


Convergent Genomic Signatures of Cashmere Traits: Evidence for Natural and Artificial Selection

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
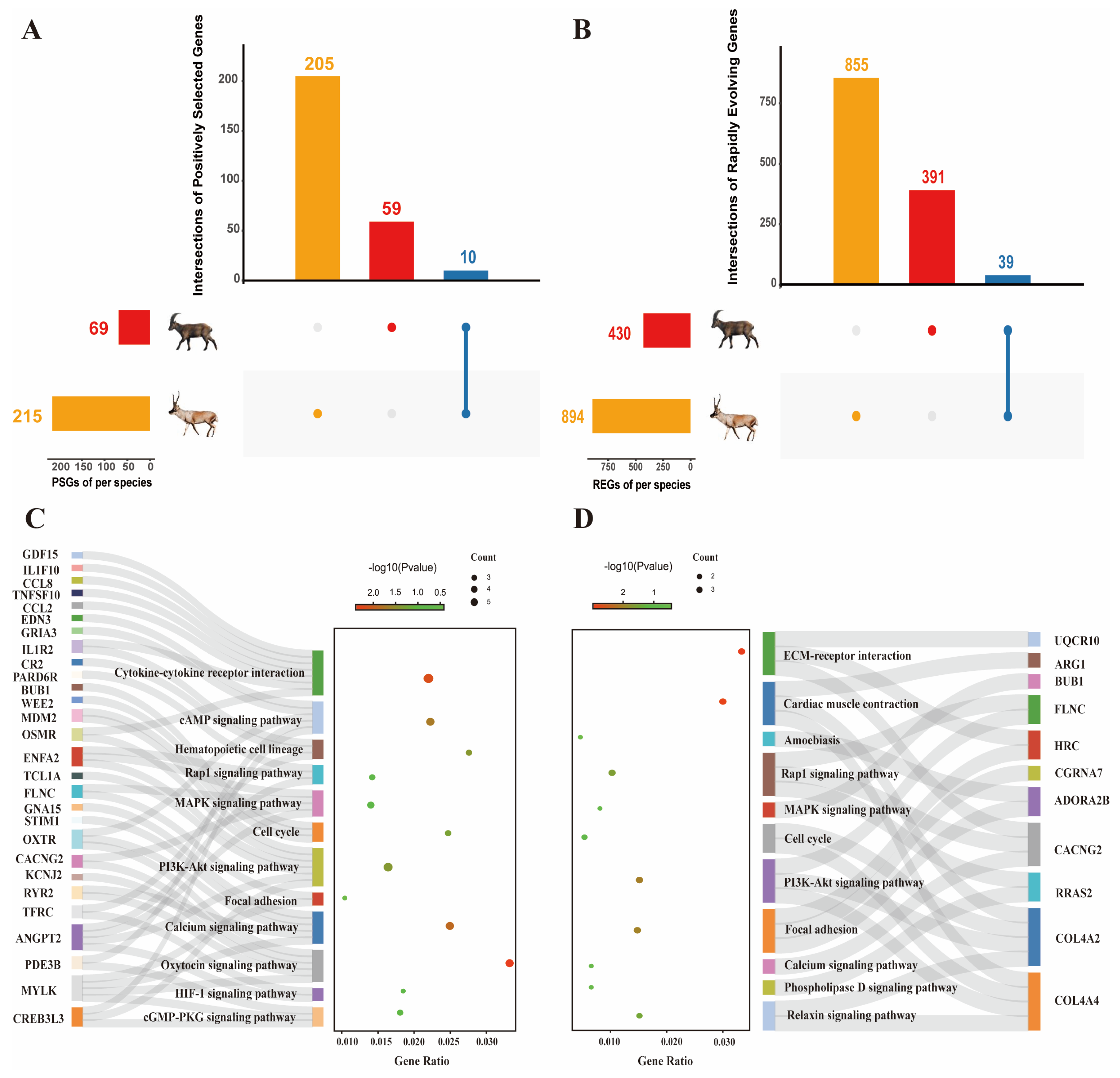
2.1. Genetic Evolution under Natural Selection Pressure
2.2. Signals of Convergent Evolution between Tibetan Antelope and Siberian Ibex
2.3. Candidate Artificial Selection Genes Screening and Enrichment Analysis of Cashmere Traits
2.4. Identification of Key Gene Modules and Differentially Expressed Genes Related to Cashmere Growth and Development
2.5. Co-Regulatory Network of Cashmere Traits
3. Discussion
4. Materials and Methods
4.1. Species Selection and Data Collection
4.2. Phylogenetic Tree and Multiple Sequence Alignment
4.3. Analysis of Positively Selected Genes and Rapidly Evolving Genes
4.4. Identification of Convergent Amino Acid Substitutions
4.5. Reads Alignment and SNP Calling
4.6. Genome-Wide Selective Sweep Test
4.7. RNA Sequencing Data Analysis
4.8. Construction of Weighted Gene Coexpression Network
4.9. Identification of Significant Modules and Key Genes Related to Hair Follicle Development
4.10. Gene Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, S.H.; He, Z.W.; Guo, Z.X.; Zhang, Z.; Wyckoff, G.J.; Greenberg, A.J.; Wu, C.I.; Shi, S.H. Genome-Wide Convergence during Evolution of Mangroves from Woody Plants. Mol. Biol. Evol. 2017, 34, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Sackton, T.B.; Clark, N.L. Convergent evolution in the genomics era: New insights and directions. Philos. Trans. R. Soc. B. 2019, 374, 20190102. [Google Scholar] [CrossRef] [PubMed]
- Christin, P.A.; Weinreich, D.M.; Besnard, G. Causes and evolutionary significance of genetic convergence. Trends Genet. TIG 2010, 26, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, Y.L.; Zhang, P.J.; Liu, C.; Wang, J.H.; Gao, H.Y.; Hoelzelg, A.R.; Seim, I.; Lv, M.Q.; Lin, M.L.; et al. Comparative genomics provides insights into the aquatic adaptations of mammals. Proc. Natl. Acad. Sci. USA 2021, 118, e2106080118. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Jin, H.; Fu, J.Z. Molecular convergent and parallel evolution among four high-elevation anuran species from the Tibetan region. BMC Genom. 2020, 21, 839. [Google Scholar] [CrossRef]
- Rocha, J.L.; Godinho, R.; Brito, J.C.; Nielsen, R. Life in Deserts: The Genetic Basis of Mammalian Desert Adaptation. Trends Ecol. Evol. 2021, 36, 637–650. [Google Scholar] [CrossRef]
- Davies, K.T.J.; Bennett, N.C.; Faulkes, C.G.; Rossiter, S.J. Limited evidence for parallel molecular adaptations associated with the subterranean niche in mammals: A comparative study of three superorders. Mol. Biol. Evol. 2018, 35, 2544–2559. [Google Scholar] [CrossRef]
- Birkeland, S.; Gustafsson, A.L.S.; Brysting, A.K.; Brochmann, C.; Nowak, M.D. Multiple genetic trajectories to extreme abiotic stress adaptation in arctic brassicaceae. Mol. Biol. Evol. 2020, 37, 2052–2068. [Google Scholar] [CrossRef]
