
Unveiling the Anti-Cancer Potential of Onoceranoid Triterpenes from Lansium domesticum Corr. cv. kokosan: An In Silico Study against Estrogen Receptor Alpha

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Molecular Docking
2.2. Prediction of Drug-Likeness
2.3. In Silico Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET)
2.4. Molecular Dynamics Simulations
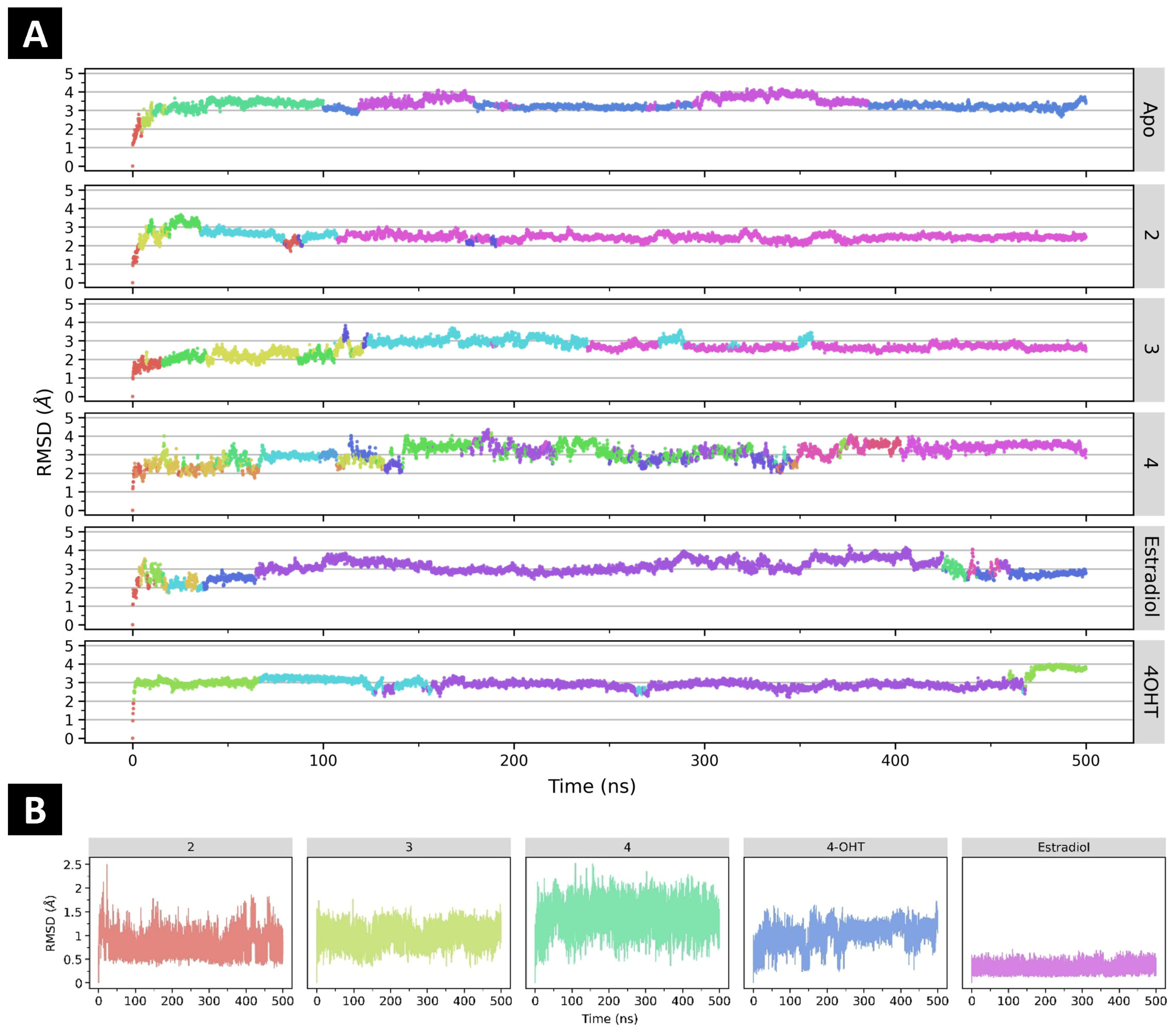
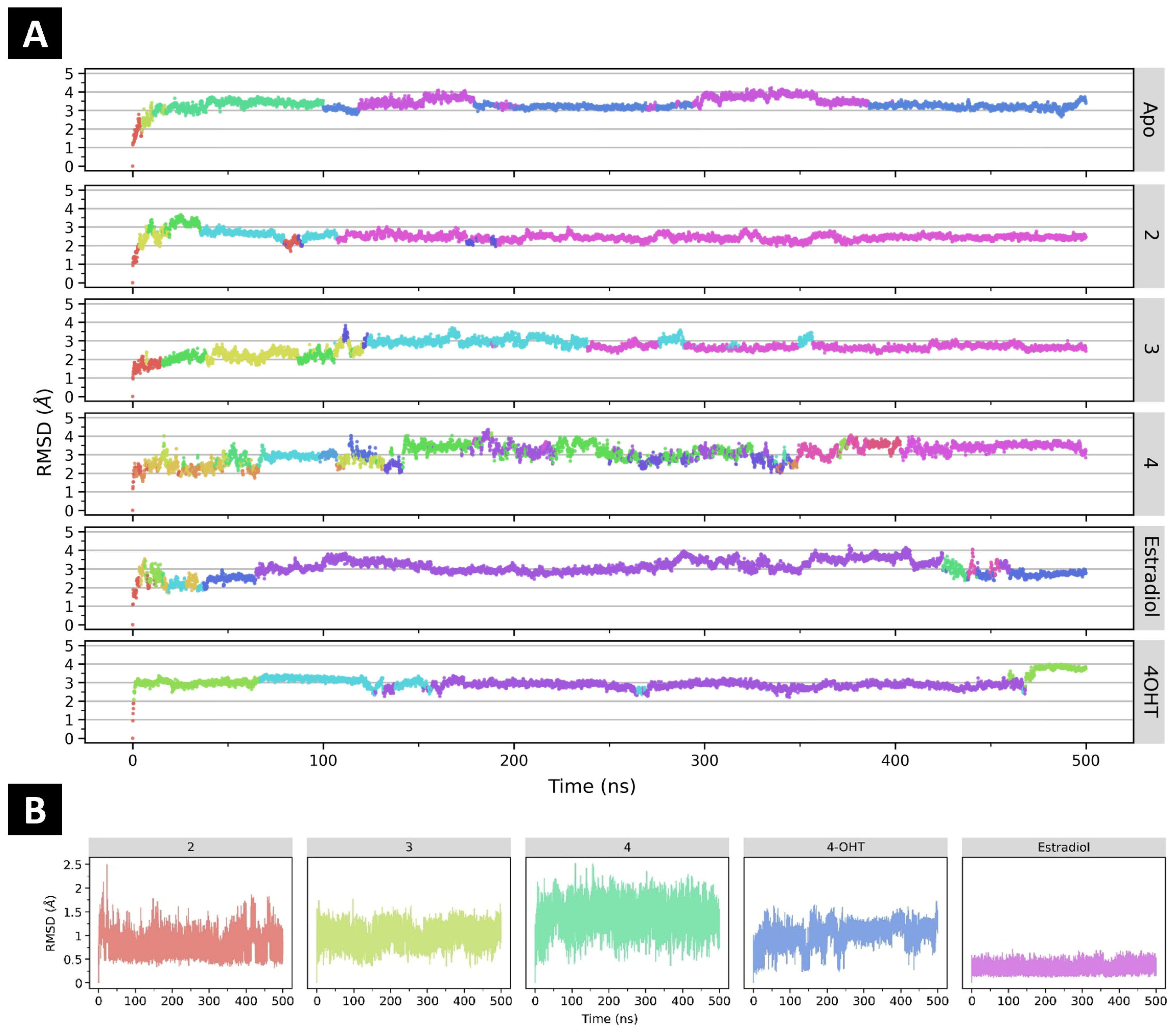
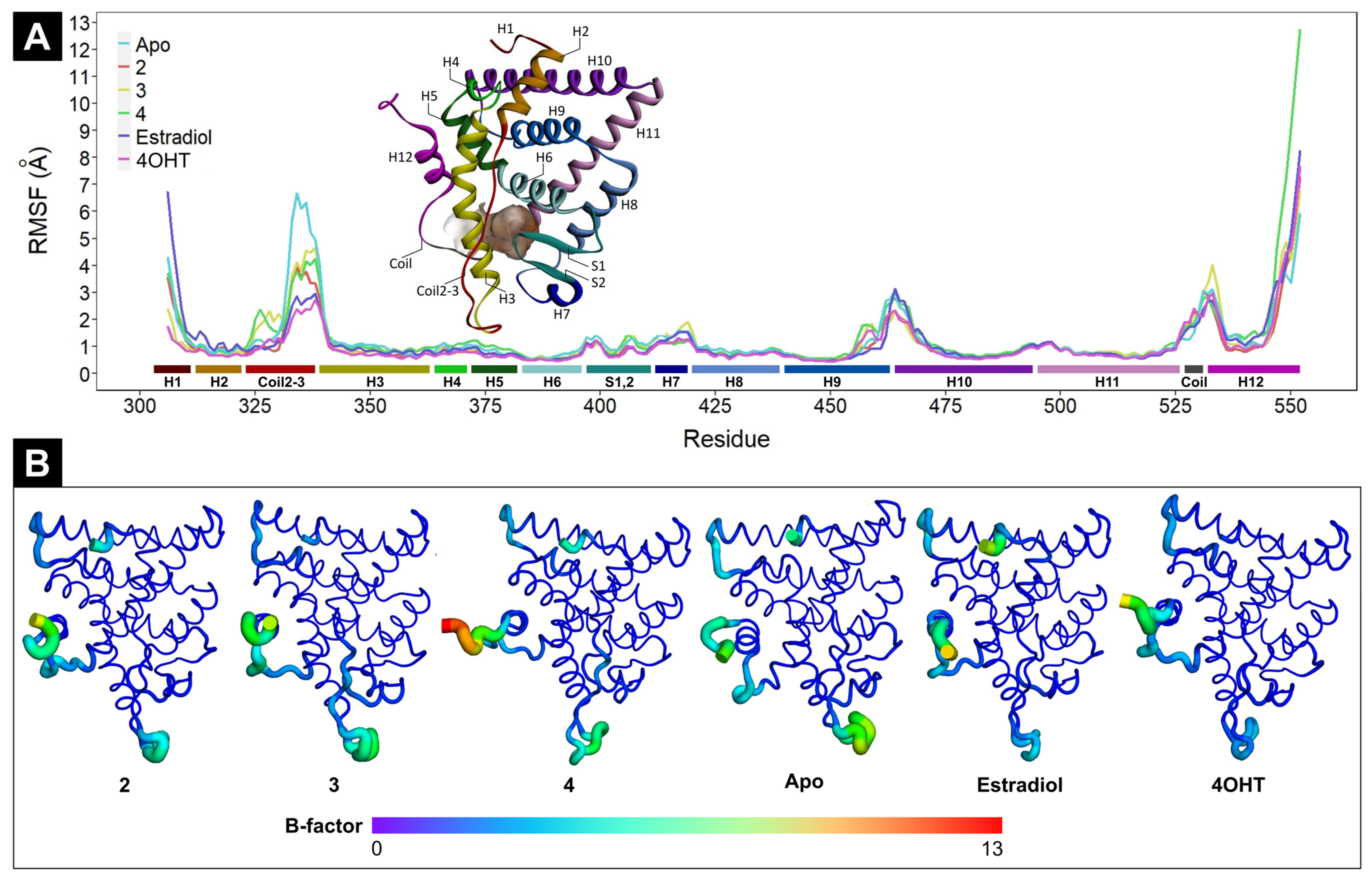
2.4.1. The Effect of Ligand Binding to ERα Conformational Stability
