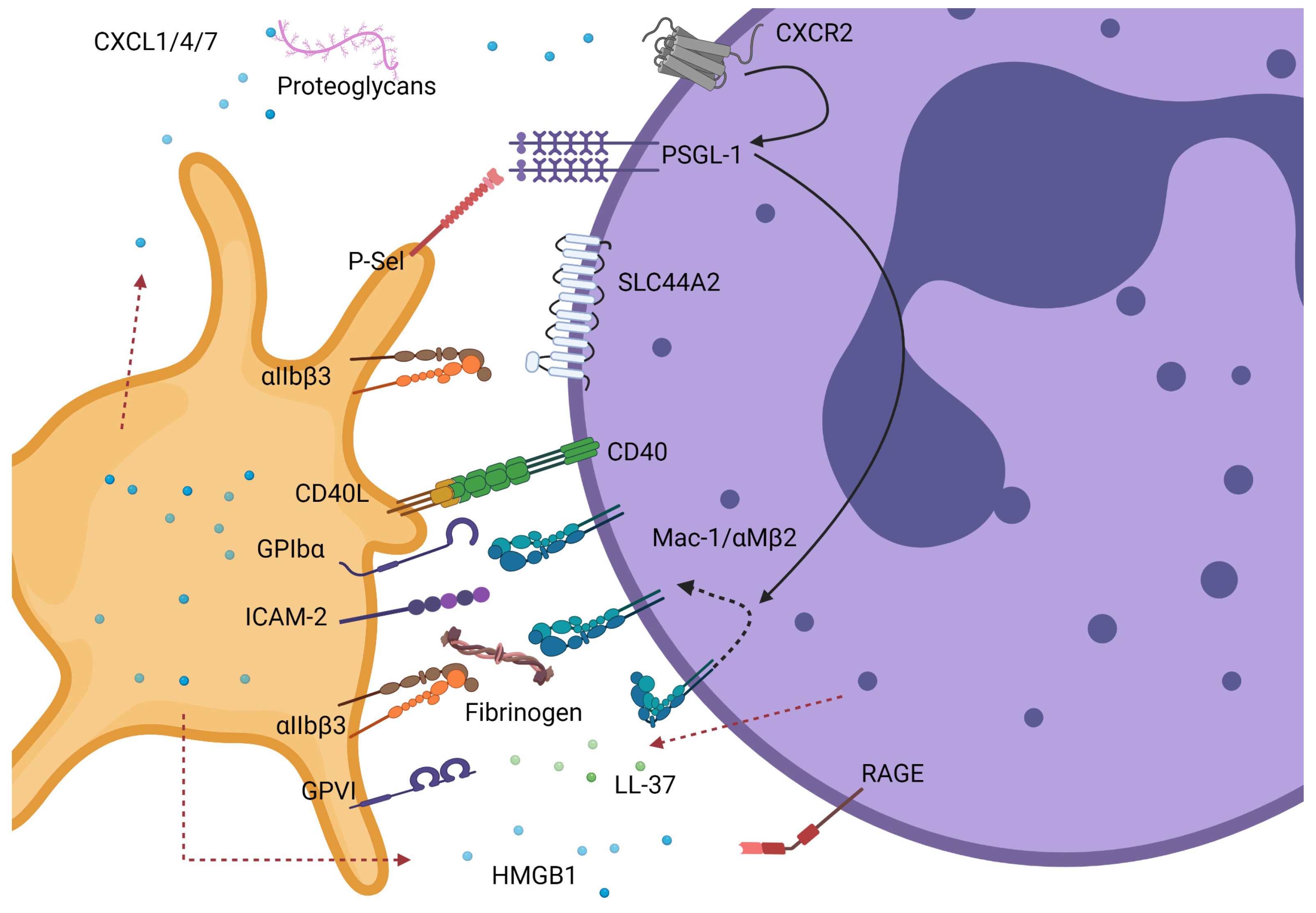
2. Platelet–Neutrophil Complexes Drive Thrombosis and Inflammation
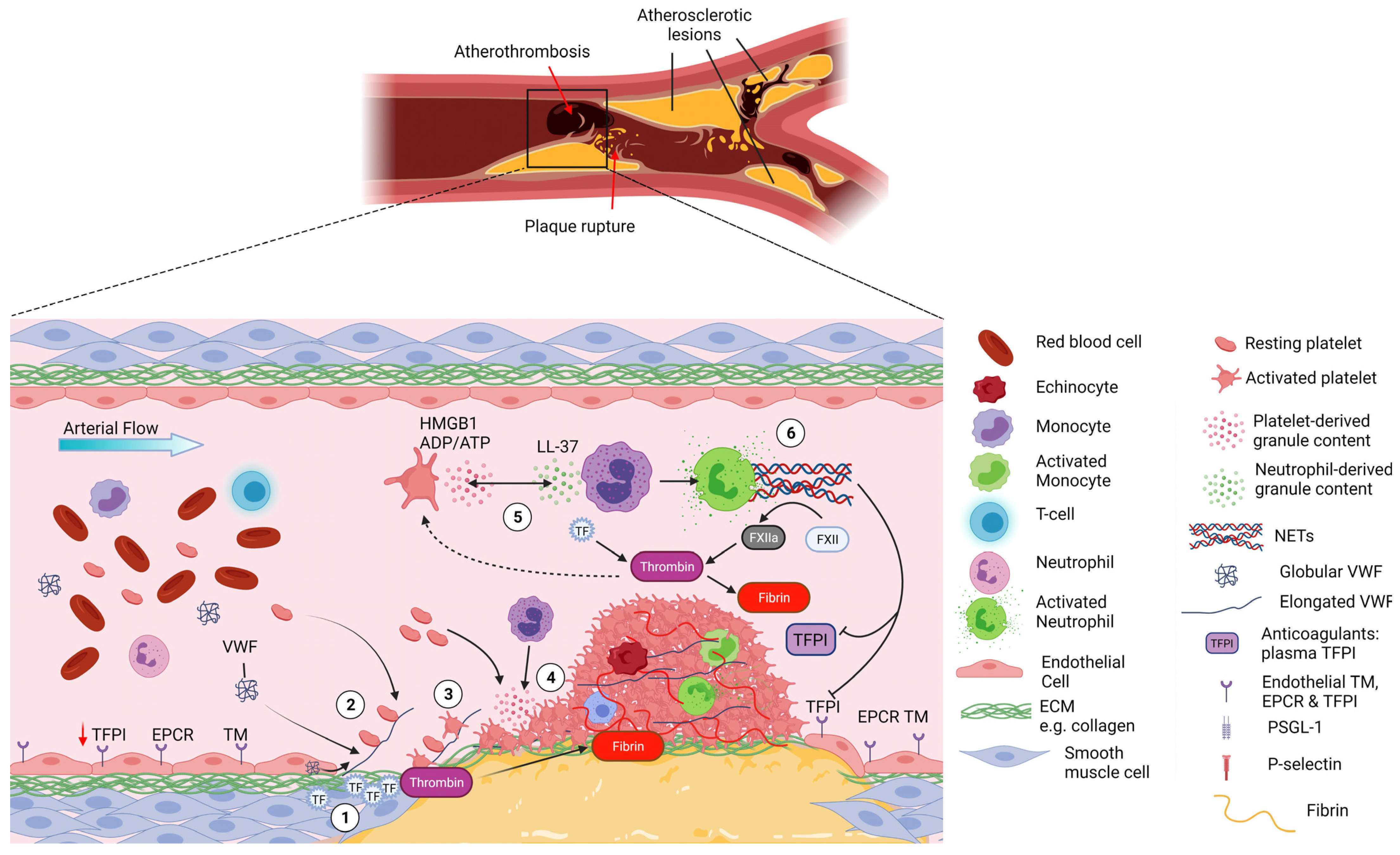
3. Arterial Thrombosis
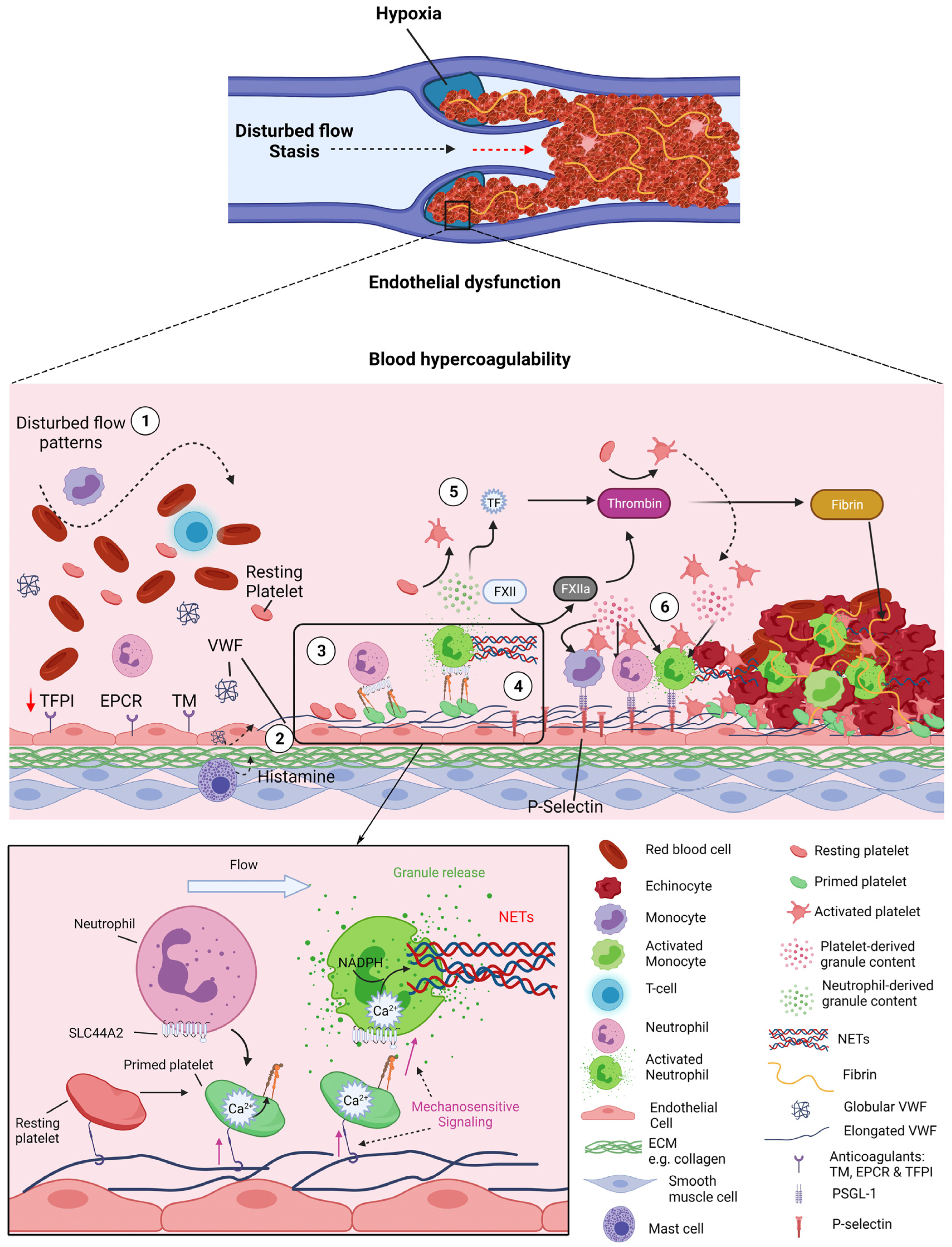
4. Deep Vein Thrombosis
5. Novel Concepts in DVT Initiation—VWF-Primed Platelets Recruit Neutrophils
6. Targeting Platelet–Neutrophil Interaction for Thrombosis Treatment
7. Platelet and Neutrophil Interactions in COVID-19
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Salles, I.I.; Feys, H.B.; Iserbyt, B.F.; De Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Inherited traits affecting platelet function. Blood Rev. 2008, 22, 155–172. [Google Scholar] [CrossRef]
- Kostmann, R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr. Suppl. 1956, 45, 1–78. [Google Scholar] [CrossRef]
- Zeidler, C.; Germeshausen, M.; Klein, C.; Welte, K. Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br. J. Haematol. 2009, 144, 459–467. [Google Scholar] [CrossRef]
- Harding, S.A.; Sommerfield, A.J.; Sarma, J.; Twomey, P.J.; Newby, D.E.; Frier, B.M.; Fox, K.A. Increased CD40 ligand and platelet-monocyte aggregates in patients with type 1 diabetes mellitus. Atherosclerosis 2004, 176, 321–325. [Google Scholar] [CrossRef]
- Lisman, T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef]
- Wrigley, B.J.; Shantsila, E.; Tapp, L.D.; Lip, G.Y. Increased formation of monocyte-platelet aggregates in ischemic heart failure. Circ. Heart Fail. 2013, 6, 127–135. [Google Scholar] [CrossRef]
- Nieswandt, B.; Kleinschnitz, C.; Stoll, G. Ischaemic stroke: A thrombo-inflammatory disease? J. Physiol. 2011, 589, 4115–4123. [Google Scholar] [CrossRef]
- Lievens, D.; Zernecke, A.; Seijkens, T.; Soehnlein, O.; Beckers, L.; Munnix, I.C.; Wijnands, E.; Goossens, P.; van Kruchten, R.; Thevissen, L.; et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 2010, 116, 4317–4327. [Google Scholar] [CrossRef]
- von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Gawaz, M.; Fateh-Moghadam, S.; Pilz, G.; Gurland, H.J.; Werdan, K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur. J. Clin. Investig. 1995, 25, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, 240–253. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B. Blood coagulation. Lancet 2000, 355, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M. Mechanisms initiating platelet thrombus formation. Thromb. Haemost. 1997, 78, 611–616. [Google Scholar] [CrossRef]
- Darbousset, R.; Delierneux, C.; Mezouar, S.; Hego, A.; Lecut, C.; Guillaumat, I.; Riederer, M.A.; Evans, R.J.; Dignat-George, F.; Panicot-Dubois, L.; et al. P2X1 expressed on polymorphonuclear neutrophils and platelets is required for thrombosis in mice. Blood 2014, 124, 2575–2585. [Google Scholar] [CrossRef]
- Darbousset, R.; Thomas, G.M.; Mezouar, S.; Frere, C.; Bonier, R.; Mackman, N.; Renne, T.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 2012, 120, 2133–2143. [Google Scholar] [CrossRef]
- Deppermann, C.; Kubes, P. Platelets and infection. Semin. Immunol. 2016, 28, 536–545. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Platelets in inflammation and infection. Platelets 2015, 26, 286–292. [Google Scholar] [CrossRef]
- Ed Rainger, G.; Chimen, M.; Harrison, M.J.; Yates, C.M.; Harrison, P.; Watson, S.P.; Lordkipanidze, M.; Nash, G.B. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 2015, 26, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Pircher, J.; Engelmann, B.; Massberg, S.; Schulz, C. Platelet-Neutrophil Crosstalk in Atherothrombosis. Thromb. Haemost. 2019, 119, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Schonbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001, 58, 4–43. [Google Scholar] [CrossRef] [PubMed]
- Laszik, Z.; Jansen, P.J.; Cummings, R.D.; Tedder, T.F.; McEver, R.P.; Moore, K.L. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood 1996, 88, 3010–3021. [Google Scholar] [CrossRef]
- Moore, K.L.; Patel, K.D.; Bruehl, R.E.; Li, F.; Johnson, D.A.; Lichenstein, H.S.; Cummings, R.D.; Bainton, D.F.; McEver, R.P. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 1995, 128, 661–671. [Google Scholar] [CrossRef]
- Vandendries, E.R.; Furie, B.C.; Furie, B. Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb. Haemost. 2004, 92, 459–466. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nacher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef]
- Evangelista, V.; Manarini, S.; Sideri, R.; Rotondo, S.; Martelli, N.; Piccoli, A.; Totani, L.; Piccardoni, P.; Vestweber, D.; de Gaetano, G.; et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: Role of PSGL-1 as a signaling molecule. Blood 1999, 93, 876–885. [Google Scholar] [CrossRef]
- Evangelista, V.; Pamuklar, Z.; Piccoli, A.; Manarini, S.; Dell’elba, G.; Pecce, R.; Martelli, N.; Federico, L.; Rojas, M.; Berton, G.; et al. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood 2007, 109, 2461–2469. [Google Scholar] [CrossRef]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef]
- Weber, C.; Springer, T.A. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J. Clin. Investig. 1997, 100, 2085–2093. [Google Scholar] [CrossRef]
- Morgan, J.; Saleem, M.; Ng, R.; Armstrong, C.; Wong, S.S.; Caulton, S.G.; Fickling, A.; Williams, H.E.L.; Munday, A.D.; Lopez, J.A.; et al. Structural basis of the leukocyte integrin Mac-1 I-domain interactions with the platelet glycoprotein Ib. Blood Adv. 2019, 3, 1450–1459. [Google Scholar] [CrossRef]
- Santoso, S.; Sachs, U.J.; Kroll, H.; Linder, M.; Ruf, A.; Preissner, K.T.; Chavakis, T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 2002, 196, 679–691. [Google Scholar] [CrossRef]
