
Liver Regeneration and Immunity: A Tale to Tell

 , , , and
, , , and 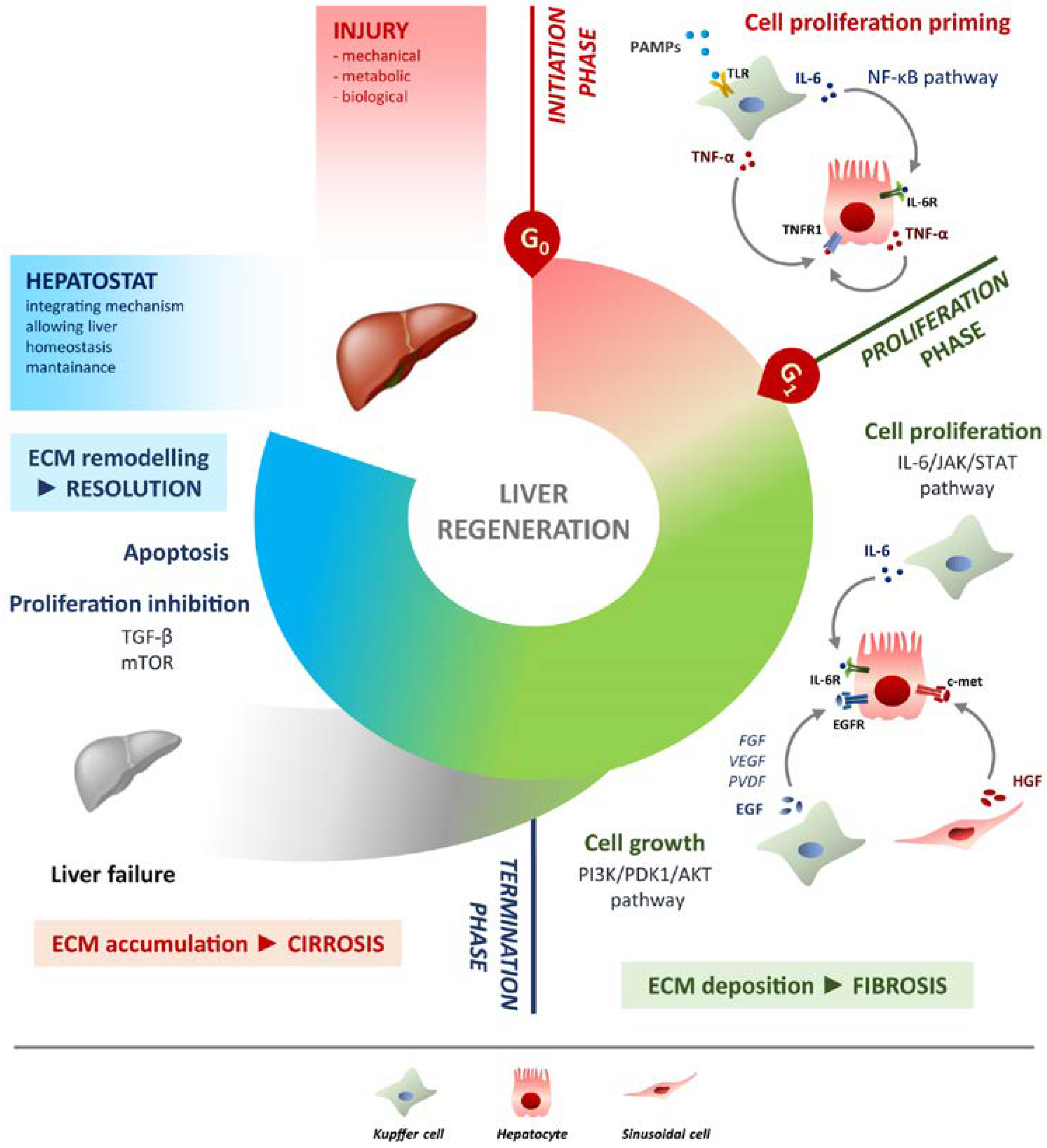{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Liver Injuries
2.1. Acute Liver Disease
2.2. Chronic Liver Diseases
3. Liver Regeneration
4. Liver Regeneration Step by Step
4.1. Initiation Phase
4.2. Proliferation Phase
4.3. Termination Phase
5. Molecular Mechanisms of Liver Regeneration
5.1. Hepatocytes Proliferation
5.2. Hepatocytes Growth
5.3. IL-17: A Balancer in Liver Regeneration
6. Recruitment of Immune Cells in Response to Liver Damage: The Balancing Process
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vlahcevic, Z.R.; Heuman, D.M.; Hylemon, P.B. Regulation of Bile Acid Synthesis. Hepatology 1991, 13, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T. Comparative Patterns of Drug Metabolism. Fed. Proc. 1967, 26, 1029–1039. [Google Scholar]
- Schreiber, G. The Synthesis and Secretion of Plasma Proteins in the Liver. Pathology 1978, 10, 394. [Google Scholar] [CrossRef]
- Grant, D.M. Detoxification Pathways in the Liver. J. Inherit. Metab. Dis. 1991, 421–430. [Google Scholar] [CrossRef]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The Liver as an Immunological Organ. Hepatology 2006, 43 (Suppl. 1), S54–S62. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Mueller, J.R.; Seidler, S.; Luedde, T.; Trautwein, C.; Tacke, F. Frequency and Phenotype of Human Circulating and Intrahepatic Natural Killer Cell Subsets Is Differentially Regulated According to Stage of Chronic Liver Disease. Digestion 2013, 88, 1–16. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F.; et al. Tissue-Resident Macrophages Originate from Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Liu, C.L.; Lo, C.M.; Lam, B.K.; Lee, E.W.; Wong, Y.; Fan, S.T. Estimating Liver Weight of Adults by Body Weight and Gender. World J. Gastroenterol. 2006, 12, 2217–2222. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Hepatostat: Liver Regeneration and Normal Liver Tissue Maintenance. Hepatology 2017, 65, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear Receptor-Dependent Bile Acid Signaling Is Required for Normal Liver Regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. The Liver as a Lymphoid Organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Calne, R.Y.; Sells, R.A.; Pena, J.R.; Davis, D.R.; Millard, P.R.; Herbertson, B.M.; Binns, R.M.; Davies, D.A.L. Induction of Immunological Tolerance by Porcine Liver Allografts. Nature 1969, 223, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, D.A.; Davidson, N.O. Functional Relationships between Lipid Metabolism and Liver Regeneration. Int. J. Hepatol. 2012, 2012, 549241. [Google Scholar] [CrossRef]
- Bartoli, D.; Piobbico, D.; Bellet, M.M.; Bennati, A.M.; Roberti, R.; Della Fazia, M.A.; Servillo, G. Impaired Cell Proliferation in Regenerating Liver of 3 β-Hydroxysterol Δ14-Reductase (TM7SF2) Knock-out Mice. Cell Cycle 2016, 15, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Hasegawa, T. Role of Neutrophils in Acute Inflammatory Liver Injury. Liver Int. 2006, 26, 912–919. [Google Scholar] [CrossRef]
- Lefkowitch, J.H. The Pathology of Acute Liver Failure. Adv. Anat. Pathol. 2016, 23, 144–158. [Google Scholar] [CrossRef]
- McDowell Torres, D.; Stevens, R.D.; Gurakar, A. Acute Liver Failure: A Management Challenge for the Practicing Gastroenterologist. Gastroenterol. Hepatol. 2010, 6, 444–450. [Google Scholar]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-Induced Liver Injury: Interactions between Drug Properties and Host Factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef]
- Rumack, B.H. Acetaminophen Hepatotoxicity: The First 35 Years. J. Toxicol. Clin. Toxicol. 2002, 40, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W.; Hyyrylainen, A.; Gera, A.; Audimoolam, V.K.; McPhail, M.J.W.; Auzinger, G.; Rela, M.; Heaton, N.; O’Grady, J.G.; Wendon, J.; et al. Lessons from Look-Back in Acute Liver Failure? A Single Centre Experience of 3300 Patients. J. Hepatol. 2013, 59, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Kwong, S.; Meyerson, C.; Zheng, W.; Kassardjian, A.; Stanzione, N.; Zhang, K.; Wang, H.L. Acute Hepatitis and Acute Liver Failure: Pathologic Diagnosis and Differential Diagnosis. Semin. Diagn. Pathol. 2019, 36, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Tapper, E.B.; Sengupta, N.; Bonder, A. The Incidence and Outcomes of Ischemic Hepatitis: A Systematic Review with Meta-Analysis. Am. J. Med. 2015, 128, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Dindo, D.; Demartines, N.; Clavien, P.A. Classification of Surgical Complications: A New Proposal with Evaluation in a Cohort of 6336 Patients and Results of a Survey. Ann. Surg. 2004, 240, 205–213. [Google Scholar] [CrossRef]
- Brempelis, K.J.; Crispe, I.N. Infiltrating Monocytes in Liver Injury and Repair. Clin. Transl. Immunol. 2016, 5, e113. [Google Scholar] [CrossRef]
- Schuppan, D.; Afdhal, N.H. Liver Cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of Liver Cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef]