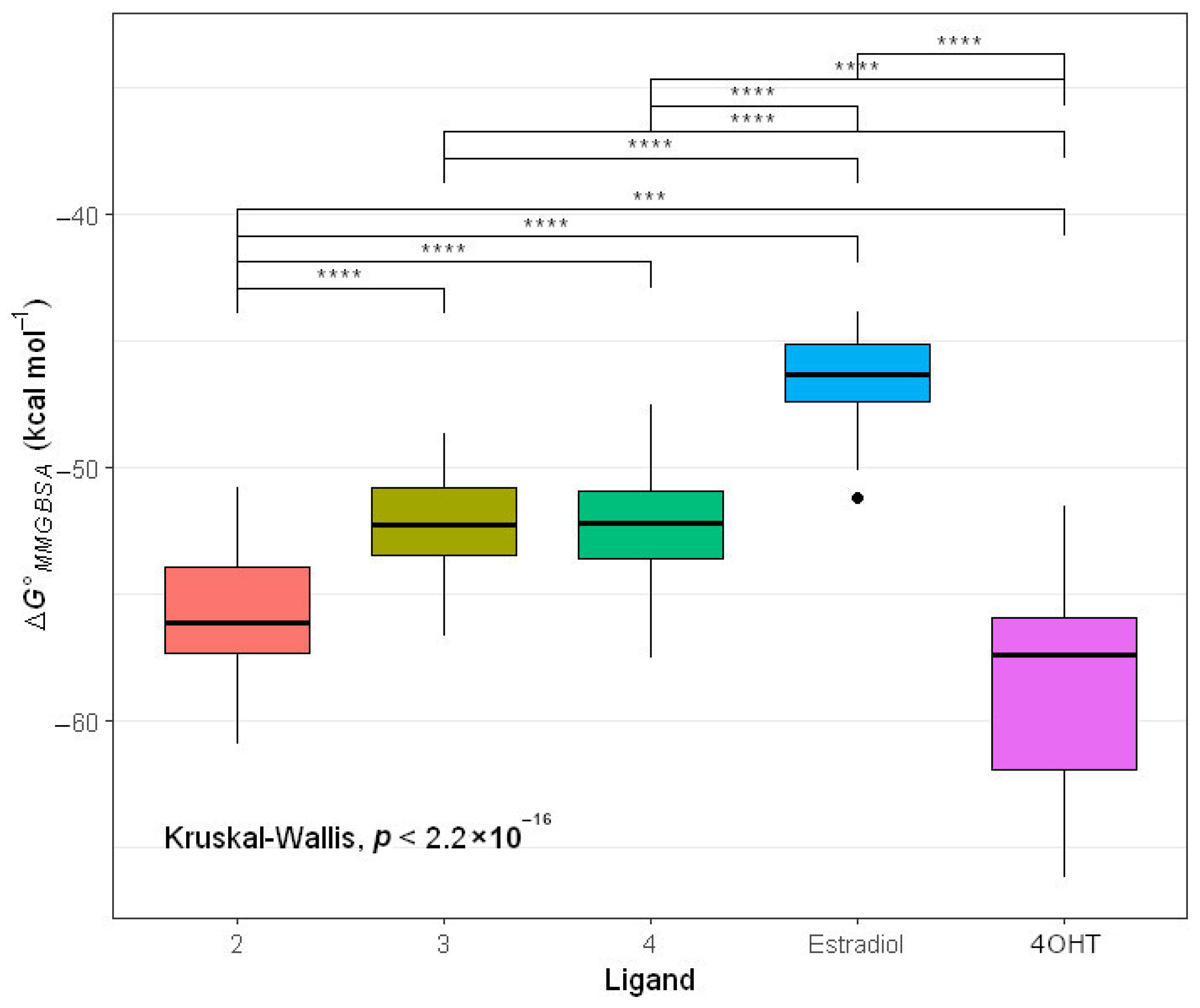
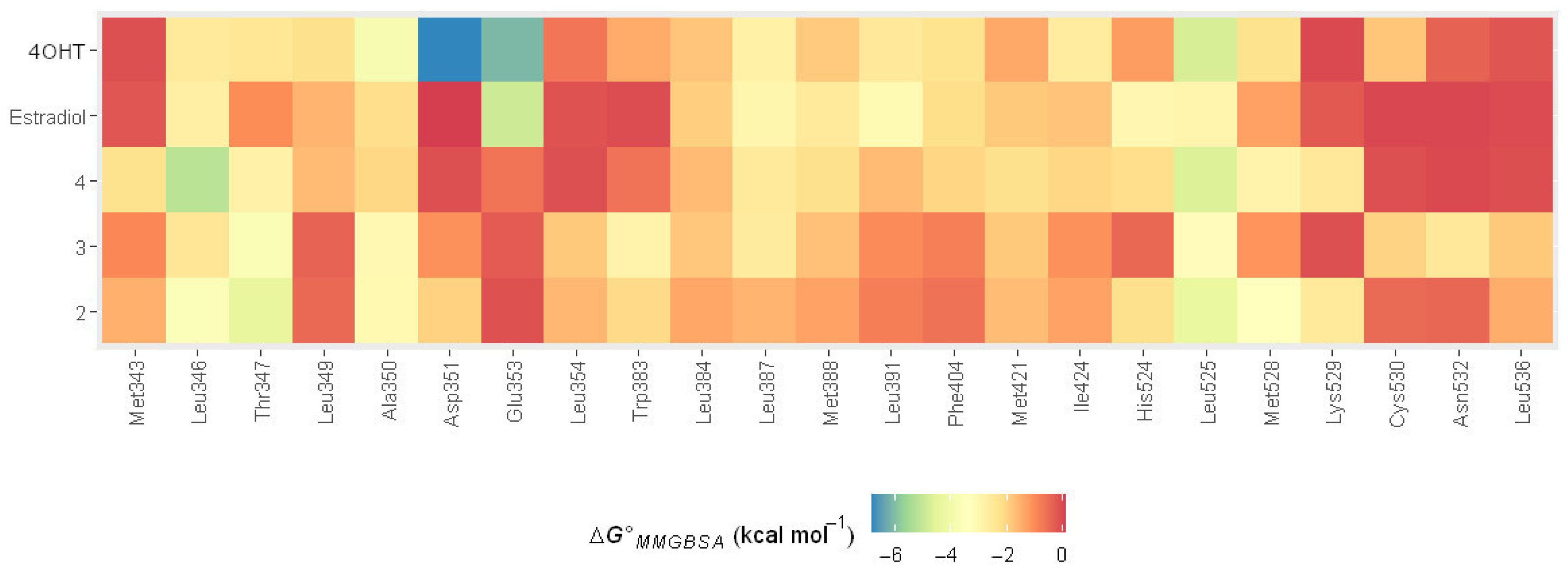
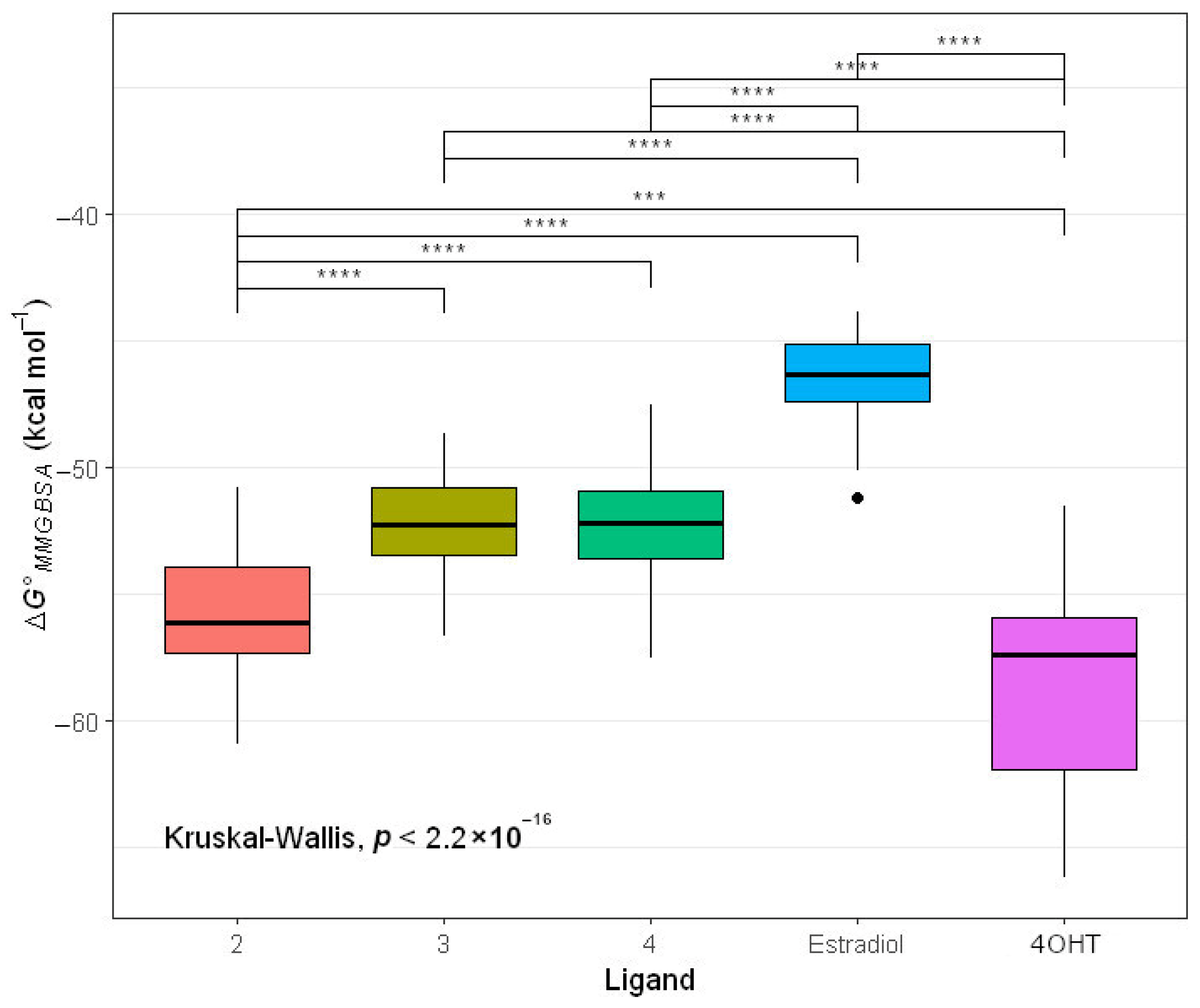
2.4.2. Molecular Mechanic Generalized Surface Area (MMGBSA) Binding Energy
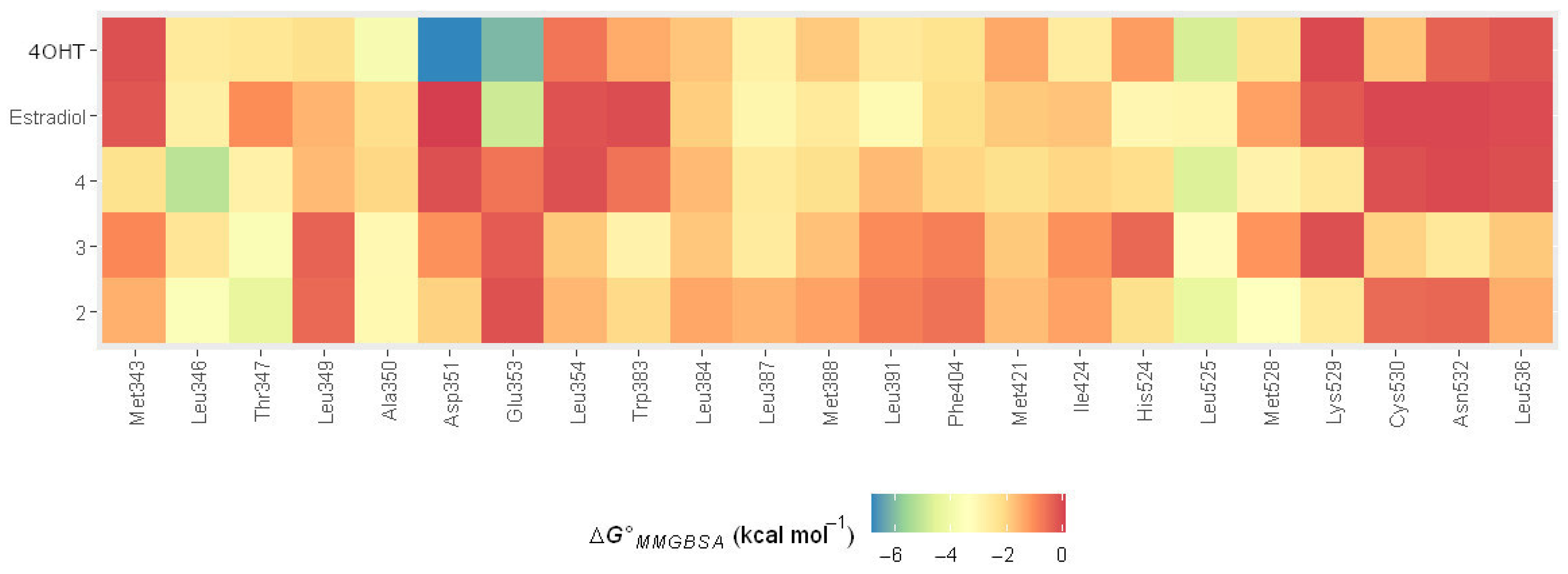
2.4.3. MMGBSA Binding Energy Decomposition