- Kuijper, P.H.; Gallardo Tores, H.I.; Lammers, J.W.; Sixma, J.J.; Koenderman, L.; Zwaginga, J.J. Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb. Haemost. 1998, 80, 443–448. [Google Scholar] [CrossRef]
- Duerschmied, D.; Suidan, G.L.; Demers, M.; Herr, N.; Carbo, C.; Brill, A.; Cifuni, S.M.; Mauler, M.; Cicko, S.; Bader, M.; et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 2013, 121, 1008–1015. [Google Scholar] [CrossRef]
- Karshovska, E.; Weber, C.; von Hundelshausen, P. Platelet chemokines in health and disease. Thromb. Haemost. 2013, 110, 894–902. [Google Scholar] [CrossRef]
- Brown, A.J.; Sepuru, K.M.; Sawant, K.V.; Rajarathnam, K. Platelet-Derived Chemokine CXCL7 Dimer Preferentially Exists in the Glycosaminoglycan-Bound Form: Implications for Neutrophil-Platelet Crosstalk. Front. Immunol. 2017, 8, 1248. [Google Scholar] [CrossRef]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Bruhl, M.L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Pircher, J.; Czermak, T.; Ehrlich, A.; Eberle, C.; Gaitzsch, E.; Margraf, A.; Grommes, J.; Saha, P.; Titova, A.; Ishikawa-Ankerhold, H.; et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat. Commun. 2018, 9, 1523. [Google Scholar] [CrossRef]
- Mandel, J.; Casari, M.; Stepanyan, M.; Martyanov, A.; Deppermann, C. Beyond Hemostasis: Platelet Innate Immune Interactions and Thromboinflammation. Int. J. Mol. Sci. 2022, 23, 3868. [Google Scholar] [CrossRef]
- Thalin, C.; Hisada, Y.; Lundstrom, S.; Mackman, N.; Wallen, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Kollnberger, M.; Wakefield, T.W.; Lammle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef]
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Wagner, D.D. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Mackman, N.; Bergmeier, W.; Stouffer, G.A.; Weitz, J.I. Therapeutic strategies for thrombosis: New targets and approaches. Nat. Rev. Drug Discov. 2020, 19, 333–352. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Burger, P.C.; Wagner, D.D. Platelet P-selectin facilitates atherosclerotic lesion development. Blood 2003, 101, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Brand, K.; Gruner, S.; Page, S.; Muller, E.; Muller, I.; Bergmeier, W.; Richter, T.; Lorenz, M.; Konrad, I.; et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J. Exp. Med. 2002, 196, 887–896. [Google Scholar] [CrossRef]
- Majithia, A.; Bhatt, D.L. Novel Antiplatelet Therapies for Atherothrombotic Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 546–557. [Google Scholar] [CrossRef]
- Kuijpers, M.J.; Gilio, K.; Reitsma, S.; Nergiz-Unal, R.; Prinzen, L.; Heeneman, S.; Lutgens, E.; van Zandvoort, M.A.; Nieswandt, B.; Egbrink, M.G.; et al. Complementary roles of platelets and coagulation in thrombus formation on plaques acutely ruptured by targeted ultrasound treatment: A novel intravital model. J. Thromb. Haemost. 2009, 7, 152–161. [Google Scholar] [CrossRef]
- Schulz, C.; Massberg, S. Platelets in atherosclerosis and thrombosis. Handb. Exp. Pharmacol. 2012, 210, 111–133. [Google Scholar] [CrossRef]
- Chernysh, I.N.; Nagaswami, C.; Kosolapova, S.; Peshkova, A.D.; Cuker, A.; Cines, D.B.; Cambor, C.L.; Litvinov, R.I.; Weisel, J.W. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci. Rep. 2020, 10, 5112. [Google Scholar] [CrossRef]
- Chia, S.; Nagurney, J.T.; Brown, D.F.; Raffel, O.C.; Bamberg, F.; Senatore, F.; Wackers, F.J.; Jang, I.K. Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST-elevation myocardial infarction. Am. J. Cardiol. 2009, 103, 333–337. [Google Scholar] [CrossRef]
- Shah, A.D.; Denaxas, S.; Nicholas, O.; Hingorani, A.D.; Hemingway, H. Neutrophil Counts and Initial Presentation of 12 Cardiovascular Diseases: A CALIBER Cohort Study. J. Am. Coll. Cardiol. 2017, 69, 1160–1169. [Google Scholar] [CrossRef]
- Ionita, M.G.; van den Borne, P.; Catanzariti, L.M.; Moll, F.L.; de Vries, J.P.; Pasterkamp, G.; Vink, A.; de Kleijn, D.P. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1842–1848. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef]
- Novotny, J.; Chandraratne, S.; Weinberger, T.; Philippi, V.; Stark, K.; Ehrlich, A.; Pircher, J.; Konrad, I.; Oberdieck, P.; Titova, A.; et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS ONE 2018, 13, e0190728. [Google Scholar] [CrossRef]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; Ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.J.; et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: A report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef]
- Joshi, A.; Schmidt, L.E.; Burnap, S.A.; Lu, R.; Chan, M.V.; Armstrong, P.C.; Baig, F.; Gutmann, C.; Willeit, P.; Santer, P.; et al. Neutrophil-Derived Protein S100A8/A9 Alters the Platelet Proteome in Acute Myocardial Infarction and Is Associated With Changes in Platelet Reactivity. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 49–62. [Google Scholar] [CrossRef]
- Thornton, P.; McColl, B.W.; Greenhalgh, A.; Denes, A.; Allan, S.M.; Rothwell, N.J. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood 2010, 115, 3632–3639. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Pozgajova, M.; Pham, M.; Bendszus, M.; Nieswandt, B.; Stoll, G. Targeting platelets in acute experimental stroke: Impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 2007, 115, 2323–2330. [Google Scholar] [CrossRef]