- Powell, E.E.; Edwards-Smith, C.J.; Hay, J.L.; Clouston, A.D.; Crawford, D.H.; Shorthouse, C.; Purdie, D.M.; Jonsson, J.R. Host Genetic Factors Influence Disease Progression in Chronic Hepatitis C. Hepatology 2000, 31, 828–833. [Google Scholar] [CrossRef]
- Poynard, T.; Mathurin, P.; Lai, C.L.; Guyader, D.; Poupon, R.; Tainturier, M.H.; Myers, R.P.; Muntenau, M.; Ratziu, V.; Manns, M.; et al. A Comparison of Fibrosis Progression in Chronic Liver Diseases. J. Hepatol. 2003, 38, 257–265. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the Prevalence of the Most Common Causes of Chronic Liver Diseases in the United States From 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530. [Google Scholar] [CrossRef]
- Della Fazia, M.A.; Servillo, G. Foie Gras and Liver Regeneration: A Fat Dilemma. Cell Stress 2018, 2, 162–175. [Google Scholar] [CrossRef]
- Rosenberg, W.M.C. Rating Fibrosis Progression in Chronic Liver Diseases. J. Hepatol. 2003, 38, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Shang, Z.; Gao, Y.; Wu, H.; Kong, X. Liver Regeneration in Chronic Liver Injuries: Basic and Clinical Applications Focusing on Macrophages and Natural Killer Cells. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Tarao, K.; Nozaki, A.; Ikeda, T.; Sato, A.; Komatsu, H.; Komatsu, T.; Taguri, M.; Tanaka, K. Real Impact of Liver Cirrhosis on the Development of Hepatocellular Carcinoma in Various Liver Diseases—Meta-Analytic Assessment. Cancer Med. 2019, 8, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Miyajima, A. Liver Regeneration and Fibrosis after Inflammation. Inflamm. Regen. 2016, 36, 19. [Google Scholar] [CrossRef]
- Macdonald, R.A. “Lifespan” of Liver Cells: Autoradiographic Study Using Tritiated Thymidine in Normal, Cirrhotic, and Partially Hepatectomized Rats. Arch. Intern. Med. 1961, 107, 335–343. [Google Scholar] [CrossRef]
- Higgins, G.M. Experimental Pathology of the Liver. Restoration of the Liver of the White Rat Following Partial Surgical Removal. Arch. Pathol. 1931, 12, 186–202. [Google Scholar]
- Nikfarjam, M.; Malcontenti-Wilson, C.; Fanartzis, M.; Daruwalla, J.; Christophi, C. A Model of Partial Hepatectomy in Mice. J. Investig. Surg. 2004, 17, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Curado, S.; Stainier, D.Y.R.; Anderson, R.M. Nitroreductase-Mediated Cell/Tissue Ablation in Zebrafish: A Spatially and Temporally Controlled Ablation Method with Applications in Developmental and Regeneration Studies. Nat. Protoc. 2008, 3, 948–954. [Google Scholar] [CrossRef]
- Farber, J.L.; Gerson, R.J. Mechanisms of Cell Injury with Hepatotoxic Chemicals. Pharmacol. Rev. 1984, 36 (Suppl. 2), 71S–75S. [Google Scholar] [PubMed]
- Fujii, H.; Hirose, T.; Oe, S.; Yasuchika, K.; Azuma, H.; Fujikawa, T.; Nagao, M.; Yamaoka, Y. Contribution of Bone Marrow Cells to Liver Regeneration after Partial Hepatectomy in Mice. J. Hepatol. 2002, 36, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and Unconventional Cell Division of Hepatocytes Underlie Liver Regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.Y.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic Progenitor Cells of Biliary Origin with Liver Repopulation Capacity. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef]
- He, J.; Lu, H.; Zou, Q.; Luo, L. Regeneration of Liver after Extreme Hepatocyte Loss Occurs Mainly via Biliary Transdifferentiation in Zebrafish. Gastroenterology 2014, 146, 789–800. [Google Scholar] [CrossRef]
- Choi, T.Y.; Ninov, N.; Stainier, D.Y.R.; Shin, D. Extensive Conversion of Hepatic Biliary Epithelial Cells to Hepatocytes after near Total Loss of Hepatocytes in Zebrafish. Gastroenterology 2014, 146, 776–788. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver Regeneration after Partial Hepatectomy: Critical Analysis of Mechanistic Dilemmas. Am. J. Pathol. 2010, 176, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Nagel, A.J.; Tanabe, K.; Fuchs, J.; Dehlke, K.; Ghamarnejad, O.; Lemekhova, A.; Mehrabi, A. Markers of Liver Regeneration—The Role of Growth Factors and Cytokines: A Systematic Review. BMC Surg. 2020, 20, 31. [Google Scholar] [CrossRef]
- Abu Rmilah, A.; Zhou, W.; Nelson, E.; Lin, L.; Amiot, B.; Nyberg, S.L. Understanding the Marvels behind Liver Regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e340. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver Regeneration. Hepatology 2006, 43 (Suppl. 1), S45–S53. [Google Scholar] [CrossRef]
- Campbell, J.S.; Riehle, K.J.; Brooling, J.T.; Bauer, R.L.; Mitchell, C.; Fausto, N. Proinflammatory Cytokine Production in Liver Regeneration Is Myd88-Dependent, but Independent of Cd14, Tlr2, and Tlr4. J. Immunol. 2006, 176, 2522–2528. [Google Scholar] [CrossRef]
- Seki, E.; Tsutsui, H.; Iimuro, Y.; Naka, T.; Son, G.; Akira, S.; Kishimoto, T.; Nakanishi, K.; Fujimoto, J. Contribution of Toll-like Receptor/Myeloid Differentiation Factor 88 Signaling to Murine Liver Regeneration. Hepatology 2005, 41, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, I.; Chaisson, M.; Fausto, N. Tumor Necrosis Factor Induces DNA Replication in Hepatic Cells through Nuclear Factor ΚB Activation. Cell Growth Differ. 1999, 10, 819–828. [Google Scholar] [PubMed]