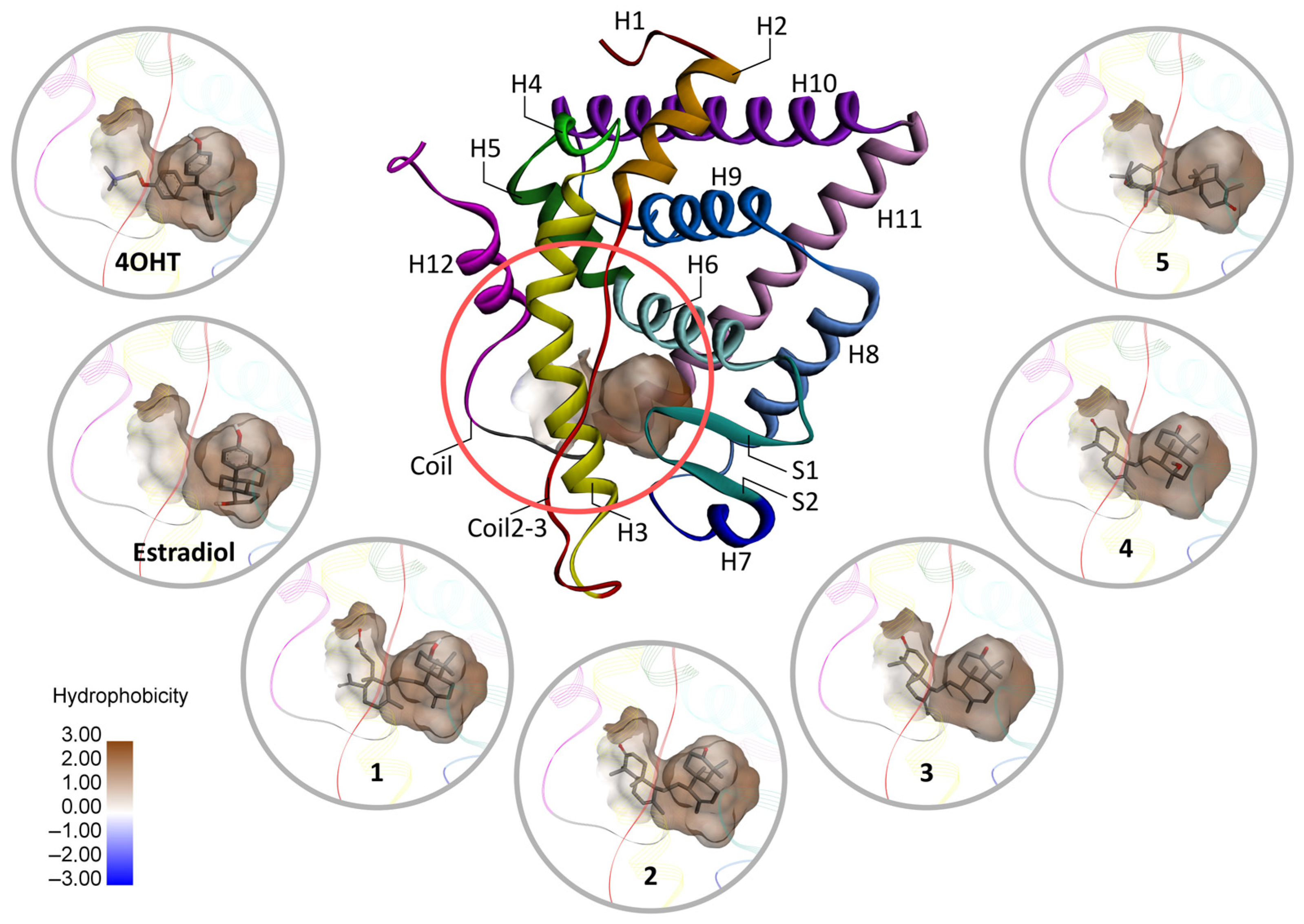
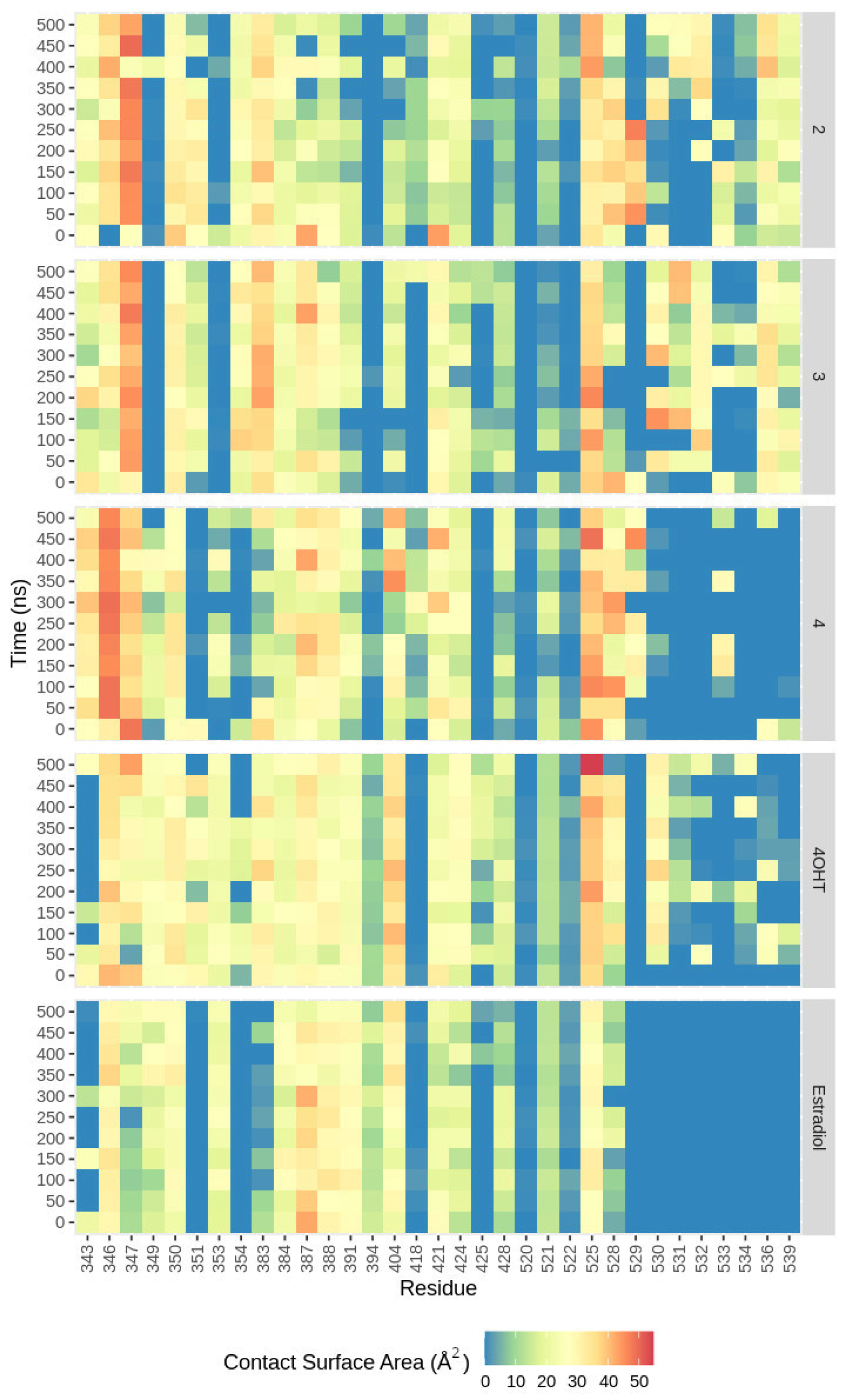
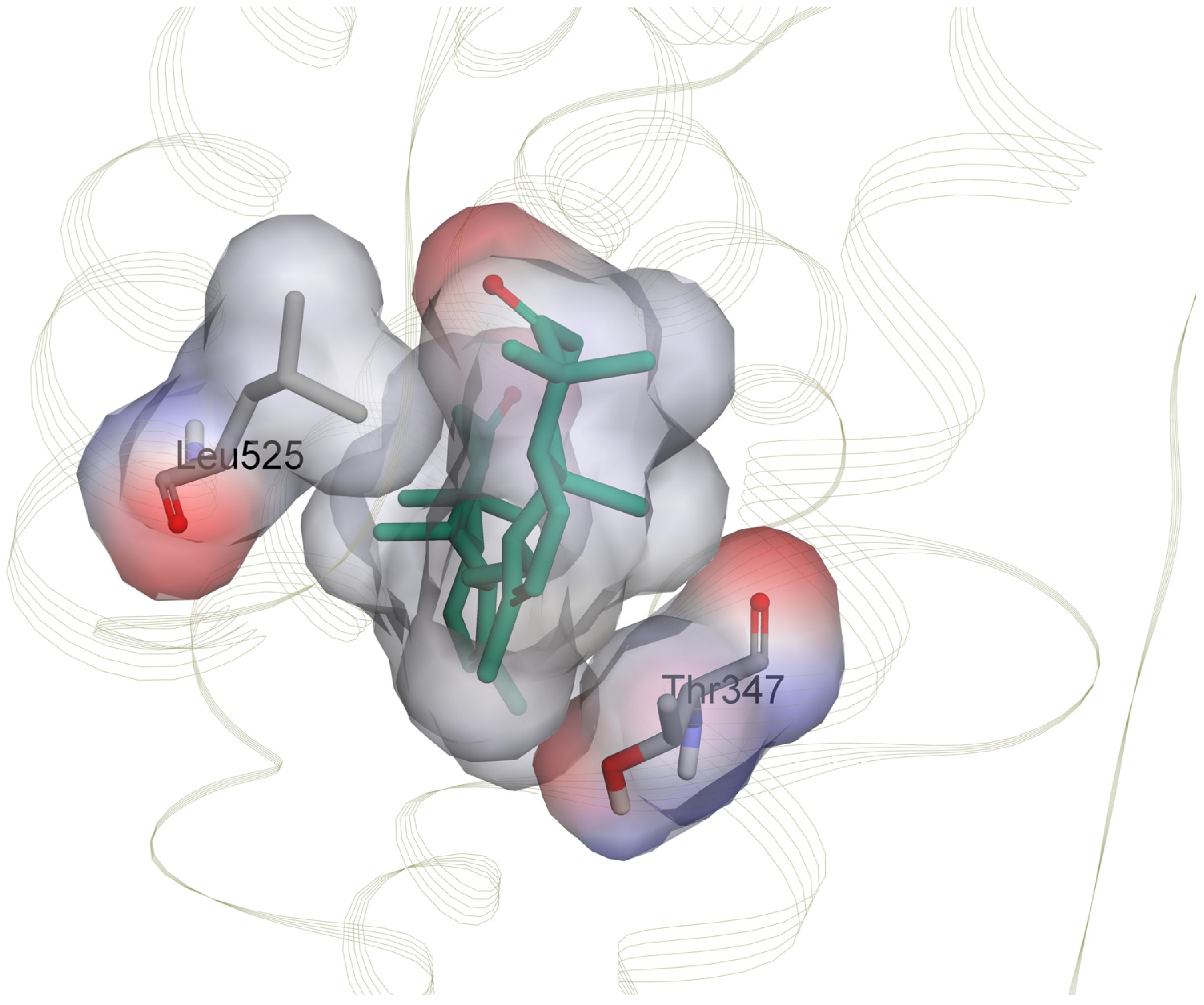
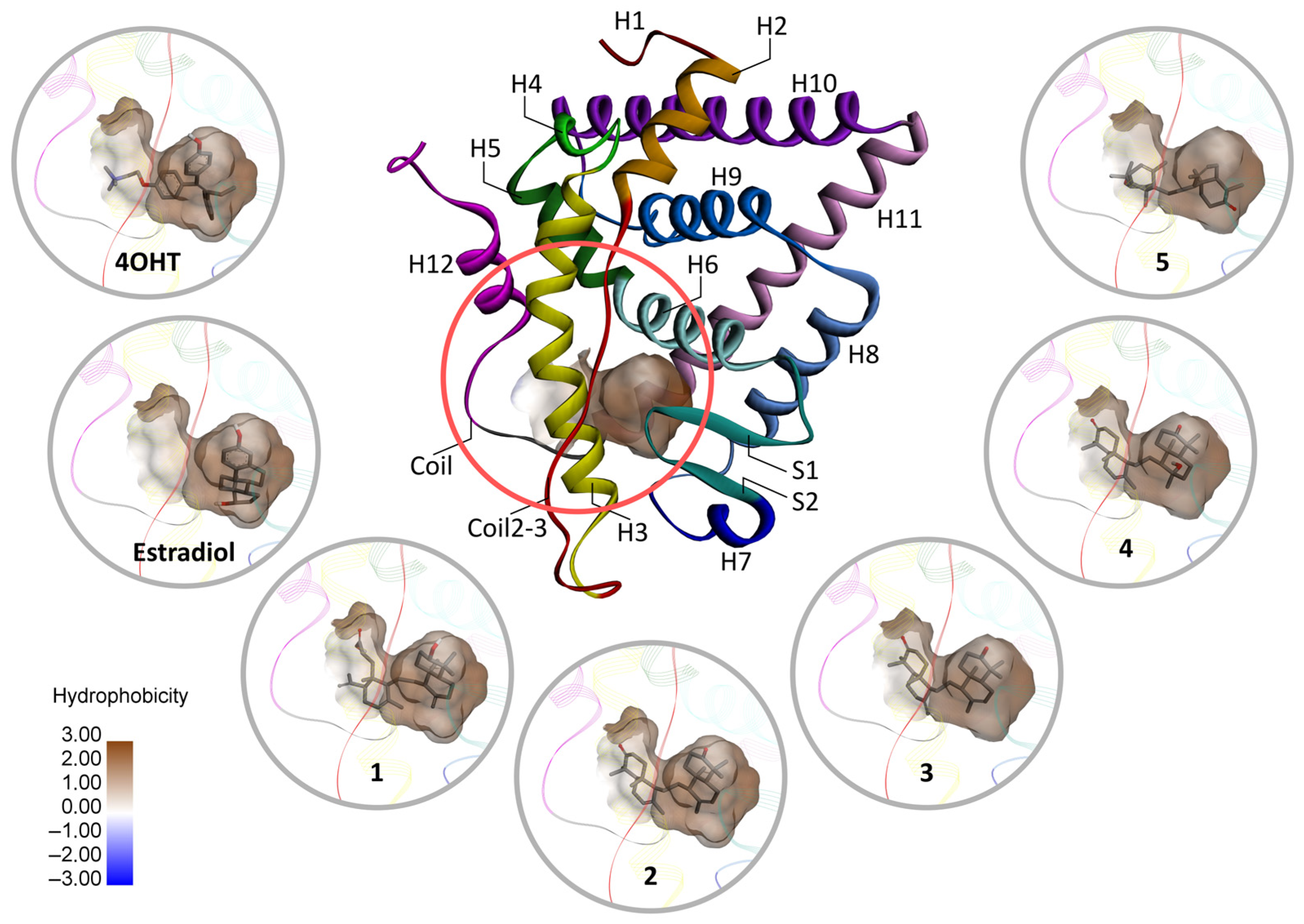
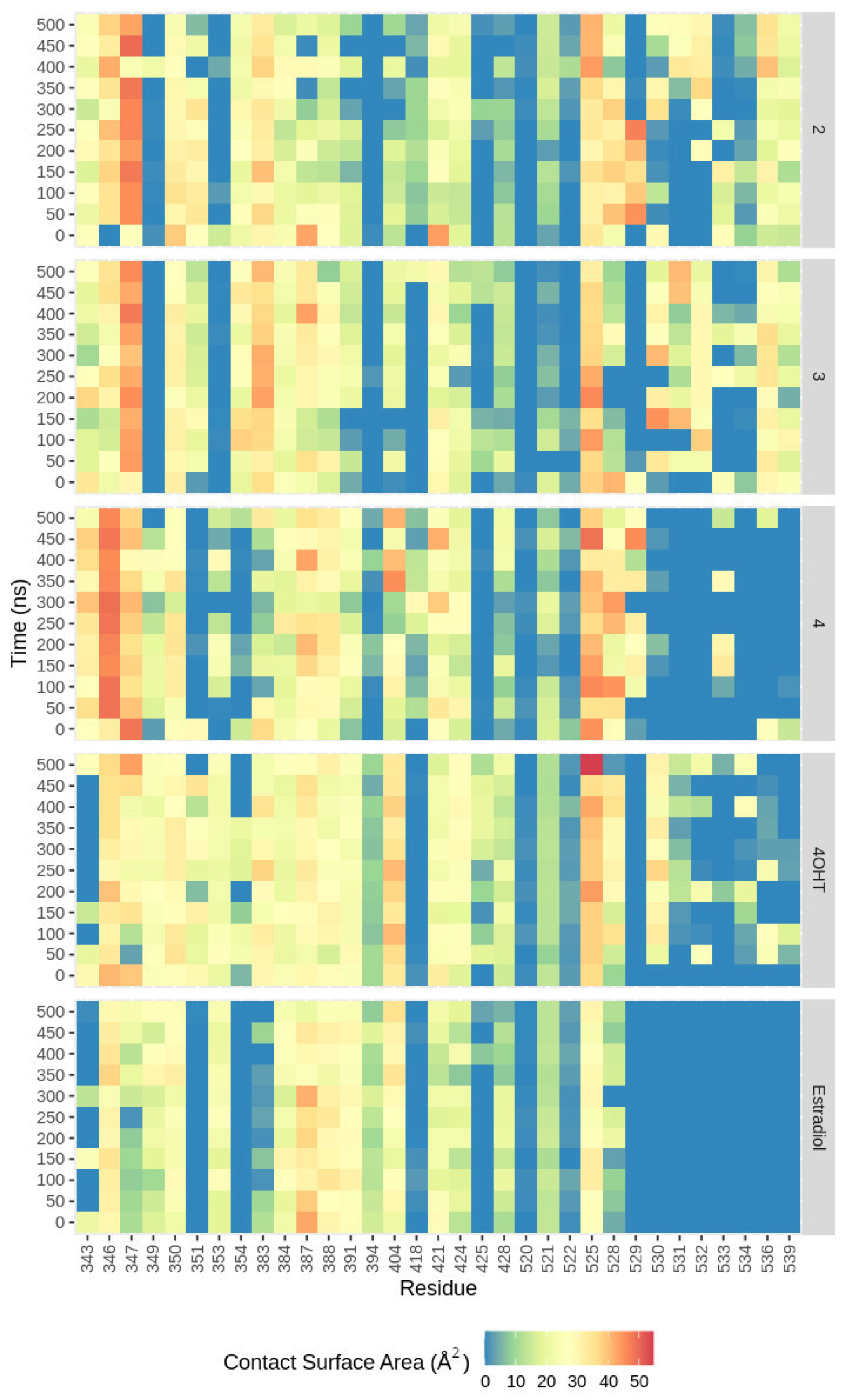
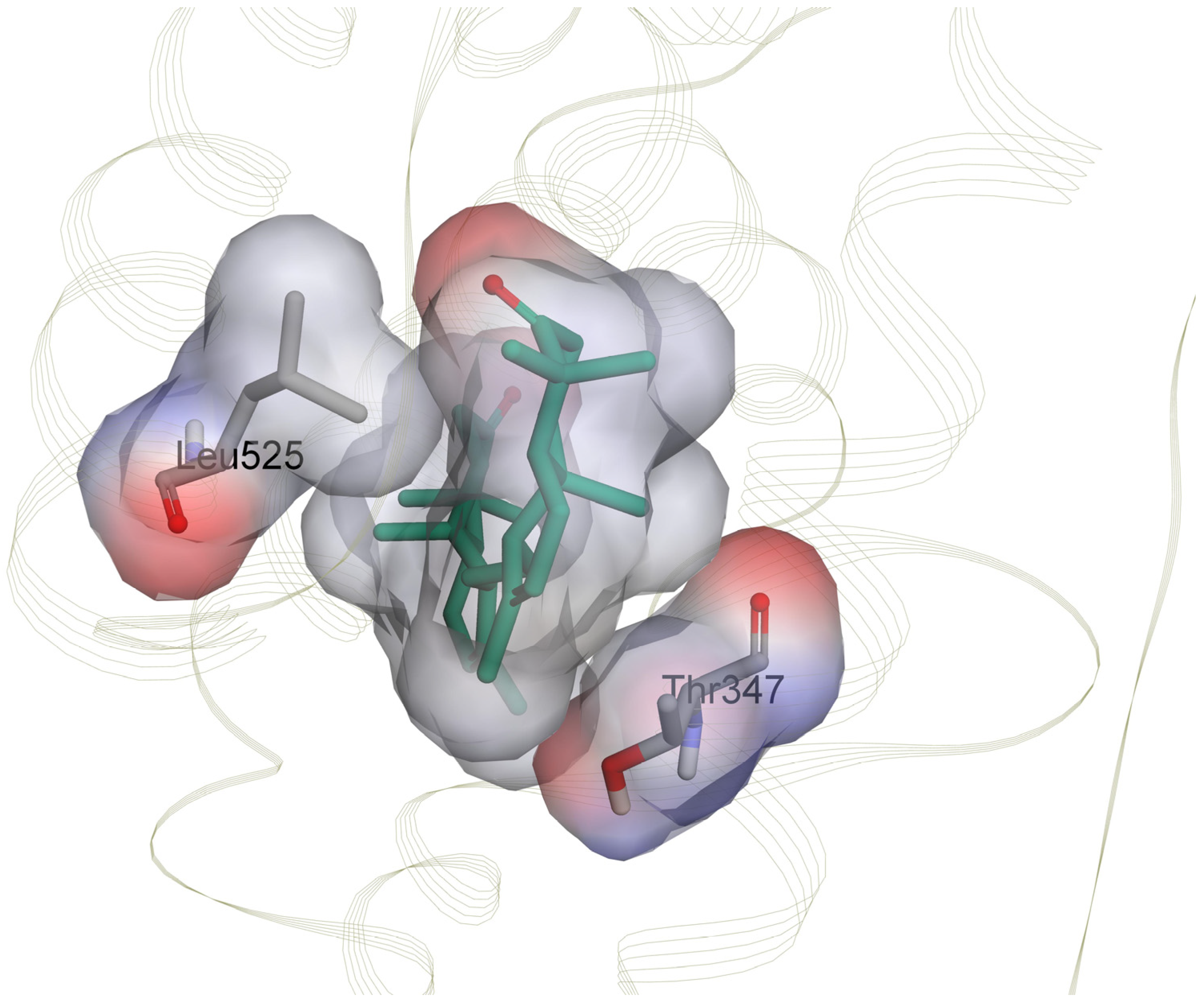
2.4.4. Nonbonded Interactions
3. Materials and Methods