- Schuhmann, M.K.; Kraft, P.; Bieber, M.; Kollikowski, A.M.; Schulze, H.; Nieswandt, B.; Pham, M.; Stegner, D.; Stoll, G. Targeting Platelet GPVI Plus rt-PA Administration but Not alpha2beta1-Mediated Collagen Binding Protects against Ischemic Brain Damage in Mice. Int. J. Mol. Sci. 2019, 20, 2019. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; De Meyer, S.F.; Schwarz, T.; Austinat, M.; Vanhoorelbeke, K.; Nieswandt, B.; Deckmyn, H.; Stoll, G. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood 2009, 113, 3600–3603. [Google Scholar] [CrossRef]
- Wichaiyo, S.; Parichatikanond, W.; Rattanavipanon, W. Glenzocimab: A GPVI (Glycoprotein VI)-Targeted Potential Antiplatelet Agent for the Treatment of Acute Ischemic Stroke. Stroke 2022, 53, 3506–3513. [Google Scholar] [CrossRef]
- Uphaus, T.; Richards, T.; Weimar, C.; Neugebauer, H.; Poli, S.; Weissenborn, K.; Imray, C.; Michalski, D.; Rashid, H.; Loftus, I.; et al. Revacept, an Inhibitor of Platelet Adhesion in Symptomatic Carotid Stenosis: A Multicenter Randomized Phase II Trial. Stroke 2022, 53, 2718–2729. [Google Scholar] [CrossRef]
- De Meyer, S.F.; Langhauser, F.; Haupeltshofer, S.; Kleinschnitz, C.; Casas, A.I. Thromboinflammation in Brain Ischemia: Recent Updates and Future Perspectives. Stroke 2022, 53, 1487–1499. [Google Scholar] [CrossRef]
- Denorme, F.; Manne, B.K.; Portier, I.; Eustes, A.S.; Kosaka, Y.; Kile, B.T.; Rondina, M.T.; Campbell, R.A. Platelet necrosis mediates ischemic stroke outcome in mice. Blood 2020, 135, 429–440. [Google Scholar] [CrossRef]
- Buck, B.H.; Liebeskind, D.S.; Saver, J.L.; Bang, O.Y.; Yun, S.W.; Starkman, S.; Ali, L.K.; Kim, D.; Villablanca, J.P.; Salamon, N.; et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 2008, 39, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.X.; Kim, H.A.; Lee, S.; Moore, J.P.; Chan, C.T.; Vinh, A.; Gelderblom, M.; Arumugam, T.V.; Broughton, B.R.; Drummond, G.R.; et al. Immune cell infiltration in malignant middle cerebral artery infarction: Comparison with transient cerebral ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 450–459. [Google Scholar] [CrossRef]
- Desilles, J.P.; Syvannarath, V.; Di Meglio, L.; Ducroux, C.; Boisseau, W.; Louedec, L.; Jandrot-Perrus, M.; Michel, J.B.; Mazighi, M.; Ho-Tin-Noe, B. Downstream Microvascular Thrombosis in Cortical Venules Is an Early Response to Proximal Cerebral Arterial Occlusion. J. Am. Heart Assoc. 2018, 7, e007804. [Google Scholar] [CrossRef] [PubMed]
- Laridan, E.; Denorme, F.; Desender, L.; Francois, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, T.; Jin, J.; Liu, Y.; Li, B.; Sun, Q.; Tian, J.; Zhao, H.; Liu, Z.; Ma, S.; et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. eBioMedicine 2020, 53, 102671. [Google Scholar] [CrossRef] [PubMed]
- Staessens, S.; Denorme, F.; Francois, O.; Desender, L.; Dewaele, T.; Vanacker, P.; Deckmyn, H.; Vanhoorelbeke, K.; Andersson, T.; De Meyer, S.F. Structural analysis of ischemic stroke thrombi: Histological indications for therapy resistance. Haematologica 2020, 105, 498–507. [Google Scholar] [CrossRef] [PubMed]
- McCabe, D.J.; Harrison, P.; Mackie, I.J.; Sidhu, P.S.; Purdy, G.; Lawrie, A.S.; Watt, H.; Machin, S.J.; Brown, M.M. Increased platelet count and leucocyte-platelet complex formation in acute symptomatic compared with asymptomatic severe carotid stenosis. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Ruan, Y.; Yuan, C.; Cheng, Q.; Cheng, H.; Zeng, Y.; Chen, Y.; Huang, G.; Chen, H.; He, J. High Neutrophil-to-Platelet Ratio Is Associated With Hemorrhagic Transformation in Patients with Acute Ischemic Stroke. Front. Neurol. 2019, 10, 1310. [Google Scholar] [CrossRef]
- Denorme, F.; Portier, I.; Rustad, J.L.; Cody, M.J.; de Araujo, C.V.; Hoki, C.; Alexander, M.D.; Grandhi, R.; Dyer, M.R.; Neal, M.D.; et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J. Clin. Investig. 2022, 132, e154225. [Google Scholar] [CrossRef]
- Oklu, R.; Albadawi, H.; Watkins, M.T.; Monestier, M.; Sillesen, M.; Wicky, S. Detection of extracellular genomic DNA scaffold in human thrombus: Implications for the use of deoxyribonuclease enzymes in thrombolysis. J. Vasc. Interv. Radiol. 2012, 23, 712–718. [Google Scholar] [CrossRef]
- Longstaff, C.; Varju, I.; Sotonyi, P.; Szabo, L.; Krumrey, M.; Hoell, A.; Bota, A.; Varga, Z.; Komorowicz, E.; Kolev, K. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J. Biol. Chem. 2013, 288, 6946–6956. [Google Scholar] [CrossRef]
- Zhang, S.; Cao, Y.; Du, J.; Liu, H.; Chen, X.; Li, M.; Xiang, M.; Wang, C.; Wu, X.; Liu, L.; et al. Neutrophil extracellular traps contribute to tissue plasminogen activator resistance in acute ischemic stroke. FASEB J. 2021, 35, e21835. [Google Scholar] [CrossRef]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 2020, 11, 2488. [Google Scholar] [CrossRef] [PubMed]
- Pena-Martinez, C.; Duran-Laforet, V.; Garcia-Culebras, A.; Ostos, F.; Hernandez-Jimenez, M.; Bravo-Ferrer, I.; Perez-Ruiz, A.; Ballenilla, F.; Diaz-Guzman, J.; Pradillo, J.M.; et al. Pharmacological Modulation of Neutrophil Extracellular Traps Reverses Thrombotic Stroke tPA (Tissue-Type Plasminogen Activator) Resistance. Stroke 2019, 50, 3228–3237. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Liu, Z.; Chang, L.; Bai, X.; Kang, L.; Cao, Y.; Yang, X.; Yu, H.; Shi, M.J.; et al. Neutrophil extracellular traps promote tPA-induced brain hemorrhage via cGAS in mice with stroke. Blood 2021, 138, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Carminita, E.; Crescence, L.; Brouilly, N.; Altie, A.; Panicot-Dubois, L.; Dubois, C. DNAse-dependent, NET-independent pathway of thrombus formation in vivo. Proc. Natl. Acad. Sci. USA 2021, 118, e2100561118. [Google Scholar] [CrossRef] [PubMed]