- Libermann, T.A.; Baltimore, D. Activation of Interleukin-6 Gene Expression through the NF-κB Transcription Factor. Mol. Cell. Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef]
- Zimmers, T.A.; McKillop, I.H.; Pierce, R.H.; Yoo, J.Y.; Koniaris, L.G. Massive Liver Growth in Mice Induced by Systemic Interleukin 6 Administration. Hepatology 2003, 38, 326–334. [Google Scholar] [CrossRef]
- Webber, E.M.; Bruix, J.; Pierce, R.H.; Fausto, N. Tumor Necrosis Factor Primes Hepatocytes for DNA Replication in the Rat. Hepatology 1998, 28, 1226–1234. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver Regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef]
- Haga, S.; Ogawa, W.; Inoue, H.; Terui, K.; Ogino, T.; Igarashi, R.; Takeda, K.; Akira, S.; Enosawa, S.; Furukawa, H.; et al. Compensatory Recovery of Liver Mass by Akt-Mediated Hepatocellular Hypertrophy in Liver-Specific STAT3-Deficient Mice. J. Hepatol. 2005, 43, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of Liver Regeneration by Growth Factors and Cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef]
- Tomiya, T.; Ogata, I.; Yamaoka, M.; Yanase, M.; Inoue, Y.; Fujiwara, K. The Mitogenic Activity of Hepatocyte Growth Factor on Rat Hepatocytes Is Dependent upon Endogenous Transforming Growth Factor-α. Am. J. Pathol. 2000, 157, 1693–1701. [Google Scholar] [CrossRef]
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met Provides Essential Signals for Liver Regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar] [CrossRef]
- Mitchell, C.; Nivison, M.; Jackson, L.F.; Fox, R.; Lee, D.C.; Campbell, J.S.; Fausto, N. Heparin-Binding Epidermal Growth Factor-like Growth Factor Links Hepatocyte Priming with Cell Cycle Progression during Liver Regeneration. J. Biol. Chem. 2005, 280, 2562–2568. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF Receptor Ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, S.; Bowen, W.C.; Mars, W.M.; Orr, A.; Haynes, M.M.; DeFrances, M.C.; Liu, S.; Tseng, G.C.; Tsagianni, A.; Michalopoulos, G.K. Combined Systemic Elimination of MET and Epidermal Growth Factor Receptor Signaling Completely Abolishes Liver Regeneration and Leads to Liver Decompensation. Hepatology 2016, 64, 1711–1724. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, M.; Chen, E.; Tang, H. Liver Regeneration: Analysis of the Main Relevant Signaling Molecules. Mediat. Inflamm. 2017, 2017, 4256352. [Google Scholar] [CrossRef] [PubMed]
- Vine, W.; Kier, A.; Starzl, T.; Warty, V. Baboon-to-Human Liver Transplantation. Lancet 1993, 341, 1158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, Y.P.; Ballou, L.M.; Lin, R.Z. Rapamycin-Insensitive Regulation of 4E-BP1 in Regenerating Rat Liver. J. Biol. Chem. 2001, 276, 10943–10951. [Google Scholar] [CrossRef] [PubMed]
- Goggin, M.M.; Nelsen, C.J.; Kimball, S.R.; Jefferson, L.S.; Morley, S.J.; Albrecht, J.H. Rapamycin-Sensitive Induction of Eukaryotic Initiation Factor 4F in Regenerating Mouse Liver. Hepatology 2004, 40, 537–544. [Google Scholar] [CrossRef]
- Kim, D.H.; Sabatini, D.M. Raptor and MTOR: Subunits of a Nutrient-Sensitive Complex. Curr. Top. Microbiol. Immunol. 2004, 279, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Braun, L.; Mead, J.E.; Panzica, M.; Mikumo, R.; Bell, G.I.; Fausto, N. Transforming Growth Factor β MRNA Increases during Liver Regeneration: A Possible Paracrine Mechanism of Growth Regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 1539–1543. [Google Scholar] [CrossRef]
- Nakamura, T.; Tomita, Y.; Hirai, R.; Yamaoka, K.; Kaji, K.; Ichihara, A. Inhibitory Effect of Transforming Growth Factor-β on DNA Synthesis of Adult Rat Hepatocytes in Primary Culture. Biochem. Biophys. Res. Commun. 1985, 133, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Apte, U.; Gkretsi, V.; Bowen, W.C.; Mars, W.M.; Luo, J.H.; Donthamsetty, S.; Orr, A.; Monga, S.P.S.; Wu, C.; Michalopoulos, G.K. Enhanced Liver Regeneration Following Changes Induced by Hepatocyte-Specific Genetic Ablation of Integrin-Linked Kinase. Hepatology 2009, 50, 844–851. [Google Scholar] [CrossRef]
- Lalli, E.; Sassone-Corsi, P. Signal Transduction and Gene Regulation: The Nuclear Response to CAMP. J. Biol. Chem. 1994, 269, 17359–17362. [Google Scholar] [CrossRef] [PubMed]
- Servillo, G.; Della Fazia, M.A.; Sassone-Corsi, P. Transcription Factor CREM Coordinates the Timing of Hepatocyte Proliferation in the Regenerating Liver. Genes Dev. 1998, 12, 3639–3643. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.P.S.; Laurance, M.E.; Lundblad, J.R.; Goldman, P.S.; Shih, H.; Connor, L.M.; Marriott, S.J.; Goodman, R.H. Control of CAMP-Regulated Enhancers by the Viral Transactivator Tax through CREB and the Co-Activator CBP. Nature 1996, 380, 642–646. [Google Scholar] [CrossRef]
- Lee, M.W.; Chanda, D.; Yang, J.; Oh, H.; Kim, S.S.; Yoon, Y.S.; Hong, S.; Park, K.G.; Lee, I.K.; Choi, C.S.; et al. Regulation of Hepatic Gluconeogenesis by an ER-Bound Transcription Factor, CREBH. Cell Metab. 2010, 11, 331–339. [Google Scholar] [CrossRef]
- Stark, R.; Guebre-Egziabher, F.; Zhao, X.; Feriod, C.; Dong, J.; Alves, T.C.; Ioja, S.; Pongratz, R.L.; Bhanot, S.; Roden, M.; et al. A Role for Mitochondrial Phosphoenolpyruvate Carboxykinase (PEPCK-M) in the Regulation of Hepatic Gluconeogenesis. J. Biol. Chem. 2014, 289, 7257–7263. [Google Scholar] [CrossRef]