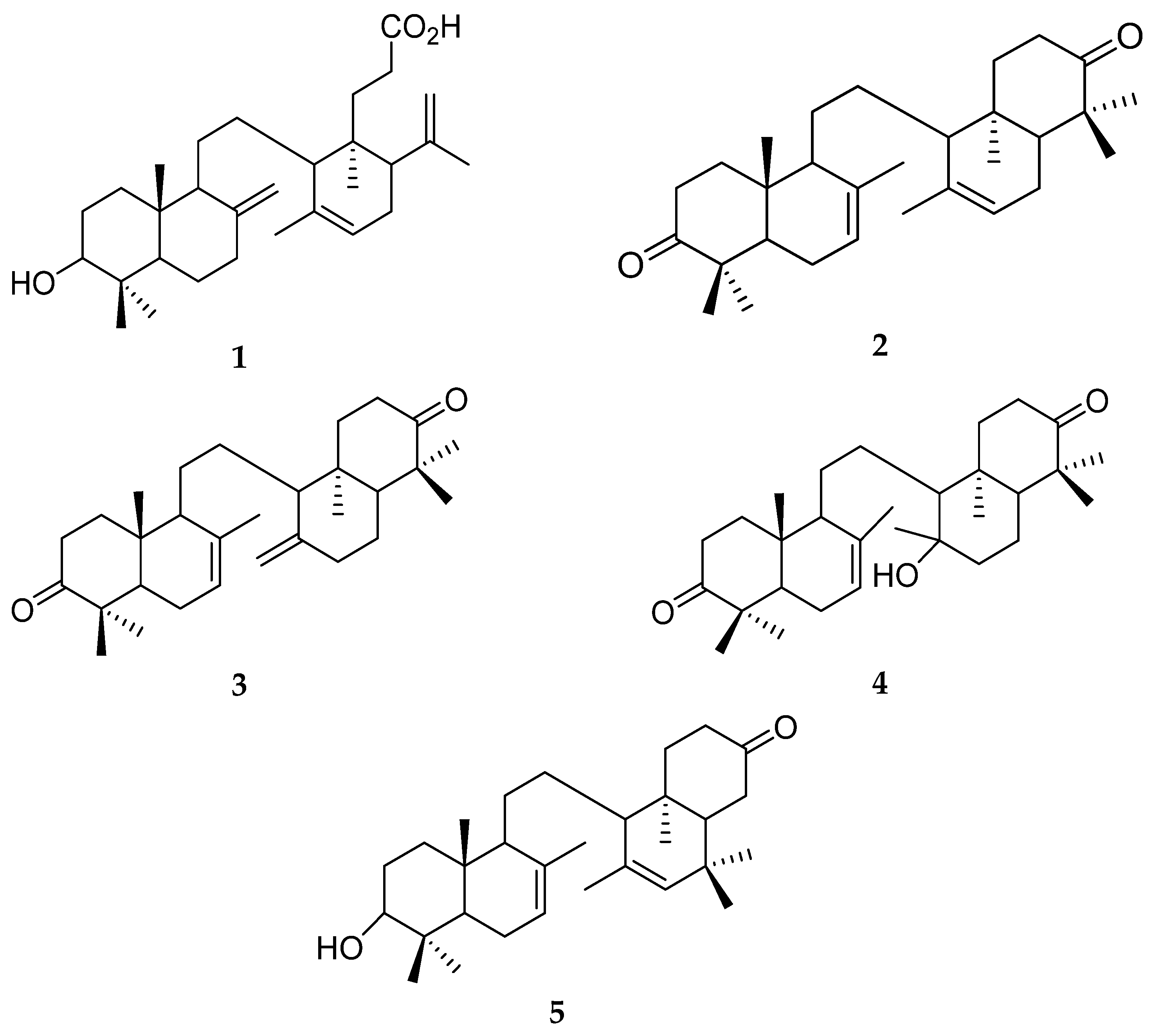
3.1. Preparation of Onoceranoid Triterpenes Structures
3.2. Molecular Docking of Onoceranoid Triterpenes against ERα
3.3. Lipinski’s Rule of Five and ADMET Analysis
3.4. Molecular Dynamics Simulations
3.5. Trajectory Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Salehiniya, H. Epidemiological Characteristics of and Risk Factors for Breast Cancer in the World. Breast Cancer Targets Ther. 2019, 11, 151–164. [Google Scholar] [CrossRef]
- WHO. Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 30 June 2023).