- Wendelboe, A.M.; Raskob, G.E. Global Burden of Thrombosis: Epidemiologic Aspects. Circ. Res. 2016, 118, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Grosse, S.D.; Nelson, R.E.; Nyarko, K.A.; Richardson, L.C.; Raskob, G.E. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb. Res. 2016, 137, 3–10. [Google Scholar] [CrossRef]
- House of Commons Health Committee. The Prevention of Venous Thromboembolism in Hospitalised Patients; Second Report of Session 2004-05; House of Commons Health Committee: London, UK, 2005. [Google Scholar]
- Heit, J.A.; Spencer, F.A.; White, R.H. The epidemiology of venous thromboembolism. J. Thromb. Thrombolysis 2016, 41, 3–14. [Google Scholar] [CrossRef]
- Wolberg, A.S.; Rosendaal, F.R.; Weitz, J.I.; Jaffer, I.H.; Agnelli, G.; Baglin, T.; Mackman, N. Venous thrombosis. Nat. Rev. Dis. Prim. 2015, 1, 15006. [Google Scholar] [CrossRef] [PubMed]
- Bagot, C.N.; Arya, R. Virchow and his triad: A question of attribution. Br. J. Haematol. 2008, 143, 180–190. [Google Scholar] [CrossRef]
- Mackman, N. New insights into the mechanisms of venous thrombosis. J. Clin. Investig. 2012, 122, 2331–2336. [Google Scholar] [CrossRef]
- Lund, F.; Diener, L.; Ericsson, J.L. Postmortem intraosseous phlebography as an aid in studies of venous thromboembolism. With application on a geriatric clientele. Angiology 1969, 20, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.G.; Trotman, W.; Wadsworth, M.P.; Taatjes, D.J.; Evans, M.F.; Ittleman, F.P.; Callas, P.W.; Esmon, C.T.; Bovill, E.G. Valves of the deep venous system: An overlooked risk factor. Blood 2009, 114, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.D.; Hoofnagle, M.H.; Bamezai, S.; Oxendine, M.; Lim, L.; Hall, J.D.; Yang, J.; Schultz, S.; Engel, J.D.; Kume, T.; et al. Hemodynamic regulation of perivalvular endothelial gene expression prevents deep venous thrombosis. J. Clin. Investig. 2019, 129, 5489–5500. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hao, H.; Leeper, N.J.; Zhu, L.; Early Career, C. Thrombotic Regulation From the Endothelial Cell Perspectives. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e90–e95. [Google Scholar] [CrossRef] [PubMed]
- Budnik, I.; Brill, A. Immune Factors in Deep Vein Thrombosis Initiation. Trends Immunol. 2018, 39, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef]
- Zapponi, K.C.S.; Orsi, F.A.; Cunha, J.L.R.; de Brito, I.R.; Romano, A.V.C.; Bittar, L.F.; De Paula, E.V.; Penteado, C.F.; Montalvao, S.; Annichino-Bizzacchi, J.M. Neutrophil activation and circulating neutrophil extracellular traps are increased in venous thromboembolism patients for at least one year after the clinical event. J. Thromb. Thrombolysis 2022, 53, 30–42. [Google Scholar] [CrossRef]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production through Interleukin-1alpha and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, J.; Zhang, G.; Wu, C.; Abdel-Latif, A.; Smyth, S.S.; Shiroishi, T.; Mackman, N.; Wei, Y.; Tao, M.; et al. Inflammasome activation promotes venous thrombosis through pyroptosis. Blood Adv. 2021, 5, 2619–2623. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; Ponomaryov, T.; De Prendergast, A.; Whitworth, K.; Smith, C.W.; Khan, A.O.; Kavanagh, D.; Brill, A. Neutrophil extracellular traps and inflammasomes cooperatively promote venous thrombosis in mice. Blood Adv. 2021, 5, 2319–2324. [Google Scholar] [CrossRef]
- Wang, X.; Smith, P.L.; Hsu, M.Y.; Gailani, D.; Schumacher, W.A.; Ogletree, M.L.; Seiffert, D.A. Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. J. Thromb. Haemost. 2006, 4, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.K.; Kisucka, J.; Lamb, C.B.; Bergmeier, W.; Wagner, D.D. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood 2007, 109, 2424–2429. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Vreys, I.; Stassen, J.M.; Yoshimoto, R.; Vermylen, J.; Hoylaerts, M.F. Antagonism of vWF inhibits both injury induced arterial and venous thrombosis in the hamster. Thromb. Haemost. 1998, 79, 202–210. [Google Scholar] [CrossRef]
- Lei, X.; Reheman, A.; Hou, Y.; Zhou, H.; Wang, Y.; Marshall, A.H.; Liang, C.; Dai, X.; Li, B.X.; Vanhoorelbeke, K.; et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb. Haemost. 2014, 111, 279–289. [Google Scholar] [CrossRef]
- Dyer, M.R.; Chen, Q.; Haldeman, S.; Yazdani, H.; Hoffman, R.; Loughran, P.; Tsung, A.; Zuckerbraun, B.S.; Simmons, R.L.; Neal, M.D. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 2018, 8, 2068. [Google Scholar] [CrossRef]
- Puhr-Westerheide, D.; Schink, S.J.; Fabritius, M.; Mittmann, L.; Hessenauer, M.E.T.; Pircher, J.; Zuchtriegel, G.; Uhl, B.; Holzer, M.; Massberg, S.; et al. Neutrophils promote venular thrombosis by shaping the rheological environment for platelet aggregation. Sci. Rep. 2019, 9, 15932. [Google Scholar] [CrossRef]
- Yago, T.; Liu, Z.; Ahamed, J.; McEver, R.P. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood 2018, 132, 1426–1437. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu-Bercu, A.; Grassi, L.; Frontini, M.; Salles-Crawley, I.; Woollard, K.; Crawley, J.T. Activated αIIbβ3 on platelets mediates flow-dependent NETosis via SLC44A2. eLife 2020, 9, e53353. [Google Scholar] [CrossRef] [PubMed]