- Wahlang, B.; Mcclain, C.; Barve, S.; Gobejishvili, L. Role of CAMP and Phosphodiesterase Signaling in Liver Health and Disease HHS Public Access. Cell. Signal. 2018, 49, 105–115. [Google Scholar] [CrossRef]
- Thoresen, G.H.; Sand, T.-E.; Refsnes, M.; Dajani, O.F.; Guren, T.K.; Gladhaug, I.P.; Killi, A.; Christoffersen, T. Dual Effects of Glucagon and Cyclic Amp on DNA Synthesis in Cultured Rat Hepatocytes: Stimulatory Regulation in Early G1 and Inhibition Shortly before the s Phase Entry. J. Cell. Physiol. 1990, 144, 523–530. [Google Scholar] [CrossRef]
- Diehl, A.M.; Yang, S.Q.; Wolfgang, D.; Wand, G. Differential Expression of Guanine Nucleotide-Binding Proteins Enhances CAMP Synthesis in Regenerating Rat Liver. J. Clin. Investig. 1992, 89, 1706–1712. [Google Scholar] [CrossRef]
- Della Fazia, M.A.; Castelli, M.; Bartoli, D.; Pieroni, S.; Pettirossi, V.; Piobbico, D.; Viola-Magni, M.; Servillo, G. HOPS: A Novel CAMP-Dependent Shutting Protein Involved in Protein Synthesis Regulation. J. Cell Sci. 2005, 118 Pt 14, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Della-Fazia, M.A.; Castelli, M.; Piobbico, D.; Pieroni, S.; Servillo, G. The Ins and Outs of HOPS/TMUB1 in Biology and Pathology. FEBS J. 2021, 288, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Della Fazia, M.A.; Pettirossi, V.; Ayroldi, E.; Riccardi, C.; Magni, M.V.; Servillo, G. Differential Expression of CD44 Isoforms during Liver Regeneration in Rats. J. Hepatol. 2001, 34, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Siapati, E.K.; Roubelakis, M.G.; Vassilopoulos, G. Liver Regeneration by Hematopoietic Stem Cells: Have We Reached the End of the Road? Cells 2022, 11, 2312. [Google Scholar] [CrossRef] [PubMed]
- Tsolaki, E.; Yannaki, E. Stem Cell-Based Regenerative Opportunities for the Liver: State of the Art and Beyond. World J. Gastroenterol. 2015, 21, 12334–12350. [Google Scholar] [CrossRef]
- Austin, T.W.; Lagasse, E. Hepatic Regeneration from Hematopoietic Stem Cells. Mech. Dev. 2003, 120, 131–135. [Google Scholar] [CrossRef]
- Kiu, H.; Nicholson, S.E. Biology and Significance of the JAK/STAT Signalling Pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef]
- Wakahara, R.; Kunimoto, H.; Tanino, K.; Kojima, H.; Inoue, A.; Shintaku, H.; Nakajima, K. Phospho-Ser727 of STAT3 Regulates STAT3 Activity by Enhancing Dephosphorylation of Phospho-Tyr705 Largely through TC45. Genes Cells 2012, 17, 132–145. [Google Scholar] [CrossRef]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver Failure and Defective Hepatocyte Regeneration in Interleukin-6- Deficient Mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef]
- Inoue, H.; Ogawa, W.; Ozaki, M.; Haga, S.; Matsumoto, M.; Furukawa, K.; Hashimoto, N.; Kido, Y.; Mori, T.; Sakaue, H.; et al. Role of STAT-3 in Regulation of Hepatic Gluconeogenic Genes and Carbohydrate Metabolism In Vivo. Nat. Med. 2004, 10, 168–174. [Google Scholar] [CrossRef]
- Moh, A.; Iwamoto, Y.; Chai, G.X.; Zhang, S.S.M.; Kano, A.; Yang, D.D.; Zhang, W.; Wang, J.; Jacoby, J.J.; Gao, B.; et al. Role of STAT3 in Liver Regeneration: Survival, DNA Synthesis, Inflammatory Reaction and Liver Mass Recovery. Lab. Investig. 2007, 87, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kirillova, I.; Peschon, J.J.; Fausto, N. Initiation of Liver Growth by Tumor Necrosis Factor: Deficient Liver Regeneration in Mice Lacking Type I Tumor Necrosis Factor Receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Bellet, M.M.; Piobbico, D.; Bartoli, D.; Castelli, M.; Pieroni, S.; Brunacci, C.; Chiacchiaretta, M.; Del Sordo, R.; Fallarino, F.; Sidoni, A.; et al. NEDD4 Controls the Expression of GUCD1, a Protein Upregulated in Proliferating Liver Cells. Cell Cycle 2014, 13, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F. Liver Proliferation: The GUCD1/NEDD4-1 Connection. Cell Cycle 2014, 13, 2022–2023. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fan, M.; Wang, X.; Xu, G.; Yan, Q.; Huang, W. Bile Acid Signaling and Liver Regeneration. Biochim. Biophys. Acta 2015, 1849, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Wallek, G.; Friedrich, N.; Ittermann, T.; Mayerle, J.; Völzke, H.; Nauck, M.; Spielhagen, C. IGF-1 and IGFBP-3 in Patients with Liver Disease/IGF-1 Und IGFBP-3 Bei Patienten Mit Lebererkrankungen. Laboratoriumsmedizin 2013, 37, 13–20. [Google Scholar] [CrossRef]
- Edinger, A.L.; Thompson, C.B. Akt Maintains Cell Size and Survival by Increasing MTOR-Dependent Nutrient Uptake. Mol. Biol. Cell 2002, 13, 2276–2288. [Google Scholar] [CrossRef]
- Latronico, M.V.G.; Costinean, S.; Lavitrano, M.L.; Peschle, C.; Condorelli, G. Regulation of Cell Size and Contractile Function by AKT in Cardiomyocytes. Ann. N. Y. Acad. Sci. 2004, 1015, 250–260. [Google Scholar] [CrossRef]