- Fares, M.Y.; Salhab, H.A.; Khachfe, H.H.; Khachfe, H.M. Breast Cancer Epidemiology among Lebanese Women: An 11-Year Analysis. Medicina 2019, 55, 463. [Google Scholar] [CrossRef]
- Mueller, C.; Haymond, A.; Davis, J.B.; Williams, A.; Espina, V. Protein Biomarkers for Subtyping Breast Cancer and Implications for Future Research. Expert Rev. Proteom. 2018, 15, 131–152. [Google Scholar] [CrossRef]
- Niemeier, L.A.; Dabbs, D.J.; Beriwal, S.; Striebel, J.M.; Bhargava, R. Androgen Receptor in Breast Cancer: Expression in Estrogen Receptor-Positive Tumors and in Estrogen Receptor-Negative Tumors with Apocrine Differentiation. Mod. Pathol. 2010, 23, 205–212. [Google Scholar] [CrossRef]
- Liu, M.-M.; Albanese, C.; Anderson, C.M.; Hilty, K.; Webb, P.; Uht, R.M.; Price, R.H.; Pestell, R.G.; Kushner, P.J. Opposing Action of Estrogen Receptors α and β on Cyclin D1 Gene Expression. J. Biol. Chem. 2002, 277, 24353–24360. [Google Scholar] [CrossRef]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmäki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain Protein BRD4 Is Required for Estrogen Receptor-Dependent Enhancer Activation and Gene Transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef]
- Xue, M.; Zhang, K.; Mu, K.; Xu, J.; Yang, H.; Liu, Y.; Wang, B.; Wang, Z.; Li, Z.; Kong, Q.; et al. Regulation of Estrogen Signaling and Breast Cancer Proliferation by an Ubiquitin Ligase TRIM56. Oncogenesis 2019, 8, 30. [Google Scholar] [CrossRef]
- Sun, X.; Ding, S.; Lu, S.; Wang, Z.; Chen, X.; Shen, K. Identification of Ten Mitosis Genes Associated with Tamoxifen Resistance in Breast Cancer. OncoTargets Ther. 2021, 14, 3611–3624. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Lorizio, W.; Wu, A.H.B.; Beattie, M.S.; Rugo, H.; Tchu, S.; Kerlikowske, K.; Ziv, E. Clinical and Biomarker Predictors of Side Effects from Tamoxifen. Breast Cancer Res. Treat. 2012, 132, 1107–1118. [Google Scholar] [CrossRef]
- Abdmouleh, F.; Arbi, M.E.; Saad, H.B.; Jellali, K.; Ketata, E.; Amara, I.B.; Pigeon, P.; Hassen, H.B.; Top, S.; Jaouen, G.; et al. Antimicrobial, Antitumor and Side Effects Assessment of a Newly Synthesized Tamoxifen Analog. Curr. Top. Med. Chem. 2020, 20, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-J.; Lim, Y.S.; Kim, M.S.; Lee, B.; Kim, B.-Y.; Kim, Z.; Lee, J.E.; Lee, M.H.; Kim, S.G.; Kim, Y.S. Risk of Fatty Liver after Long-Term Use of Tamoxifen in Patients with Breast Cancer. PLoS ONE 2020, 15, e0236506. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Clappier, C.; Kleiter, I.; Heuchel, R. Tamoxifen Affects Chronic Pancreatitis-Related Fibrogenesis in an Experimental Mouse Model: An Effect beyond Cre Recombination. FEBS Open Bio 2019, 9, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Isobe, H.; Shimoda, M.; Kan, Y.; Tatsumi, F.; Katakura, Y.; Kimura, T.; Obata, A.; Kohara, K.; Nakanishi, S.; Mune, T.; et al. A Case of Tamoxifen-Induced Hypertriglyceridemia Monitoring the Changes in Lipoprotein Fractions over Time. BMC Endocr. Disord. 2021, 21, 115. [Google Scholar] [CrossRef]
- Daurio, N.A.; Tuttle, S.W.; Worth, A.J.; Song, E.Y.; Davis, J.M.; Snyder, N.W.; Blair, I.A.; Koumenis, C. AMPK Activation and Metabolic Reprogramming by Tamoxifen through Estrogen Receptor–Independent Mechanisms Suggests New Uses for This Therapeutic Modality in Cancer Treatment. Cancer Res. 2016, 76, 3295–3306. [Google Scholar] [CrossRef]
- Bishayee, A.; Ahmed, S.; Brankov, N.; Perloff, M. Triterpenoids as Potential Agents for the Chemoprevention and Therapy of Breast Cancer. Front. Biosci. 2011, 16, 980–996. [Google Scholar] [CrossRef]
- Mitra, S.; Dash, R. Natural Products for the Management and Prevention of Breast Cancer. Evid.-Based Complement. Altern. Med. 2018, 2018, e8324696. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Meng, X.; Gan, R.-Y.; Zhang, J.-J.; Li, H.-B. Dietary Natural Products for Prevention and Treatment of Breast Cancer. Nutrients 2017, 9, 728. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, C.; Bao, J.; Jia, X.; Liang, Y.; Wang, X.; Chen, M.; Su, H.; Li, P.; Wan, J.-B.; et al. Synergistic Chemopreventive Effects of Curcumin and Berberine on Human Breast Cancer Cells through Induction of Apoptosis and Autophagic Cell Death. Sci. Rep. 2016, 6, 26064. [Google Scholar] [CrossRef]
- Ranjan, A.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Wright, S.; Srivastava, S.; Das, H.; Srivastava, S.; Prasad, S.; Srivastava, S.K. Role of Phytochemicals in Cancer Prevention. Int. J. Mol. Sci. 2019, 20, 4981. [Google Scholar] [CrossRef]
- Abrahim, N.N.; Kanthimathi, M.S.; Abdul-Aziz, A. Piper Betle Shows Antioxidant Activities, Inhibits MCF-7 Cell Proliferation and Increases Activities of Catalase and Superoxide Dismutase. BMC Complement. Altern. Med. 2012, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Bimonte, S.; Barbieri, A.; Palma, G.; Rea, D.; Luciano, A.; D’Aiuto, M.; Arra, C.; Izzo, F. Dissecting the Role of Curcumin in Tumour Growth and Angiogenesis in Mouse Model of Human Breast Cancer. BioMed Res. Int. 2015, 2015, e878134. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.M.; Sekar, M.; Seow, L.J.; Gan, S.H.; Bonam, S.R.; Rani, N.N.I.M.; Lum, P.T.; Subramaniyan, V.; Wu, Y.S.; Fuloria, N.K.; et al. Mangifera indica (Mango): A Promising Medicinal Plant for Breast Cancer Therapy and Understanding Its Potential Mechanisms of Action. Breast Cancer Targets Ther. 2021, 13, 471–503. [Google Scholar] [CrossRef]
- Ko, E.-Y.; Moon, A. Natural Products for Chemoprevention of Breast Cancer. J. Cancer Prev. 2015, 20, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Mayanti, T.; Sianturi, J.; Harneti, D.; Darwati; Supratman, U.; Rosli, M.M.; Fun, H.-K. 9,19-Cyclolanost-24-En-3-One,21,23-Epoxy-21,22-Dihydroxy (21R, 22S, 23S) from the Leaves of Lansium domesticum Corr cv kokossan. Molbank 2015, 2015, M880. [Google Scholar] [CrossRef]
- Mayanti, T.; Supratman, U.; Mukhtar, M.R.; Awang, K.; Ng, S.W. Kokosanolide from the Seed of Lansium domesticum Corr. Acta Crystallogr. Sect. E Crystallogr. Commun. 2009, 65, o750. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Gordon, M.A.; Babbs, B.; Spoelstra, N.S.; Carson Butterfield, K.T.; Torkko, K.C.; Phan, V.T.; Barton, V.N.; Rogers, T.J.; Sartorius, C.A.; et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol. Cancer Res. 2016, 14, 1054–1067. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D’Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the Androgen Receptor in Breast Cancer and Preclinical Analysis of Enzalutamide. Breast Cancer Res. 2014, 16, R7. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Z.; Gao, Y.; Wang, L.; Chen, C.; Wang, X. TP53 Mutations Promote Immunogenic Activity in Breast Cancer. J. Oncol. 2019, 2019, e5952836. [Google Scholar] [CrossRef]
- Nishizawa, M.; Emura, M.; Yamada, H.; Shiro, M.; Chairul; Hayashi, Y.; Tokuda, V.H. Isolation of a New Cycloartanoid Triterpene from Leaves of Lansium domesticum Novel Skin-Tumor Promotion Inhibitors. Tetrahedron Lett. 1989, 30, 5615–5618. [Google Scholar] [CrossRef]
- Tjokronegero, R.; Mayanti, T.; Supratman, U.; Mukhtar, M.R.; Ng, S.W. 8,14-Secogammacera-7,14(27)-Diene-3,21-Dione–8,14-Secogammacera-7,14-Diene-3,21-Dione (1.5/0.5) from the Bark of Lansium domesticum Corr. Acta Crystallogr. Sect. E Crystallogr. Commun. 2009, 65, o1448. [Google Scholar] [CrossRef]
- Zulfikar; Putri, N.K.; Fajriah, S.; Yusuf, M.; Maharani, R.; Anshori, J.A.; Supratman, U.; Mayanti, T. 3-Hydroxy-8,14-Secogammacera-7,14-Dien-21-One: A New Onoceranoid Triterpenes from Lansium domesticum Corr. cv kokossan. Molbank 2020, 2020, M1157. [Google Scholar] [CrossRef]
- Nugroho, A.E.; Inoue, D.; Wong, C.P.; Hirasawa, Y.; Kaneda, T.; Shirota, O.; Hadi, A.H.A.; Morita, H. Reinereins A and B, New Onocerane Triterpenoids from Reinwardtiodendron cinereum. J. Nat. Med. 2018, 72, 588–592. [Google Scholar] [CrossRef]
- Fadhilah, K.; Wahyuono, S.; Astuti, P. A Bioactive Compound Isolated from Duku (Lansium domesticum Corr) Fruit Peels Exhibits Cytotoxicity against T47D Cell Line. F1000Research 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Hardianto, A.; Yusuf, M.; Hidayat, I.W.; Ishmayana, S.; Soedjanaatmadja, U.M.S. Exploring the Potency of Nigella Sativa Seed in Inhibiting SARS-CoV-2 Main Protease Using Molecular Docking and Molecular Dynamics Simulations. Indones. J. Chem. 2021, 21, 1252–1262. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for Molecular Docking: A Review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Sharma, D.; Kumar, S.; Narasimhan, B. Estrogen Alpha Receptor Antagonists for the Treatment of Breast Cancer: A Review. Chem. Cent. J. 2018, 12, 107. [Google Scholar] [CrossRef]