- Ge, R.L.; Cai, Q.L.; Shen, Y.Y.; San, A.; Ma, L.; Zhang, Y.; Yi, X.; Chen, Y.; Yang, L.F.; Huang, Y.; et al. Draft genome sequence of the Tibetan antelope. Nat. Commun. 2013, 4, 1858. [Google Scholar] [CrossRef]
- Fedosenko, A.K.; Blank, D.A. Capra sibirica. Mamm. Species 2001, 675, 1–13. [Google Scholar] [CrossRef]
- Salvatori, M.; Tenan, S.; Oberosler, V.; Augugliaro, C.; Christe, P.; Groff, C.; Krofel, M.; Zimmermann, F.; Rovero, F. Co-occurrence of snow leopard.; wolf and Siberian ibex under livestock encroachment into protected areas across the Mongolian Altai. Biol. Conserv. 2021, 261, 109294. [Google Scholar] [CrossRef]
- Han, L.; Blanks, D.; Wang, M.Y.; Yang, W.K.; Alves Da Silva, A.; Alves, J. Grouping patterns and social organization in Siberian ibex (Capra sibirica): Feeding strategy matters. Folia Zool. 2019, 68, 35–42. [Google Scholar]
- Chen, L.; Qiu, Q.; Jiang, Y.; Wang, K.; Lin, Z.S.; Li, Z.P.; Bibi, F.; Yang, Y.Z.; Wang, J.H.; Nie, W.H.; et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 2019, 364, eaav6202. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Yang, J.; Zhou, H.; Tang, M.F.; Lu, W.M.; Yan, A.; Hou, Y.; Zhang, S.Y. A novel method for identifying shahtoosh. J. Forensic Sci. 2014, 59, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Tonin, C.; Bianchetto, M.; Vineis, C.; Festa, B.M. Differentiating fine hairs from wild and domestic species: Investigations of shatoosh.; yangir.; and cashmere fibers. Text. Res. J. 2002, 72, 701–705. [Google Scholar] [CrossRef]
- Vineis, C.; Aluigi, A.; Tonin, C. Outstanding traits and thermal behaviour for the identification of speciality animal fibres. Text. Res. J. 2011, 81, 264–272. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Wang, Z.X.; Liu, Y.; Wang, R.J.; Zhang, Y.J.; Su, R.; Li, J.Q. Genetic evaluation of fiber length and fiber diameter from Inner Mongolia White Cashmere goats at different ages. Small Rumin. Res. 2015, 123, 22–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, D.Y.; Xu, Y.N.; Qin, Y.T.; Gu, M.; Cai, W.D.; Bai, Z.X.; Zhang, X.J.; Chen, R.; Sun, Y.G.; et al. Selection of Cashmere Fineness Functional Genes by Translatomics. Front. Genet. 2021, 12, 775499. [Google Scholar] [CrossRef]
- Zeder, M.A.; Hesse, B. The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago. Science 2000, 287, 2254–2257. [Google Scholar] [CrossRef]
- Wang, X.L.; Liu, J.; Zhou, G.X.; Guo, J.Z.; Yan, H.L.; Niu, Y.Y.; Li, Y.; Yuan, C.; Geng, R.Q.; Lan, X.Y.; et al. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 2016, 6, 38932. [Google Scholar] [CrossRef]
- Cai, Y.D.; Fu, W.W.; Cai, D.W.; Heller, R.; Zheng, Z.Q.; Wen, J.; Li, H.; Wang, X.L.; Alshawi, A.; Sun, Z.Y.; et al. Ancient Genomes Reveal the Evolutionary History and Origin of Cashmere-Producing Goats in China. Mol. Biol. Evol. 2020, 37, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Antonini, M.; Wang, J.; Lou, Y.J.; Tang, P.R.; Renieri, C.; Pazzaglia, I.; Valbonesi, A. Effects of year and sampling site on mean fibre diameter of Alashan cashmere goat. Small Rumin. Res. 2016, 137, 71–72. [Google Scholar] [CrossRef]