- Hawkins, P.T.; Anderson, K.E.; Davidson, K.; Stephens, L.R. Signalling through Class I PI3Ks in Mammalian Cells. Biochem. Soc. Trans. 2006, 34 Pt 5, 647–662. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The Evolution of Phosphatidylinositol 3-Kinases as Regulators of Growth and Metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Puri, K.D.; Doggett, T.A.; Huang, C.Y.; Douangpanya, J.; Hayflick, J.S.; Turner, M.; Penninger, J.; Diacovo, T.G. The Role of Endothelial PI3Kγ Activity in Neutrophil Trafficking. Blood 2005, 106, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Banno, Y.; Nagaki, M.; Brenner, D.A.; Naiki, T.; Nozawa, Y.; Nakashima, S.; Moriwaki, H. TNF-α-Induced Sphingosine 1-Phosphate Inhibits Apoptosis through a Phosphatidylinositol 3-Kinase/Akt Pathway in Human Hepatocytes. J. Immunol. 2001, 167, 173–180. [Google Scholar] [CrossRef]
- Desmots, F.; Rissel, M.; Gilot, D.; Lagadic-Gossmann, D.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A.; Loyer, P. Pro-Inflammatory Cytokines Tumor Necrosis Factor α and Interleukin-6 and Survival Factor Epidermal Growth Factor Positively Regulate the Murine GSTA4 Enzyme in Hepatocytes. J. Biol. Chem. 2002, 277, 17892–17900. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Jain, S.K. Phosphatidylinositol-3,4,5-Triphosphate and Cellular Signaling: Implications for Obesity and Diabetes. Cell. Physiol. Biochem. 2015, 35, 1253–1275. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Chou, M.M.; Hou, W.; Johnson, J.; Graham, L.K.; Lee, M.H.; Chen, C.S.; Newton, A.C.; Schaffhausen, B.S.; Toker, A. Regulation of Protein Kinase C ζ by PI 3-Kinase and PDK-1. Curr. Biol. 1998, 8, 1069–1077. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of Activation of Protein Kinase B by Insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, X.; Wang, J.; Liu, W.; Liu, Q.; Ye, Y.; Dai, B.; Guo, D.; Zhang, P.; Yang, P.; et al. Translationally Controlled Tumor Protein Promotes Liver Regeneration by Activating MTORC2/AKT Signaling. Cell Death Dis. 2020, 11, 58. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and MTOR Signalling Controls Tumour Cell Growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Shahbazian, D.; Roux, P.P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.B.; Blenis, J.; Pende, M.; Sonenberg, N. The MTOR/PI3K and MAPK Pathways Converge on EIF4B to Control Its Phosphorylation and Activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef]
- Haga, S.; Ozaki, M.; Inoue, H.; Okamoto, Y.; Ogawa, W.; Takeda, K.; Akira, S.; Todo, S. The Survival Pathways Phosphatidylinositol-3 Kinase (PI3-K)/Phosphoinositide-Dependent Protein Kinase 1 (PDK1)/Akt Modulate Liver Regeneration through Hepatocyte Size Rather than Proliferation. Hepatology 2009, 49, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Gadd, V.L.; Aleksieva, N.; Forbes, S.J. Epithelial Plasticity during Liver Injury and Regeneration. Cell Stem Cell 2020, 27, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Raven, A.; Lu, W.Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes Act as Facultative Liver Stem Cells during Impaired Hepatocyte Regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef]
- Sahoo, S.; Mishra, A.; Diehl, A.M.; Jolly, M.K. Dynamics of Hepatocyte-Cholangiocyte Cell-Fate Decisions during Liver Development and Regeneration. iScience 2022, 25, 104955. [Google Scholar] [CrossRef] [PubMed]
- Dickson, I. Cholangiocytes Regenerate Hepatocytes during Severe Liver Injury. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 503. [Google Scholar] [CrossRef]
- Huang, R.; Zhang, X.; Gracia-Sancho, J.; Xie, W.F. Liver Regeneration: Cellular Origin and Molecular Mechanisms. Liver Int. 2022, 42, 1486–1495. [Google Scholar] [CrossRef]
- Miossec, P.; Korn, T.; Kuchroo, V.K. Interleukin-17 and Type 17 Helper T Cells. N. Engl. J. Med. 2009, 361, 888–898. [Google Scholar] [CrossRef]
- Shalom-Barak, T.; Quach, J.; Lotz, M. Interleukin-17-Induced Gene Expression in Articular Chondrocytes Is Associated with Activation of Mitogen-Activated Protein Kinases and NF-ΚB. J. Biol. Chem. 1998, 273, 27467–27473. [Google Scholar] [CrossRef]
- Schwandner, R.; Yamaguchi, K.; Cao, Z. Requirement of Tumor Necrosis Factor Receptor-Associated Factor (TRAF)6 in Interleukin 17 Signal Transduction. J. Exp. Med. 2000, 191, 1233–1240. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Steward-Tharp, S.M.; Laurence, A.; Watford, W.T.; Wei, L.; Adamson, A.S.; Fan, S. Signal Transduction and Th17 Cell Differentiation. Microbes Infect. 2009, 11, 599–611. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wu, L.; Xie, W.; Shao, Y.; Jiang, J.; Zhao, Z.; Yan, M.; Chen, Z.; Cui, D. The Imbalance of Th17/Treg Cells Is Involved in the Progression of Nonalcoholic Fatty Liver Disease in Mice. BMC Immunol. 2017, 18, 33. [Google Scholar] [CrossRef]
- Roh, Y.S.; Park, S.; Lim, C.W.; Kim, B. Depletion of Foxp3+ Regulatory T Cells Promotes Profibrogenic Milieu of Cholestasis-Induced Liver Injury. Dig. Dis. Sci. 2015, 60, 2009–2018. [Google Scholar] [CrossRef]
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Tato, C.M. Innate IL-17-Producing Cells: The Sentinels of the Immune System. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.-G.; Wang, T.; Zheng, J.; et al. Pivotal Role of Dermal IL-17-Producing γδ T Cells in Skin Inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Beringer, A.; Thiam, N.; Molle, J.; Bartosch, B.; Miossec, P. Synergistic Effect of Interleukin-17 and Tumour Necrosis Factor-α on Inflammatory Response in Hepatocytes through Interleukin-6-Dependent and Independent Pathways. Clin. Exp. Immunol. 2018, 193, 221–233. [Google Scholar] [CrossRef]