- Egeland, N.G.; Lunde, S.; Jonsdottir, K.; Lende, T.H.; Cronin-Fenton, D.; Gilje, B.; Janssen, E.A.M.; Søiland, H. The Role of MicroRNAs as Predictors of Response to Tamoxifen Treatment in Breast Cancer Patients. Int. J. Mol. Sci. 2015, 16, 24243–24275. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Singh, D. Defining Desirable Natural Product Derived Anticancer Drug Space: Optimization of Molecular Physicochemical Properties and ADMET Attributes. ADMET DMPK 2016, 4, 98–113. [Google Scholar] [CrossRef]
- Wei, F.; Yang, C.; Wu, L.; Sun, J.; Wang, Z.; Wang, Z. Simultaneous Determination and Pharmacokinetics Study of Three Triterpenes from Sanguisorba officinalis L. in Rats by UHPLC–MS/MS. Molecules 2022, 27, 5412. [Google Scholar] [CrossRef] [PubMed]
- Nishinarizki, V.; Hardianto, A.; Gaffar, S.; Muchtaridi, M.; Herlina, T. Virtual Screening Campaigns and ADMET Evaluation to Unlock the Potency of Flavonoids from Erythrina as 3CLpro SARS-COV-2 Inhibitors. J. Appl. Pharm. Sci. 2023, 13, 78–88. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- De Sá, A.G.C.; Long, Y.; Portelli, S.; Pires, D.E.V.; Ascher, D.B. ToxCSM: Comprehensive Prediction of Small Molecule Toxicity Profiles. Brief. Bioinform. 2022, 23, bbac337. [Google Scholar] [CrossRef]
- Vidal-Limon, A.; Aguilar-Toalá, J.E.; Liceaga, A.M. Integration of Molecular Docking Analysis and Molecular Dynamics Simulations for Studying Food Proteins and Bioactive Peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef]
- Hardianto, A.; Khanna, V.; Liu, F.; Ranganathan, S. Diverse Dynamics Features of Novel Protein Kinase C (PKC) Isozymes Determine the Selectivity of a Fluorinated Balanol Analogue for PKCε. BMC Bioinform. 2019, 19, 342. [Google Scholar] [CrossRef]
- Hardianto, A.; Liu, F.; Ranganathan, S. Molecular Dynamics Pinpoint the Global Fluorine Effect in Balanoid Binding to PKCε and PKA. J. Chem. Inf. Model. 2018, 58, 511–519. [Google Scholar] [CrossRef]
- Paris, R.D.; Quevedo, C.V.; Ruiz, D.D.A.; de Souza, O.N. An Effective Approach for Clustering InhA Molecular Dynamics Trajectory Using Substrate-Binding Cavity Features. PLoS ONE 2015, 10, e0133172. [Google Scholar] [CrossRef]
- Pollard, T.D.; Earnshaw, W.C.; Lippincott-Schwartz, J.; Johnson, G.T. (Eds.) Chapter 4—Biophysical Principles. In Cell Biology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 53–62. ISBN 978-0-323-34126-4. [Google Scholar]
- Ribeiro, J.; Ríos-Vera, C.; Melo, F.; Schüller, A. Calculation of Accurate Interatomic Contact Surface Areas for the Quantitative Analysis of Non-Bonded Molecular Interactions. Bioinformatics 2019, 35, 3499–3501. [Google Scholar] [CrossRef]
- Ouellette, R.J.; Rawn, J.D. 4-Alkanes and Cycloalkanes: Structures and Reactions. In Organic Chemistry; Ouellette, R.J., Rawn, J.D., Eds.; Elsevier: Boston, MA, USA, 2014; pp. 111–161. ISBN 978-0-12-800780-8. [Google Scholar]
- Rayan, A.; Raiyn, J.; Falah, M. Nature Is the Best Source of Anticancer Drugs: Indexing Natural Products for Their Anticancer Bioactivity. PLoS ONE 2017, 12, e0187925. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Gandhi, A.; Fimognari, C.; Atanasov, A.G.; Bishayee, A. Alkaloids for Cancer Prevention and Therapy: Current Progress and Future Perspectives. Eur. J. Pharmacol. 2019, 858, 172472. [Google Scholar] [CrossRef] [PubMed]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as Anticancer Agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-H.; Li, T.; Fong, C.M.V.; Chen, X.; Chen, X.-J.; Wang, Y.-T.; Huang, M.-Q.; Lu, J.-J. Saponins from Chinese Medicines as Anticancer Agents. Molecules 2016, 21, 1326. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Sanner, M.F.; Olson, A.J.; Forli, S. The AutoDock Suite at 30. Protein Sci. 2021, 30, 31–43. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California—San Francisco: San Francisco, CA, USA, 2020. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Granger, B.E.; Pérez, F. Jupyter: Thinking and Storytelling with Code and Data. Comput. Sci. Eng. 2021, 23, 7–14. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Kassambara, A. Rstatix: Pipe-Friendly Framework for Basic Statistical Tests; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Grant, B.J.; Skjærven, L.; Yao, X. The Bio3D Packages for Structural Bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: A Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. Tidyr: Tidy Messy Data; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Inkscape Project. 2023. Inkscape. Available online: https://inkscape.org (accessed on 1 May 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Binding Energy (kcal mol−1) | Intermolecular Energy (kcal mol−1) | Torsional Energy (kcal mol−1) |
|---|---|---|---|
| 4OHT | −11.50 | −14.18 | 2.68 |
| Estradiol | −9.63 | −9.63 | 0 |
| 1 | −9.37 | −11.45 | 2.09 |
| 2 | −11.43 | −12.33 | 0.89 |
| 3 | −11.60 | −12.49 | 0.89 |
| 4 | −11.05 | −12.24 | 1.19 |
| 5 | −10.03 | −10.92 | 0.89 |
| Ligand | Attractive Charge | Conventional H-Bond | Carbon H-Bond | Hydrophobic | Unfavorable Interaction | |
|---|---|---|---|---|---|---|
| Alkyl | π–Alkyl | |||||
| 4OHT | 1 | 2 | 2 | 2 | 7 | 0 |
| Estradiol | 0 | 3 | 0 | 3 | 5 | 0 |
| 1 | 0 | 0 | 0 | 16 | 1 | 1 |
| 2 | 0 | 1 | 0 | 0 | 0 | 1 |
| 3 | 0 | 1 | 0 | 2 | 0 | 0 |
| 4 | 0 | 1 | 0 | 0 | 0 | 1 |
| 5 | 0 | 0 | 0 | 2 | 0 | 0 |
| Compound | MW (g mol−1) | logP | HBA | HBD | Ro5 Violations |
|---|---|---|---|---|---|
| Estradiol | 272.38 | 3.60 | 2 | 2 | 0 |
| 4OHT | 371.52 | 5.99 | 2 | 0 | 1 |
| 2 | 452.72 | 8.11 | 2 | 0 | 1 |
| 3 | 452.72 | 8.11 | 2 | 0 | 1 |
| 4 | 470.73 | 7.30 | 3 | 1 | 1 |
| Criterion | Onoceranoid Triterpene | ||
|---|---|---|---|
| 2 | 3 | 4 | |
| Absorption | |||
| Water solubility (log mol L−1) | −7.21 | −7.18 | −6.55 |
| Caco2 permeability (log cm s−1) | 1.22 | 1.24 | 1.21 |
| Human intestinal absorption (%) | 96.41 | 97.33 | 95.01 |
| P-glycoprotein substrate | No | No | Yes |
| P-glycoprotein I inhibitor | Yes | Yes | Yes |
| P-glycoprotein II inhibitor | Yes | Yes | Yes |
| Distribution | |||
| VDss in humans (log L kg−1) | 0.5 | 0.33 | 0.3 |
| Fraction unbound | 0 | 0 | 0 |
| BBB permeability (log blood–brain barrier permeability) | 0.14 | 0.04 | −0.20 |
| CNS permeability (log blood–brain permeability surface) area | −2.53 | −2.25 | −1.23 |
| Metabolism | |||
| CYP2D6 substrate | No | No | No |
| CYP3A4 substrate | Yes | Yes | Yes |
| CYP1A2 inhibitor | No | No | No |
| CYP2C19 inhibitor | No | No | No |
| CYP2C9 inhibitor | No | No | No |
| CYP2D6 inhibitor | No | No | No |
| CYP3A4 inhibitor | No | No | No |
| Excretion | |||
| Total clearance (log mL min−1 kg−1) | 0.37 | 0.29 | 0.24 |
| Renal OCT2 substrate | No | No | No |
| Toxicity | |||
| AMES toxicity | No | No | No |
| hERG I inhibitor | No | No | No |
| hERG II inhibitor | Yes | Yes | No |
| Hepatotoxicity | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardianto, A.; Mardetia, S.S.; Destiarani, W.; Budiman, Y.P.; Kurnia, D.; Mayanti, T. Unveiling the Anti-Cancer Potential of Onoceranoid Triterpenes from Lansium domesticum Corr. cv. kokosan: An In Silico Study against Estrogen Receptor Alpha. Int. J. Mol. Sci. 2023, 24, 15033. https://doi.org/10.3390/ijms241915033
Hardianto A, Mardetia SS, Destiarani W, Budiman YP, Kurnia D, Mayanti T. Unveiling the Anti-Cancer Potential of Onoceranoid Triterpenes from Lansium domesticum Corr. cv. kokosan: An In Silico Study against Estrogen Receptor Alpha. International Journal of Molecular Sciences. 2023; 24(19):15033. https://doi.org/10.3390/ijms241915033
Chicago/Turabian StyleHardianto, Ari, Sarah Syifa Mardetia, Wanda Destiarani, Yudha Prawira Budiman, Dikdik Kurnia, and Tri Mayanti. 2023. "Unveiling the Anti-Cancer Potential of Onoceranoid Triterpenes from Lansium domesticum Corr. cv. kokosan: An In Silico Study against Estrogen Receptor Alpha" International Journal of Molecular Sciences 24, no. 19: 15033. https://doi.org/10.3390/ijms241915033
APA StyleHardianto, A., Mardetia, S. S., Destiarani, W., Budiman, Y. P., Kurnia, D., & Mayanti, T. (2023). Unveiling the Anti-Cancer Potential of Onoceranoid Triterpenes from Lansium domesticum Corr. cv. kokosan: An In Silico Study against Estrogen Receptor Alpha. International Journal of Molecular Sciences, 24(19), 15033. https://doi.org/10.3390/ijms241915033