- Nair, T.S.; Kommareddi, P.K.; Galano, M.M.; Miller, D.M.; Kakaraparthi, B.N.; Telian, S.A.; Arts, H.A.; El-Kashlan, H.; Kilijanczyk, A.; Lassig, A.A.; et al. SLC44A2 single nucleotide polymorphisms, isoforms, and expression: Association with severity of Meniere’s disease? Genomics 2016, 108, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Nair, T.S.; Kozma, K.E.; Hoefling, N.L.; Kommareddi, P.K.; Ueda, Y.; Gong, T.W.; Lomax, M.I.; Lansford, C.D.; Telian, S.A.; Satar, B.; et al. Identification and characterization of choline transporter-like protein 2, an inner ear glycoprotein of 68 and 72 kDa that is the target of antibody-induced hearing loss. J. Neurosci. 2004, 24, 1772–1779. [Google Scholar] [CrossRef]
- Nakamura, T.; Fujiwara, R.; Ishiguro, N.; Oyabu, M.; Nakanishi, T.; Shirasaka, Y.; Maeda, T.; Tamai, I. Involvement of choline transporter-like proteins, CTL1 and CTL2, in glucocorticoid-induced acceleration of phosphatidylcholine synthesis via increased choline uptake. Biol. Pharm. Bull. 2010, 33, 691–696. [Google Scholar] [CrossRef]
- Iwao, B.; Yara, M.; Hara, N.; Kawai, Y.; Yamanaka, T.; Nishihara, H.; Inoue, T.; Inazu, M. Functional expression of choline transporter like-protein 1 (CTL1) and CTL2 in human brain microvascular endothelial cells. Neurochem. Int. 2016, 93, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.A.; Mastrangelo, M.A.; Ture, S.K.; Smith, C.O.; Loelius, S.G.; Berg, R.A.; Shi, X.; Burke, R.M.; Spinelli, S.L.; Cameron, S.J.; et al. The choline transporter Slc44a2 controls platelet activation and thrombosis by regulating mitochondrial function. Nat. Commun. 2020, 11, 3479. [Google Scholar] [CrossRef] [PubMed]
- Nair, T.S.; Kakaraparthi, B.N.; Yang, L.; Lu, L.; Thomas, T.B.; Morris, A.C.; Kommareddi, P.; Kanicki, A.; Carey, T.E. Slc44a2 deletion alters tetraspanin and N-cadherin expression: Reduced adhesion and enhanced proliferation in cultured mesenchymal lung cells. Tissue Cell 2021, 73, 101599. [Google Scholar] [CrossRef]
- Kommareddi, P.; Nair, T.; Kakaraparthi, B.N.; Galano, M.M.; Miller, D.; Laczkovich, I.; Thomas, T.; Lu, L.; Rule, K.; Kabara, L.; et al. Hair Cell Loss, Spiral Ganglion Degeneration, and Progressive Sensorineural Hearing Loss in Mice with Targeted Deletion of Slc44a2/Ctl2. J. Assoc. Res. Otolaryngol. 2015, 16, 695–712. [Google Scholar] [CrossRef]
- Bayat, B.; Tjahjono, Y.; Berghofer, H.; Werth, S.; Deckmyn, H.; De Meyer, S.F.; Sachs, U.J.; Santoso, S. Choline Transporter-Like Protein-2: New von Willebrand Factor-Binding Partner Involved in Antibody-Mediated Neutrophil Activation and Transfusion-Related Acute Lung Injury. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1616–1622. [Google Scholar] [CrossRef]
- Chiaretti, S.; Burton, M.; Hassel, P.; Radenkovic, F.; Devikashri, N.; Sultana, A.J.; Temple, F.T.; Dean, M.M.; Tung, J.P. Human neutrophil antigen 3 genotype impacts neutrophil-mediated endothelial cell cytotoxicity in a two-event model of TRALI. Blood Transfus. 2022, 20, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Chasman, D.I.; de Haan, H.; Tang, W.; Lindstrom, S.; Weng, L.C.; de Andrade, M.; de Visser, M.C.; Wiggins, K.L.; Suchon, P.; et al. Meta-analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am. J. Hum. Genet. 2015, 96, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Saut, N.; Greliche, N.; Dina, C.; Lambert, J.C.; Perret, C.; Cohen, W.; Oudot-Mellakh, T.; Antoni, G.; Alessi, M.C.; et al. Genetics of venous thrombosis: Insights from a new genome wide association study. PLoS ONE 2011, 6, e25581. [Google Scholar] [CrossRef] [PubMed]
- Hinds, D.A.; Buil, A.; Ziemek, D.; Martinez-Perez, A.; Malik, R.; Folkersen, L.; Germain, M.; Malarstig, A.; Brown, A.; Soria, J.M.; et al. Genome-wide association analysis of self-reported events in 6135 individuals and 252 827 controls identifies 8 loci associated with thrombosis. Hum. Mol. Genet. 2016, 25, 1867–1874. [Google Scholar] [CrossRef]
- Tilburg, J.; Adili, R.; Nair, T.S.; Hawley, M.E.; Tuk, D.C.; Jackson, M.; Spronk, H.M.; Versteeg, H.H.; Carey, T.E.; van Vlijmen, B.J.M.; et al. Characterization of hemostasis in mice lacking the novel thrombosis susceptibility gene Slc44a2. Thromb. Res. 2018, 171, 155–159. [Google Scholar] [CrossRef]
- Tilburg, J.; Coenen, D.M.; Zirka, G.; Dolleman, S.; van Oeveren-Rietdijk, A.M.; Karel, M.F.A.; de Boer, H.C.; Cosemans, J.; Versteeg, H.H.; Morange, P.E.; et al. SLC44A2 deficient mice have a reduced response in stenosis but not in hypercoagulability driven venous thrombosis. J. Thromb. Haemost. 2020, 18, 1714–1727. [Google Scholar] [CrossRef]
- Apipongrat, D.; Numbenjapon, T.; Prayoonwiwat, W.; Arnutti, P.; Nathalang, O. Association between SLC44A2 rs2288904 polymorphism and risk of recurrent venous thromboembolism among Thai patients. Thromb. Res. 2019, 174, 163–165. [Google Scholar] [CrossRef]
- Lindstrom, S.; Wang, L.; Smith, E.N.; Gordon, W.; van Hylckama Vlieg, A.; de Andrade, M.; Brody, J.A.; Pattee, J.W.; Haessler, J.; Brumpton, B.M.; et al. Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood 2019, 134, 1645–1657. [Google Scholar] [CrossRef]
- Zhi, L.; Feng, W.; Liang, J.; Zhong, Q.; Ren, L.; Ma, J.; Yao, S. The Effect of Common Variants in SLC44A2 on the Contribution to the Risk of Deep Cein Thrombosis after Orthopedic Surgery. J. Atheroscler. Thromb. 2021, 28, 293–303. [Google Scholar] [CrossRef]
- Zirka, G.; Robert, P.; Tilburg, J.; Tishkova, V.; Maracle, C.X.; Legendre, P.; van Vlijmen, B.J.M.; Alessi, M.C.; Lenting, P.J.; Morange, P.E.; et al. Impaired adhesion of neutrophils expressing Slc44a2/HNA-3b to VWF protects against NETosis under venous shear rates. Blood 2021, 137, 2256–2266. [Google Scholar] [CrossRef]