- Wang, F.H.; Zhang, L.; Gong, G.; Yan, X.C.; Zhang, L.; Zhang, F.T.; Liu, H.F.; Lv, Q.; Wang, Z.Y.; Wang, R.J.; et al. Genome-wide association study of fleece traits in Inner Mongolia Cashmere goats. Anim. Genet. 2021, 52, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Li, X.K.; Su, R.; Wan, W.T.; Zhang, W.G.; Jiang, H.Z.; Qiao, X.; Fan, Y.X.; Zhang, Y.J.; Wang, R.J.; Liu, Z.H.; et al. Identification of selection signals by large-scale whole-genome resequencing of cashmere goats. Sci. Rep. 2017, 7, 15142. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, Z.H.; Zhao, M.; Mu, Q.; Che, T.Y.; Xie, Y.C.; Ma, L.N.; Mi, L.; Li, J.Q.; Zhao, Y.H. Skin transcriptome reveals the periodic changes in genes underlying cashmere (ground hair) follicle transition in cashmere goats. BMC Genom. 2020, 21, 392. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Vanhoutte, E.K.; Claes, G.R.F.; Helderman-van den Enden, A.T.J.M.; Hoeijmakers, J.G.J.; Hellebrekers, D.M.E.I.; Haan, A.D.; Christiaans, I.; Deprez, R.H.L.; Boen, H.M.; et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum. Mutat. 2020, 41, 1091–1111. [Google Scholar] [CrossRef]
- Rey, C.; Gueguen, L.; Sémon, M.; Boussau, B. Accurate detection of convergent amino-acid evolution with pcoc. Mol. Biol. Evol. 2018, 35, 2296–2306. [Google Scholar] [CrossRef]
- Pesch, M.; Koenig, S.; Aumailley, M. Targeted disruption of the lama3 gene in adult mice is sufficient to induce skin inflammation and fibrosis. J. Investig. Dermatol. 2017, 137, 332–340. [Google Scholar] [CrossRef]
- Graves, K.T.; Henney, P.J.; Ennis, R.B. Partial deletion of the LAMA3 gene is responsible for hereditary junctional epidermolysis bullosa in the American Saddlebred Horse. Anim. Genet. 2009, 40, 35–41. [Google Scholar] [CrossRef]
- Sartelet, A.; Harland, C.; Tamma, N.; Karim, L.; Bayrou, C.; Li, W.B.; Ahariz, N.; Coppieters, W.; Georges, M.; Charlier, C. A stop-gain in the laminin.; alpha 3 gene causes recessive junctional epidermolysis bullosa in Belgian Blue cattle. Anim. Genet. 2015, 46, 566–570. [Google Scholar] [CrossRef]
- Tayem, R.; Niemann, C.; Pesch, M.; Morgner, J.; Niessen, C.M.; Wickström, S.A.; Aumailley, M. Laminin 332 is indispensable for homeostatic epidermal differentiation programs. J. Investig. Dermatol. 2021, 141, 2602–2610. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks.; and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Han, W.J.; Yang, F.; Wu, Z.H.; Guo, F.Q.; Zhang, J.J.; Hai, E.H.; Shang, F.Z.; Su, R.; Wang, R.J.; Wang, Z.Y.; et al. Inner Mongolian cashmere goat secondary follicle development regulation research based on mRNA-miRNA co-analysis. Sci. Rep. 2020, 10, 4519. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome-an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.K.; Burridge, K. Focal adhesions: A nexus for intracellular signaling and cytoskeletal dynamics. Exp. Cell Res. 2000, 261, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins bidirectional.; allosteric signaling machines. Cell. 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.Q.; Costell, M.; Fässler, R. Integrin activation by talin.; kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.Q.; Wen, Z.K.; Sun, Q.Y.; Zhao, X.H.; Yang, H.; Shi, X.Y.; Xin, T. Apatinib suppresses the proliferation and apoptosis of gastric cancer cells via the PI3K/AKT signaling pathway. J. BUON Off. J. Balk. Union Oncol. 2019, 24, 1985–1991. [Google Scholar]