- Sparna, T.; Rétey, J.; Schmich, K.; Albrecht, U.; Naumann, K.; Gretz, N.; Fischer, H.P.; Bode, J.G.; Merfort, I. Genome-Wide Comparison between IL-17 and Combined TNF-α/IL-17 Induced Genes in Primary Murine Hepatocytes. BMC Genom. 2010, 11, 226. [Google Scholar] [CrossRef]
- Gu, F.M.; Li, Q.L.; Gao, Q.; Jiang, J.H.; Zhu, K.; Huang, X.Y.; Pan, J.F.; Yan, J.; Hu, J.H.; Wang, Z.; et al. IL-17 Induces AKT-Dependent IL-6/JAK2/STAT3 Activation and Tumor Progression in Hepatocellular Carcinoma. Mol. Cancer 2011, 10, 150. [Google Scholar] [CrossRef]
- Zhao, L.; Tang, Y.; You, Z.; Wang, Q.; Liang, S.; Han, X.; Qiu, D.; Wei, J.; Liu, Y.; Shen, L.; et al. Interleukin-17 Contributes to the Pathogenesis of Autoimmune Hepatitis through Inducing Hepatic Interleukin-6 Expression. PLoS ONE 2011, 6, e18909. [Google Scholar] [CrossRef]
- Amara, S.; Lopez, K.; Banan, B.; Brown, S.K.; Whalen, M.; Myles, E.; Ivy, M.T.; Johnson, T.; Schey, K.L.; Tiriveedhi, V. Synergistic Effect of Pro-Inflammatory TNFα and IL-17 in Periostin Mediated Collagen Deposition: Potential Role in Liver Fibrosis. Mol. Immunol. 2015, 64, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 Signaling in Inflammatory, Kupffer Cells, and Hepatic Stellate Cells Exacerbates Liver Fibrosis in Mice. Gastroenterology 2012, 143, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Furuya, S.; Kono, H.; Hara, M.; Hirayama, K.; Tsuchiya, M.; Fujii, H. Interleukin-17A Plays a Pivotal Role after Partial Hepatectomy in Mice. J. Surg. Res. 2013, 184, 838–846. [Google Scholar] [CrossRef]
- Rao, R.; Graffeo, C.S.; Gulati, R.; Jamal, M.; Narayan, S.; Zambirinis, C.P.; Barilla, R.; Deutsch, M.; Greco, S.H.; Ochi, A.; et al. Interleukin 17-Producing γδT Cells Promote Hepatic Regeneration in Mice. Gastroenterology 2014, 147, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Piobbico, D.; Bartoli, D.; Pieroni, S.; De Luca, A.; Castelli, M.; Romani, L.; Servillo, G.; Della-Fazia, M.A. Role of IL-17RA in the Proliferative Priming of Hepatocytes in Liver Regeneration. Cell Cycle 2018, 17, 2423–2435. [Google Scholar] [CrossRef]
- De Luca, A.; Pariano, M.; Cellini, B.; Costantini, C.; Villella, V.R.; Jose, S.S.; Palmieri, M.; Borghi, M.; Galosi, C.; Paolicelli, G.; et al. The IL-17F/IL-17RC Axis Promotes Respiratory Allergy in the Proximal Airways. Cell Rep. 2017, 20, 1667–1680. [Google Scholar] [CrossRef]
- Longo, C.R.; Patel, V.I.; Shrikhande, G.V.; Scali, S.T.; Csizmadia, E.; Daniel, S.; Sun, D.W.; Grey, S.T.; Arvelo, M.B.; Ferran, C. A20 Protects Mice from Lethal Radical Hepatectomy by Promoting Hepatocyte Proliferation via a P21waf1-Dependent Mechanism. Hepatology 2005, 42, 156–164. [Google Scholar] [CrossRef]
- Da Silva, C.G.; Studer, P.; Skroch, M.; Mahiou, J.; Minussi, D.C.; Peterson, C.R.; Wilson, S.W.; Patel, V.I.; Ma, A.; Csizmadia, E.; et al. A20 Promotes Liver Regeneration by Decreasing SOCS3 Expression to Enhance IL-6/STAT3 Proliferative Signals. Hepatology 2013, 57, 2014–2025. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Stankovic, S.; Baxter, A.G. Raising the NKT Cell Family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef]
- Sun, R.; Gao, B. Negative Regulation of Liver Regeneration by Innate Immunity (Natural Killer Cells/Interferon-γ). Gastroenterology 2004, 127, 1525–1539. [Google Scholar] [CrossRef]
- Dambacher, J.; Beigel, F.; Zitzmann, K.; Olszak, T.; Prüfer, T.; Steib, C.J.; Storr, M.; Göke, B.; Diepolder, H.; Bilzer, M.; et al. IL-22 Mediated Liver Cell Regeneration Is Abrogated by SOCS-1/3 Overexpression. Z. Gastroenterol. 2006, 44, 43. [Google Scholar] [CrossRef]
- Ren, X.; Hu, B.; Colletti, L.M. IL-22 Is Involved in Liver Regeneration after Hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, 74–80. [Google Scholar] [CrossRef]
- Han, X.; Yang, Q.; Lin, L.; Xu, C.; Zheng, C.; Chen, X.; Han, Y.; Li, M.; Cao, W.; Cao, K.; et al. Interleukin-17 Enhances Immunosuppression by Mesenchymal Stem Cells. Cell Death Differ. 2014, 21, 1758–1768. [Google Scholar] [CrossRef]
- Tian, J.; Rui, K.; Tang, X.; Wang, W.; Ma, J.; Tian, X.; Wang, Y.; Xu, H.; Lu, L.; Wang, S.; et al. IL-17 down-Regulates the Immunosuppressive Capacity of Olfactory Ecto-Mesenchymal Stem Cells in Murine Collagen-Induced Arthritis. Oncotarget 2016, 7, 42953–42962. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer Cell Engulfment of Apoptotic Bodies Stimulates Death Ligand and Cytokine Expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Selzner, N.; Selzner, M.; Odermatt, B.; Tian, Y.; Van Rooijen, N.; Clavien, P.A. ICAM-1 Triggers Liver Regeneration through Leukocyte Recruitment and Kupffer Cell-Dependent Release of TNF-α/IL-6 in Mice. Gastroenterology 2003, 124, 692–700. [Google Scholar] [CrossRef]