- Denis, C.; Methia, N.; Frenette, P.S.; Rayburn, H.; Ullman-Cullere, M.; Hynes, R.O.; Wagner, D.D. A mouse model of severe von Willebrand disease: Defects in hemostasis and thrombosis. Proc. Natl. Acad. Sci. USA 1998, 95, 9524–9529. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.; Russell, S.; Ruggeri, Z.M. Generation and rescue of a murine model of platelet dysfunction: The Bernard-Soulier syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 2803–2808. [Google Scholar] [CrossRef] [PubMed]
- Poujol, C.; Ware, J.; Nieswandt, B.; Nurden, A.T.; Nurden, P. Absence of GPIbalpha is responsible for aberrant membrane development during megakaryocyte maturation: Ultrastructural study using a transgenic model. Exp. Hematol. 2002, 30, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F. Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet J. Rare Dis. 2006, 1, 46. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, T.; Russell, S.; Ware, J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood 2002, 100, 2102–2107. [Google Scholar] [CrossRef]
- Jain, S.; Zuka, M.; Liu, J.; Russell, S.; Dent, J.; Guerrero, J.A.; Forsyth, J.; Maruszak, B.; Gartner, T.K.; Felding-Habermann, B.; et al. Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 9024–9028. [Google Scholar] [CrossRef]
- Constantinescu-Bercu, A.; Wang, Y.A.; Woollard, K.J.; Mangin, P.; Vanhoorelbeke, K.; Crawley, J.T.B.; Salles-Crawley, I.I. The GPIbalpha intracellular tail—Role in transducing VWF- and collagen/GPVI-mediated signaling. Haematologica 2022, 107, 933–946. [Google Scholar] [CrossRef]
- Thomas, A.A.; Wright, H.; Chan, K.; Ross, H.; Prasad, P.; Goodwin, A.; Holmes, C.E. Safety of apixaban for venous thromboembolic primary prophylaxis in patients with newly diagnosed malignant glioma. J. Thromb. Thrombolysis 2022, 53, 479–484. [Google Scholar] [CrossRef]
- Chan, N.C.; Eikelboom, J.W.; Weitz, J.I. Evolving Treatments for Arterial and Venous Thrombosis: Role of the Direct Oral Anticoagulants. Circ. Res. 2016, 118, 1409–1424. [Google Scholar] [CrossRef]
- Schulman, S.; Re, M.; Investigators, R.-S.T. Extended anticoagulation in venous thromboembolism. N. Engl. J. Med. 2013, 368, 2329. [Google Scholar] [CrossRef]
- Schulman, S.; Kakkar, A.K.; Goldhaber, S.Z.; Schellong, S.; Eriksson, H.; Mismetti, P.; Christiansen, A.V.; Friedman, J.; Le Maulf, F.; Peter, N.; et al. Treatment of acute venous thromboembolism with dabigatran or warfarin and pooled analysis. Circulation 2014, 129, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Candeloro, M.; van Es, N.; Cantor, N.; Schulman, S.; Carrier, M.; Ageno, W.; Aibar, J.; Donadini, M.P.; Bavalia, R.; Arsenault, M.P.; et al. Recurrent bleeding and thrombotic events after resumption of oral anticoagulants following gastrointestinal bleeding: Communication from the ISTH SSC Subcommittee on Control of Anticoagulation. J. Thromb. Haemost. 2021, 19, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Spronk, H.M.H.; Stouffer, G.A.; Ten Cate, H. Dual Anticoagulant and Antiplatelet Therapy for Coronary Artery Disease and Peripheral Artery Disease Patients. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 726–732. [Google Scholar] [CrossRef]
- Floyd, C.N. Dual Antiplatelet Therapy in Coronary Artery Disease: Comparison Between ACC/AHA 2016 and ESC 2017 Guidelines. Eur. Cardiol. 2020, 15, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, R.; Wu, O.; Briere, J.B.; Bowrin, K.; Borkowska, K.; Jakubowska, A.; Taieb, V.; Toumi, M.; Huelsebeck, M. Antithrombotic Treatments in Patients with Chronic Coronary Artery Disease or Peripheral Artery Disease: A Systematic Review of Randomised Controlled Trials. Cardiovasc. Ther. 2020, 2020, 3057168. [Google Scholar] [CrossRef]
- Tzoran, I.; Brenner, B.; Sakharov, G.; Trujillo-Santos, J.; Lorenzo, A.; Madridano, O.; Lopez-Saez, J.B.; Monreal, M.; RIETE Investigators. Clinical outcome in patients with venous thromboembolism receiving concomitant anticoagulant and antiplatelet therapy. Eur. J. Intern. Med. 2014, 25, 821–825. [Google Scholar] [CrossRef]
- Diep, R.; Garcia, D. Does aspirin prevent venous thromboembolism? Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 634–641. [Google Scholar] [CrossRef]
- Becattini, C.; Agnelli, G.; Schenone, A.; Eichinger, S.; Bucherini, E.; Silingardi, M.; Bianchi, M.; Moia, M.; Ageno, W.; Vandelli, M.R.; et al. Aspirin for preventing the recurrence of venous thromboembolism. N. Engl. J. Med. 2012, 366, 1959–1967. [Google Scholar] [CrossRef]
- Paciullo, F.; Bury, L.; Noris, P.; Falcinelli, E.; Melazzini, F.; Orsini, S.; Zaninetti, C.; Abdul-Kadir, R.; Obeng-Tuudah, D.; Heller, P.G.; et al. Antithrombotic prophylaxis for surgery-associated venous thromboembolism risk in patients with inherited platelet disorders. The SPATA-DVT Study. Haematologica 2020, 105, 1948–1956. [Google Scholar] [CrossRef]
- Weitz, J.I.; Chan, N.C. Novel antithrombotic strategies for treatment of venous thromboembolism. Blood 2020, 135, 351–359. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Intrinsic Pathway of Coagulation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 331–338. [Google Scholar] [CrossRef] [PubMed]
- David, T.; Kim, Y.C.; Ely, L.K.; Rondon, I.; Gao, H.; O’Brien, P.; Bolt, M.W.; Coyle, A.J.; Garcia, J.L.; Flounders, E.A.; et al. Factor XIa-specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Sci. Transl. Med. 2016, 8, 353ra112. [Google Scholar] [CrossRef]
- Jordan, K.R.; Wyatt, C.R.; Fallon, M.E.; Woltjer, R.; Neuwelt, E.A.; Cheng, Q.; Gailani, D.; Lorentz, C.; Tucker, E.I.; McCarty, O.J.T.; et al. Pharmacological reduction of coagulation factor XI reduces macrophage accumulation and accelerates deep vein thrombosis resolution in a mouse model of venous thrombosis. J. Thromb. Haemost. 2022, 20, 2035–2045. [Google Scholar] [CrossRef] [PubMed]