- Sun, Y.; Dai, J.; Jiao, R.; Jiang, Q.; Wang, J. Homoharringtonine inhibits fibroblasts proliferation, extracellular matrix production and reduces surgery-induced knee arthrofibrosis via PI3K/AKT/mTOR pathway-mediated apoptosis. J. Orthop. Surg. Res. 2021, 16, 9. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.H.; Shi, X.; Ji, J.; Zhan, F.Q.; Leng, H. Sortilin regulates keratinocyte proliferation and apoptosis through the PI3K-AKT signaling pathway. Life Sci. 2021, 278, 119630. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.Z.; Wang, N.N.; Li, Z.P.; Heller, R.; Liu, R.; Zhao, Y.; Han, J.G.; Pan, X.Y.; Zheng, Z.Q.; et al. Genetic basis of ruminant headgear and rapid antler regeneration. Science 2019, 364, eaav6335. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.S.; Chen, L.; Chen, X.Q.; Zhong, Y.B.; Yang, Y.; Xia, W.H.; Liu, C.; Zhu, W.B.; Wang, H.; Yan, B.Y.; et al. Biological adaptations in the Arctic cervid.; the reindeer (Rangifer tarandus). Science 2019, 364, eaav6312. [Google Scholar] [CrossRef]
- Liu, C.; Gao, J.B.; Cui, X.X.; Li, Z.P.; Chen, L.; Yuan, Y.; Zhang, Y.L.; Mei, L.W.; Zhao, L.; Cai, D.; et al. A towering genome: Experimentally validated adaptations to high blood pressure and extreme stature in the giraffe. Sci. Adv. 2021, 7, eabe9459. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, X.L.; Yan, H.L.; Zeng, J.; Ma, S.; Niu, Y.Y.; Zhou, G.X.; Jiang, Y.; Chen, Y.L. Comparative transcriptome analysis of fetal skin reveals key genes related to hair follicle morphogenesis in cashmere goats. PLoS ONE 2016, 11, e0151118. [Google Scholar] [CrossRef]
- Wu, C.L.; Qin, C.K.; Fu, X.F.; Huang, X.X.; Tian, K.C. Integrated analysis of lncRNAs and mRNAs by RNA-Seq in secondary hair follicle development and cycling (anagen.; catagen and telogen) of Jiangnan cashmere goat (Capra hircus). BMC Vet. Res. 2022, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.L.; Lu, J.; Fei, X.J.; Lu, Z.K.; Quan, K.; Liu, Y.B.; Chu, M.X.; Di, R.; Wang, H.H.; Wei, C.H.; et al. Genetic signatures of selection for cashmere traits in chinese goats. Animals 2020, 10, 1905. [Google Scholar] [CrossRef]
- Liu, B.; Gao, F.Q.; Guo, J.; Wu, D.; Hao, B.; Li, Y.R.; Zhao, C.F. A microarray-based analysis reveals that a short photoperiod promotes hair growth in the arbas cashmere goat. PLoS ONE 2016, 11, e0147124. [Google Scholar] [CrossRef]
- Wu, Z.H.; Hai, E.H.; Di, Z.Y.; Ma, R.; Shang, F.Z.; Wang, Y.; Wang, M.; Liang, L.L.; Rong, Y.J.; Pan, J.F.; et al. Using WGCNA (weighted gene co-expression network analysis) to identify the hub genes of skin hair follicle development in fetus stage of Inner Mongolia cashmere goat. PLoS ONE 2020, 15, e0243507. [Google Scholar] [CrossRef]