- Tang, C.; Chen, H.; Jiang, L.; Liu, L. Liver Regeneration: Changes in Oxidative Stress, Immune System, Cytokines, and Epigenetic Modifications Associated with Aging. Oxid. Med. Cell. Longev. 2022, 2022, 9018811. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive Angiocrine Signals from Sinusoidal Endothelium Are Required for Liver Regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, B.G.; Pestana, R.C.; Abugabal, Y.I.; Krishnan, S.; Chen, J.; Hassan, M.M.; Wolff, R.A.; Rashid, A.; Amin, H.M.; Kaseb, A.O. Origin and Role of Hepatic Myofibroblasts in Hepatocellular Carcinoma. Oncotarget 2020, 11, 1186–1201. [Google Scholar] [CrossRef]
- Brenner, D.A.; Kisseleva, T.; Scholten, D.; Paik, Y.H.; Iwaisako, K.; Inokuchi, S.; Schnabl, B.; Seki, E.; De Minicis, S.; Oesterreicher, C.; et al. Origin of Myofibroblasts in Liver Fibrosis. Fibrogenes. Tissue Repair 2012, 5 (Suppl. 1), S17. [Google Scholar] [CrossRef]
- Kallis, Y.N.; Robson, A.J.; Fallowfield, J.A.; Thomas, H.C.; Alison, M.R.; Wright, N.A.; Goldin, R.D.; Iredale, J.P.; Forbes, S.J. Remodelling of Extracellular Matrix Is a Requirement for the Hepatic Progenitor Cell Response. Gut 2011, 60, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Cheever, A.W.; Dragana, D.; Poindexter, R.W.; Caspar, P.; Lewis, F.A.; Sher, A. An IL-12-Based Vaccination Method for Preventing Fibrosis Induced by Schistosome Infection. Nature 1995, 376, 594–596. [Google Scholar] [CrossRef]
- Friedman, S.L. Molecular Regulation of Hepatic Fibrosis, an Integrated Cellular Response to Tissue Injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Venter, J.; Standeford, H.; Onori, P.; Marzioni, M.; Alvaro, D.; Franchitto, A.; et al. The Secretin/Secretin Receptor Axis Modulates Liver Fibrosis through Changes in Transforming Growth Factor-Β1 Biliary Secretion in Mice. Hepatology 2016, 64, 865–879. [Google Scholar] [CrossRef]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a Modulator of Hepatic Fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef]
- Robert, S.; Gicquel, T.; Bodin, A.; Lagente, V.; Boichot, E. Characterization of the MMP/TIMP Imbalance and Collagen Production Induced by IL-1 β or TNF-α Release from Human Hepatic Stellate Cells. PLoS One 2016, 11, e0153118. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone Marrow-Derived Monocytes Give Rise to Self-Renewing and Fully Differentiated Kupffer Cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating Monocyte-Derived Macrophages and Resident Kupffer Cells Display Different Ontogeny and Functions in Acute Liver Injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic Recruitment of the Inflammatory Gr1+ Monocyte Subset upon Liver Injury Promotes Hepatic Fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [PubMed]
- Graubardt, N.; Vugman, M.; Mouhadeb, O.; Caliari, G.; Pasmanik-Chor, M.; Reuveni, D.; Zigmond, E.; Brazowski, E.; David, E.; Chappell-Maor, L.; et al. Ly6Chi Monocytes and Their Macrophage Descendants Regulate Neutrophil Function and Clearance in Acetaminophen-Induced Liver Injury. Front. Immunol. 2017, 8, 626. [Google Scholar] [CrossRef]
- Campana, L.; Starkey Lewis, P.J.; Pellicoro, A.; Aucott, R.L.; Man, J.; O’Duibhir, E.; Mok, S.E.; Ferreira-Gonzalez, S.; Livingstone, E.; Greenhalgh, S.N.; et al. The STAT3–IL-10–IL-6 Pathway Is a Novel Regulator of Macrophage Efferocytosis and Phenotypic Conversion in Sterile Liver Injury. J. Immunol. 2018, 200, 1169–1187. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective Depletion of Macrophages Reveals Distinct, Opposing Roles during Liver Injury and Repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, M.G.; Donaldson, D.D.; Cheever, A.W.; Wynn, T.A. An IL-13 Inhibitor Blocks the Development of Hepatic Fibrosis during a T-Helper Type 2-Dominated Inflammatory Response. J. Clin. Investig. 1999, 104, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent Angiocrine Signals from Vascular Niche Balance Liver Regeneration and Fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Yen, J.H.; Khayrullina, T.; Ganea, D. PGE2-Induced Metalloproteinase-9 Is Essential for Dendritic Cell Migration. Blood 2008, 111, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Sastre, D.; Fiel, M.I.; Lee, U.E.; Ghiassi-Nejad, Z.; Ginhoux, F.; Vivier, E.; Friedman, S.L.; Merad, M.; Aloman, C. Dendritic Cell Regulation of Carbon Tetrachloride-Induced Murine Liver Fibrosis Regression. Hepatology 2012, 55, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Perelmutter, M.; Gal, H.; Burton, D.G.A.; Ovadya, Y.; Vadai, E.; Geiger, T.; Krizhanovsky, V. Senescent Cells Communicate via Intercellular Protein Transfer. Genes Dev. 2015, 29, 791–802. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.G.A.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D Ligands Mediate Immunosurveillance of Senescent Cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef]