- Verhamme, P.; Yi, B.A.; Segers, A.; Salter, J.; Bloomfield, D.; Buller, H.R.; Raskob, G.E.; Weitz, J.I.; Investigators, A.-T. Abelacimab for Prevention of Venous Thromboembolism. N. Engl. J. Med. 2021, 385, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Strony, J.; Ageno, W.; Gailani, D.; Hylek, E.M.; Lassen, M.R.; Mahaffey, K.W.; Notani, R.S.; Roberts, R.; Segers, A.; et al. Milvexian for the Prevention of Venous Thromboembolism. N. Engl. J. Med. 2021, 385, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Revenko, A.S.; Gao, D.; Crosby, J.R.; Bhattacharjee, G.; Zhao, C.; May, C.; Gailani, D.; Monia, B.P.; MacLeod, A.R. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood 2011, 118, 5302–5311. [Google Scholar] [CrossRef]
- Zhang, H.; Lowenberg, E.C.; Crosby, J.R.; MacLeod, A.R.; Zhao, C.; Gao, D.; Black, C.; Revenko, A.S.; Meijers, J.C.; Stroes, E.S.; et al. Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: A novel antithrombotic strategy with lowered bleeding risk. Blood 2010, 116, 4684–4692. [Google Scholar] [CrossRef]
- Weitz, J.I.; Bauersachs, R.; Becker, B.; Berkowitz, S.D.; Freitas, M.C.S.; Lassen, M.R.; Metzig, C.; Raskob, G.E. Effect of Osocimab in Preventing Venous Thromboembolism Among Patients Undergoing Knee Arthroplasty: The FOXTROT Randomized Clinical Trial. JAMA 2020, 323, 130–139. [Google Scholar] [CrossRef]
- Perera, V.; Luettgen, J.M.; Wang, Z.; Frost, C.E.; Yones, C.; Russo, C.; Lee, J.; Zhao, Y.; LaCreta, F.P.; Ma, X.; et al. First-in-human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS-962212, a direct, reversible, small molecule factor XIa inhibitor in non-Japanese and Japanese healthy subjects. Br. J. Clin. Pharmacol. 2018, 84, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Guan, X.; Hu, P.; Dong, Y.; Zhu, Y.; Zhang, T.; Zou, J.; Zhang, S. First-In-Human Study to Assess the Safety, Pharmacokinetics, and Pharmacodynamics of SHR2285, a Small-Molecule Factor XIa Inhibitor in Healthy Subjects. Front. Pharmacol. 2022, 13, 821363. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, H.; Shi, C.; Erhardt, P.W.; Pavlovsky, A.; Soloviev, D.A.; Bledzka, K.; Ustinov, V.; Zhu, L.; Qin, J.; et al. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat. Commun. 2017, 8, 15559. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.J.; Park, D.D.; Park, S.S.; Haller, C.A.; Chen, J.; Dai, E.; Liu, L.; Mandhapati, A.R.; Eradi, P.; Dhakal, B.; et al. A PSGL-1 glycomimetic reduces thrombus burden without affecting hemostasis. Blood 2021, 138, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.A.; Wrobleski, S.K.; Alvarado, C.M.; Hawley, A.E.; Doornbos, N.K.; Lester, P.A.; Lowe, S.E.; Gabriel, J.E.; Roelofs, K.J.; Henke, P.K.; et al. P-selectin inhibition therapeutically promotes thrombus resolution and prevents vein wall fibrosis better than enoxaparin and an inhibitor to von Willebrand factor. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.R.; Myers, D.D., Jr.; Wrobleski, S.K.; Zajkowski, P.J.; Hawley, A.E.; Bedard, P.W.; Ballard, N.E.; Londy, F.J.; Kaila, N.; Vlasuk, G.P.; et al. Prophylactic P-selectin inhibition with PSI-421 promotes resolution of venous thrombosis without anticoagulation. Thromb. Haemost. 2008, 99, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Ramacciotti, E.; Myers, D.D., Jr.; Wrobleski, S.K.; Deatrick, K.B.; Londy, F.J.; Rectenwald, J.E.; Henke, P.K.; Schaub, R.G.; Wakefield, T.W. P-selectin/ PSGL-1 inhibitors versus enoxaparin in the resolution of venous thrombosis: A meta-analysis. Thromb. Res. 2010, 125, e138–e142. [Google Scholar] [CrossRef]
- Mertens, P.; Maes, A.; Nuyts, J.; Belmans, A.; Desmet, W.; Esplugas, E.; Charlier, F.; Figueras, J.; Sambuceti, G.; Schwaiger, M.; et al. Recombinant P-selectin glycoprotein ligand-immunoglobulin, a P-selectin antagonist, as an adjunct to thrombolysis in acute myocardial infarction. The P-Selectin Antagonist Limiting Myonecrosis (PSALM) trial. Am. Heart J. 2006, 152, 125.e1–125.e8. [Google Scholar] [CrossRef]
- Tardif, J.C.; Tanguay, J.F.; Wright, S.R.; Duchatelle, V.; Petroni, T.; Gregoire, J.C.; Ibrahim, R.; Heinonen, T.M.; Robb, S.; Bertrand, O.F.; et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: Results of the SELECT-ACS trial. J. Am. Coll. Cardiol. 2013, 61, 2048–2055. [Google Scholar] [CrossRef]
- Stahli, B.E.; Gebhard, C.; Duchatelle, V.; Cournoyer, D.; Petroni, T.; Tanguay, J.F.; Robb, S.; Mann, J.; Guertin, M.C.; Wright, R.S.; et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage After Percutaneous Coronary Intervention According to Timing of Infusion: Insights From the SELECT-ACS Trial. J. Am. Heart Assoc. 2016, 5, e004255. [Google Scholar] [CrossRef]
- Nayak, L.; Sweet, D.R.; Thomas, A.; Lapping, S.D.; Kalikasingh, K.; Madera, A.; Vinayachandran, V.; Padmanabhan, R.; Vasudevan, N.T.; Myers, J.T.; et al. A targetable pathway in neutrophils mitigates both arterial and venous thrombosis. Sci. Transl. Med. 2022, 14, eabj7465. [Google Scholar] [CrossRef]