- Wang, J.F.; Sui, J.; Mao, C.; Li, X.R.; Chen, X.Y.; Liang, C.C.; Wang, X.H.; Wang, S.H.; Jia, C.L. Identification of key pathways and genes related to the development of hair follicle cycle in cashmere goats. Genes 2021, 12, 180. [Google Scholar] [CrossRef]
- Fu, W.W.; Wang, R.; Nanaei, H.A.; Wang, J.X.; Hu, D.X.; Jiang, Y. RGD v2.0: A major update of the ruminant functional and evolutionary genomics database. Nucleic Acids Res. 2021, 50, D1091–D1099. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.Q.; Wang, X.h.; Li, M.; Li, Y.J.; Yang, Z.R.; Wang, X.L.; Pan, X.Y.; Gong, M.; Zhang, Y.; Guo, Y.W.; et al. The origin of domestication genes in goats. Sci. Adv. 2020, 6, eaaz5216. [Google Scholar] [CrossRef] [PubMed]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Kent, W.J.; Riemer, C.; Elnitski, L.; Smit, A.F.A.; Roskin, K.M.; Baertsch, R.; Rosenbloom, K.; Clawson, H.; Green, E.D.; et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004, 14, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; de Koning, A.P.J.; Kim, H.; Gu, W.J.; Noonan, B.P.; Naylor, G.; Jiang, Z.J.; Parkinson, C.L.; Pollock, D.D. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8986–8991. [Google Scholar] [CrossRef] [PubMed]
- Giannoulatou, E.; Park, S.H.; Humphreys, D.T.; Ho Joshua, W.K. Verification and validation of bioinformatics software without a gold standard: A case study of BWA and Bowtie. BMC Bioinform. 2014, 15, S15. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation dna sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.Y.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.L.B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 29, 559. [Google Scholar] [CrossRef]
- Bu, D.C.; Luo, H.T.; Huo, P.P.; Wang, Z.H.; Zhang, S.; He, Z.H.; Wu, Y.; Zhao, L.H.; Liu, J.J.; Guo, J.C.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Li, Z.; Xie, G.; Li, X.; Wu, Z.; Li, M.; Liu, A.; Xiong, Y.; Wang, Y. Convergent Genomic Signatures of Cashmere Traits: Evidence for Natural and Artificial Selection. Int. J. Mol. Sci. 2023, 24, 1165. https://doi.org/10.3390/ijms24021165
Wang W, Li Z, Xie G, Li X, Wu Z, Li M, Liu A, Xiong Y, Wang Y. Convergent Genomic Signatures of Cashmere Traits: Evidence for Natural and Artificial Selection. International Journal of Molecular Sciences. 2023; 24(2):1165. https://doi.org/10.3390/ijms24021165
Chicago/Turabian StyleWang, Wei, Zhuohui Li, Guoxiang Xie, Xinmei Li, Zhipei Wu, Manman Li, Anguo Liu, Yan Xiong, and Yu Wang. 2023. "Convergent Genomic Signatures of Cashmere Traits: Evidence for Natural and Artificial Selection" International Journal of Molecular Sciences 24, no. 2: 1165. https://doi.org/10.3390/ijms24021165
APA StyleWang, W., Li, Z., Xie, G., Li, X., Wu, Z., Li, M., Liu, A., Xiong, Y., & Wang, Y. (2023). Convergent Genomic Signatures of Cashmere Traits: Evidence for Natural and Artificial Selection. International Journal of Molecular Sciences, 24(2), 1165. https://doi.org/10.3390/ijms24021165