- Gur, C.; Doron, S.; Kfir-Erenfeld, S.; Horwitz, E.; Abu-tair, L.; Safadi, R.; Mandelboim, O. NKp46-Mediated Killing of Human and Mouse Hepatic Stellate Cells Attenuates Liver Fibrosis. Gut 2011, 61, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine Receptor CCR6-Dependent Accumulation of γδ T Cells in Injured Liver Restricts Hepatic Inflammation and Fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.S.; Lee, Y.S.; Park, S.H.; Jang, M.J.; Jo, E.; Kim, S.C.; Han, Y.M.; et al. Exosome-Mediated Activation of Toll-like Receptor 3 in Stellate Cells Stimulates Interleukin-17 Production by γδ T Cells in Liver Fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.-H.; Wu, F.-T.; Pang, L.-T.; Zhang, T.-B.; Chen, Z. Role of γδT Cells in Liver Diseases and Its Relationship with Intestinal Microbiota Conflict-of-Interest Statement. World J. Gastroenterol. 2020, 26, 2559–2569. [Google Scholar] [CrossRef]
- González-Regueiro, J.A.; Moreno-Castañeda, L.; Uribe, M.; Chávez-Tapia, N.C. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16, S21–S26. [Google Scholar] [CrossRef]
- Ohtani, N.; Kawada, N. Role of the Gut–Liver Axis in Liver Inflammation, Fibrosis, and Cancer: A Special Focus on the Gut Microbiota Relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Chu, H.; Duan, Y.; Schnabl, B. Gut Microbiota in Liver Disease: Too Much Is Harmful, Nothing at All Is Not Helpful Either. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G563–G573. [Google Scholar] [CrossRef]
- Acharya, C.; Bajaj, J.S. Chronic Liver Diseases and the Microbiome—Translating Our Knowledge of Gut Microbiota to Management of Chronic Liver Disease. Gastroenterology 2021, 160, 556–572. [Google Scholar] [CrossRef]
- Jiang, L.; Schnabl, B. Gut Microbiota in Liver Disease: What Do We Know and What Do We Not Know? Physiology 2020, 35, 261–274. [Google Scholar] [CrossRef]
- Andria, B.; Bracco, A.; Cirino, G.; Chamuleau, R.A.F.M. Liver Cell Culture Devices. Cell Med. 2010, 1, 55–70. [Google Scholar] [CrossRef]
- Yen, M.-H.; Wu, Y.-Y.; Liu, Y.-S.; Rimando, M.; Ho, J.H.-C.; Lee, O.K.-S. Efficient Generation of Hepatic Cells from Mesenchymal Stromal Cells by an Innovative Bio-Microfluidic Cell Culture Device. Stem Cell Res. Ther. 2016, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, A.; Greco Song, H.-H.; Grzelak, K.A.; Polacheck, W.J.; Fleming, H.E.; Chen, C.S.; Bhatia, S.N.; Duncan, S.; Parashurama, N. A Vascularized Model of the Human Liver Mimics Regenerative Responses. Proc. Natl. Acad. Sci. USA 2022, 119, e2115867119. [Google Scholar] [CrossRef]
- Polidoro, M.A.; Ferrari, E.; Marzorati, S.; Lleo, A.; Rasponi, M. Experimental Liver Models: From Cell Culture Techniques to Microfluidic Organs-on-Chip. Liver Int. 2021, 41, 1744–1761. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wu, Z.; Li, L. Mesenchymal Stromal Cells Promote Liver Regeneration through Regulation of Immune Cells. Int. J. Biol. Sci. 2020, 16, 893–903. [Google Scholar] [CrossRef]
- Dalsbecker, P.; Adiels, C.B.; Goksör, M. Liver-on-a-Chip Devices: The Pros and Cons of Complexity. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G188–G204. [Google Scholar] [CrossRef]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef]
- Bulutoglu, B.; Rey-Bedón, C.; Kang, Y.B.; Mert, S.; Yarmush, M.L.; Usta, O.B. A Microfluidic Patterned Model of Non-Alcoholic Fatty Liver Disease: Applications to Disease Progression and Zonation. Lab Chip 2019, 19, 3022–3031. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Deng, P.; Tao, T.; Liu, H.; Wu, S.; Chen, W.; Qin, J. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, B.; No, D.Y.; Lee, G.; Lee, S.R.; Oh, H.; Lee, S.H. A 3D Alcoholic Liver Disease Model on a Chip. Integr. Biol. 2016, 8, 302–308. [Google Scholar] [CrossRef]
- Deng, J.; Chen, Z.; Zhang, X.; Luo, Y.; Wu, Z.; Lu, Y.; Liu, T.; Zhao, W.; Lin, B. A Liver-Chip-Based Alcoholic Liver Disease Model Featuring Multi-Non-Parenchymal Cells. Biomed. Microdevices 2019, 21, 57. [Google Scholar] [CrossRef]
- Nawroth, J.C.; Petropolis, D.B.; Manatakis, D.V.; Maulana, T.I.; Burchett, G.; Schlünder, K.; Witt, A.; Shukla, A.; Kodella, K.; Ronxhi, J.; et al. Modeling Alcohol-Associated Liver Disease in a Human Liver-Chip. Cell Rep. 2021, 36, 109393. [Google Scholar] [CrossRef] [PubMed]
- McCarty, W.J.; Usta, O.B.; Yarmush, M.L. A Microfabricated Platform for Generating Physiologically-Relevant Hepatocyte Zonation. Sci. Rep. 2016, 6, 26868. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di-Iacovo, N.; Pieroni, S.; Piobbico, D.; Castelli, M.; Scopetti, D.; Ferracchiato, S.; Della-Fazia, M.A.; Servillo, G. Liver Regeneration and Immunity: A Tale to Tell. Int. J. Mol. Sci. 2023, 24, 1176. https://doi.org/10.3390/ijms24021176
Di-Iacovo N, Pieroni S, Piobbico D, Castelli M, Scopetti D, Ferracchiato S, Della-Fazia MA, Servillo G. Liver Regeneration and Immunity: A Tale to Tell. International Journal of Molecular Sciences. 2023; 24(2):1176. https://doi.org/10.3390/ijms24021176
Chicago/Turabian StyleDi-Iacovo, Nicola, Stefania Pieroni, Danilo Piobbico, Marilena Castelli, Damiano Scopetti, Simona Ferracchiato, Maria Agnese Della-Fazia, and Giuseppe Servillo. 2023. "Liver Regeneration and Immunity: A Tale to Tell" International Journal of Molecular Sciences 24, no. 2: 1176. https://doi.org/10.3390/ijms24021176
APA StyleDi-Iacovo, N., Pieroni, S., Piobbico, D., Castelli, M., Scopetti, D., Ferracchiato, S., Della-Fazia, M. A., & Servillo, G. (2023). Liver Regeneration and Immunity: A Tale to Tell. International Journal of Molecular Sciences, 24(2), 1176. https://doi.org/10.3390/ijms24021176