- Sudo, M.; Xu, J.; Mitani, K.; Jimbo, M.; Tsutsui, H.; Hatano, E.; Fujimoto, J. Antithrombin Together with NETs Inhibitor Protected Against Postoperative Adhesion Formation in Mice. Cell. Physiol. Biochem. 2021, 55, 400–412. [Google Scholar] [CrossRef]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.M.; Voorberg, J.; Nicolaes, G.A.F. PAD4 takes charge during neutrophil activation: Impact of PAD4 mediated NET formation on immune-mediated disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sonkar, V.K.; Swamy, J.; Ahmed, A.; Sharathkumar, A.A.; Pierce, G.L.; Dayal, S. DNase 1 Protects From Increased Thrombin Generation and Venous Thrombosis During Aging: Cross-Sectional Study in Mice and Humans. J. Am. Heart Assoc. 2022, 11, e021188. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, X.; Lv, K.; Zheng, G.; Wang, H.; Chen, S.; Huang, L.; Liu, Y.; Zhang, Y.; Tang, Z.; et al. Asebogenin suppresses thrombus formation via inhibition of Syk phosphorylation. Br. J. Pharmacol. 2023, 180, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knobl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Dai, K.; Bodnar, R.; Berndt, M.C.; Du, X. A critical role for 14-3-3zeta protein in regulating the VWF binding function of platelet glycoprotein Ib-IX and its therapeutic implications. Blood 2005, 106, 1975–1981. [Google Scholar] [CrossRef]
- Chen, Y.; Ruggeri, Z.M.; Du, X. 14-3-3 proteins in platelet biology and glycoprotein Ib-IX signaling. Blood 2018, 131, 2436–2448. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. eClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 2021, 88, 15–27. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Barrett, T.J.; Cornwell, M.; Myndzar, K.; Rolling, C.C.; Xia, Y.; Drenkova, K.; Biebuyck, A.; Fields, A.T.; Tawil, M.; Luttrell-Williams, E.; et al. Platelets amplify endotheliopathy in COVID-19. Sci. Adv. 2021, 7, eabh2434. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; De Lorenzo, R.; Clementi, N.; Antonia Diotti, R.; Criscuolo, E.; Godino, C.; Tresoldi, C.; Angels For Covid-Bio, B.S.G.B.; Bonini, C.; Clementi, M.; et al. Unconventional CD147-dependent platelet activation elicited by SARS-CoV-2 in COVID-19. J. Thromb. Haemost. 2022, 20, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Joppich, M.; Hoffknecht, M.L.; Gold, C.; Engel, A.; Polewka, V.; Muenchhoff, M.; et al. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 2021, 19, 574–581. [Google Scholar] [CrossRef]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets contribute to disease severity in COVID-19. J. Thromb. Haemost. 2021, 19, 3139–3153. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Sturzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. eBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and COVID-19: Nots, NETs, and knots. J. Exp. Med. 2020, 217, e20201439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890.e882. [Google Scholar] [CrossRef] [PubMed]
- Colicchia, M.; Schrottmaier, W.C.; Perrella, G.; Reyat, J.S.; Begum, J.; Slater, A.; Price, J.; Clark, J.C.; Zhi, Z.; Simpson, M.; et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbalpha. Blood 2022, 140, 2626–2643. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, M.; Luo, G.; Wu, X.; Su, B.; Zhao, L.; Zhang, S.; Chen, X.; Jia, M.; Zhu, J.; et al. The Inflammatory Factors Associated with Disease Severity to Predict COVID-19 Progression. J. Immunol. 2021, 206, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Navaz, S.A.; Hoy, C.K.; Harbaugh, A.; Gockman, K.; Zuo, M.; Madison, J.A.; Shi, H.; Kanthi, Y.; et al. Autoantibodies stabilize neutrophil extracellular traps in COVID-19. JCI Insight 2021, 6, e150111. [Google Scholar] [CrossRef] [PubMed]
- Le Joncour, A.; Biard, L.; Vautier, M.; Bugaut, H.; Mekinian, A.; Maalouf, G.; Vieira, M.; Marcelin, A.G.; Rosenzwajg, M.; Klatzmann, D.; et al. Neutrophil-Platelet and Monocyte-Platelet Aggregates in COVID-19 Patients. Thromb. Haemost. 2020, 120, 1733–1735. [Google Scholar] [CrossRef]
- Koupenova, M. Potential role of platelets in COVID-19: Implications for thrombosis. Res. Pract. Thromb. Haemost. 2020, 4, 737–740. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Tanriverdi, K.; Somasundaran, M.; Liu, P.; Soofi, S.; Bhandari, R.; Godwin, M.; Parsi, K.M.; et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ. Res. 2021, 129, 631–646. [Google Scholar] [CrossRef]
- Bury, L.; Camilloni, B.; Castronari, R.; Piselli, E.; Malvestiti, M.; Borghi, M.; KuchiBotla, H.; Falcinelli, E.; Petito, E.; Amato, F.; et al. Search for SARS-CoV-2 RNA in platelets from COVID-19 patients. Platelets 2021, 32, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Mellett, L.; Khader, S.A. S100A8/A9 in COVID-19 pathogenesis: Impact on clinical outcomes. Cytokine Growth Factor Rev. 2022, 63, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zuo, Y.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Woodward, W.; Lezak, S.P.; Lugogo, N.L.; et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 2021, 109, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mereweather, L.J.; Constantinescu-Bercu, A.; Crawley, J.T.B.; Salles-Crawley, I.I. Platelet–Neutrophil Crosstalk in Thrombosis. Int. J. Mol. Sci. 2023, 24, 1266. https://doi.org/10.3390/ijms24021266
Mereweather LJ, Constantinescu-Bercu A, Crawley JTB, Salles-Crawley II. Platelet–Neutrophil Crosstalk in Thrombosis. International Journal of Molecular Sciences. 2023; 24(2):1266. https://doi.org/10.3390/ijms24021266
Chicago/Turabian StyleMereweather, Laura J., Adela Constantinescu-Bercu, James T. B. Crawley, and Isabelle I. Salles-Crawley. 2023. "Platelet–Neutrophil Crosstalk in Thrombosis" International Journal of Molecular Sciences 24, no. 2: 1266. https://doi.org/10.3390/ijms24021266
APA StyleMereweather, L. J., Constantinescu-Bercu, A., Crawley, J. T. B., & Salles-Crawley, I. I. (2023). Platelet–Neutrophil Crosstalk in Thrombosis. International Journal of Molecular Sciences, 24(2), 1266. https://doi.org/10.3390/ijms24021266