

Iron-Dependent Cell Death: A New Treatment Approach against Pancreatic Ductal Adenocarcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
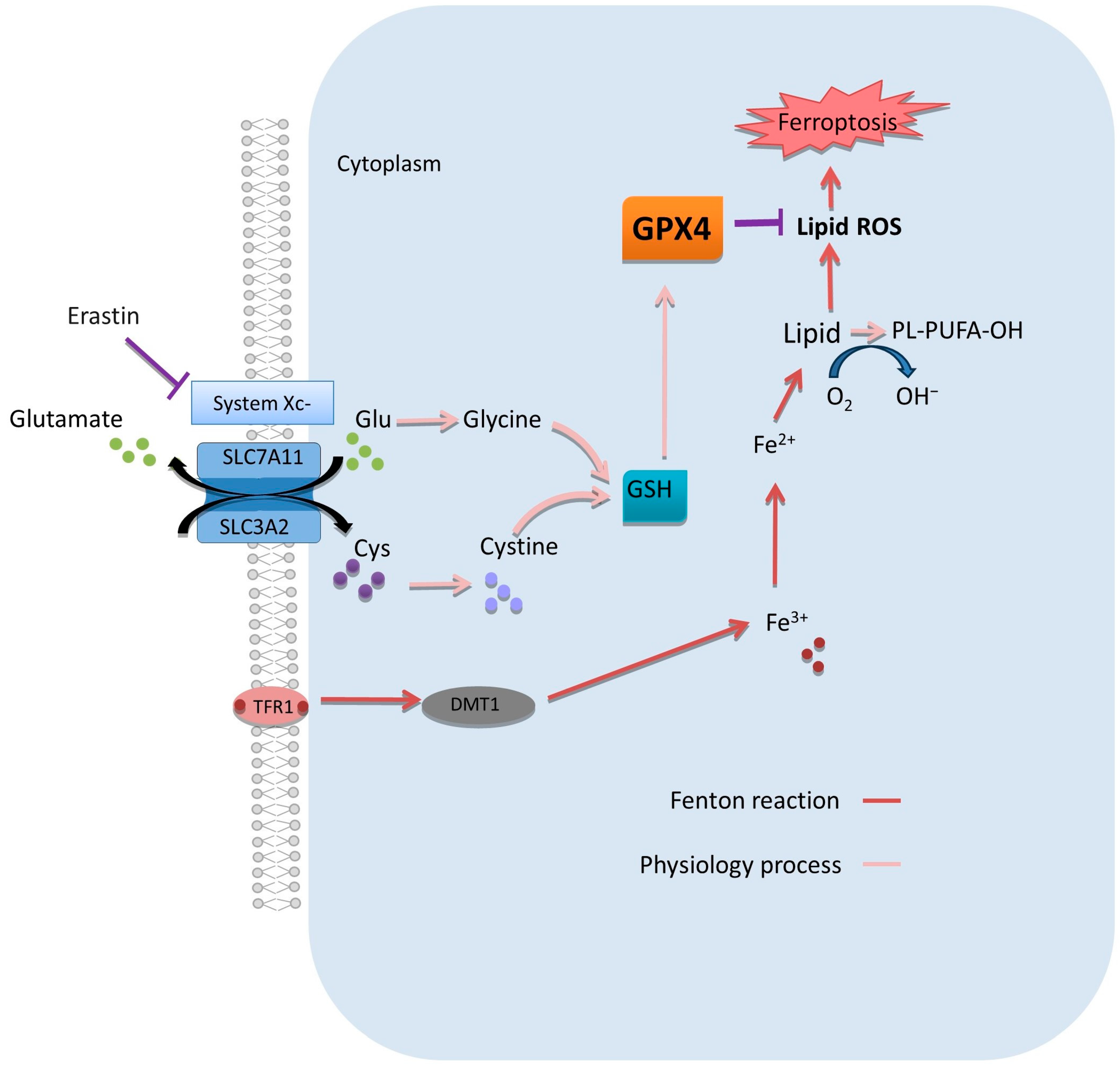
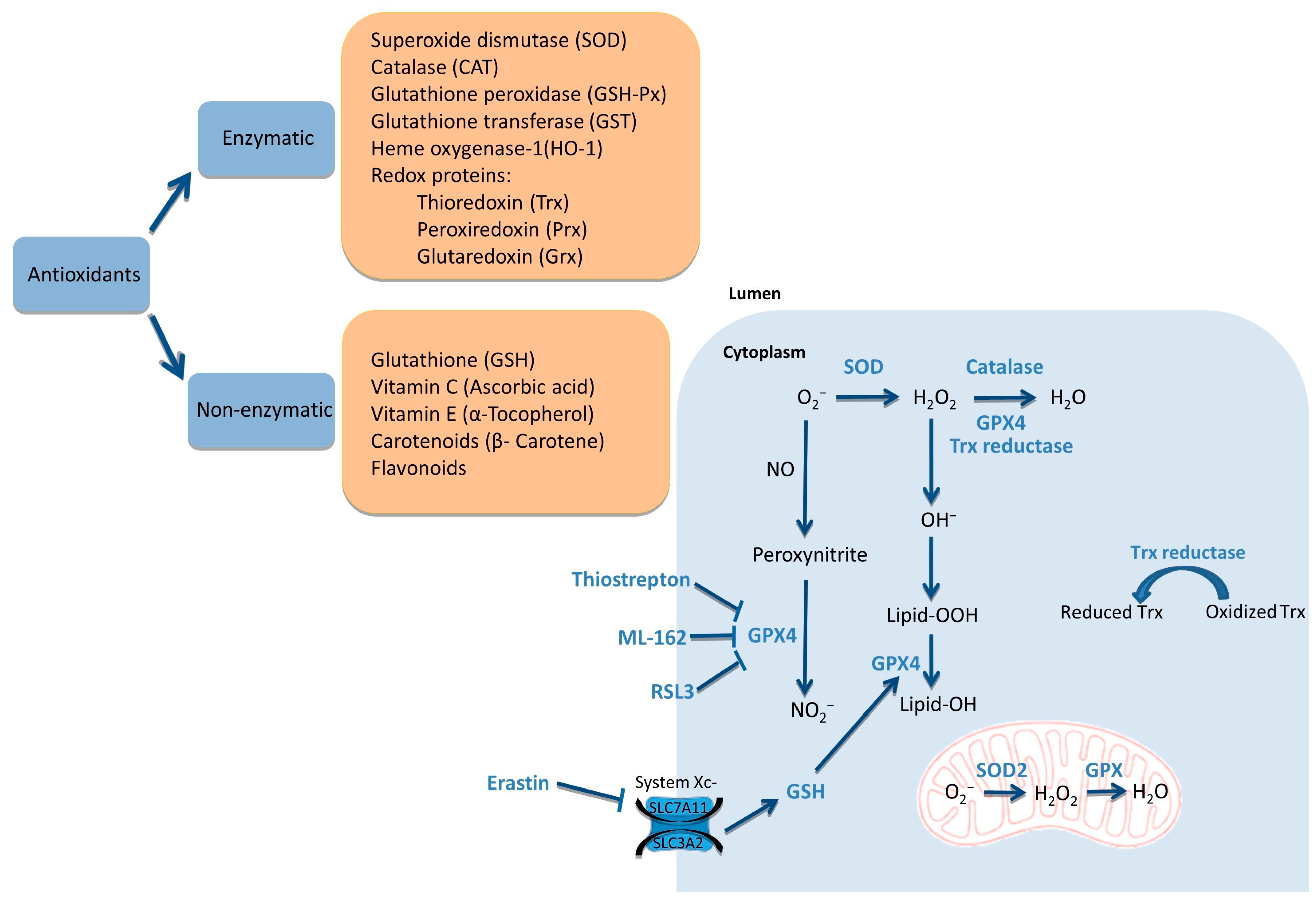
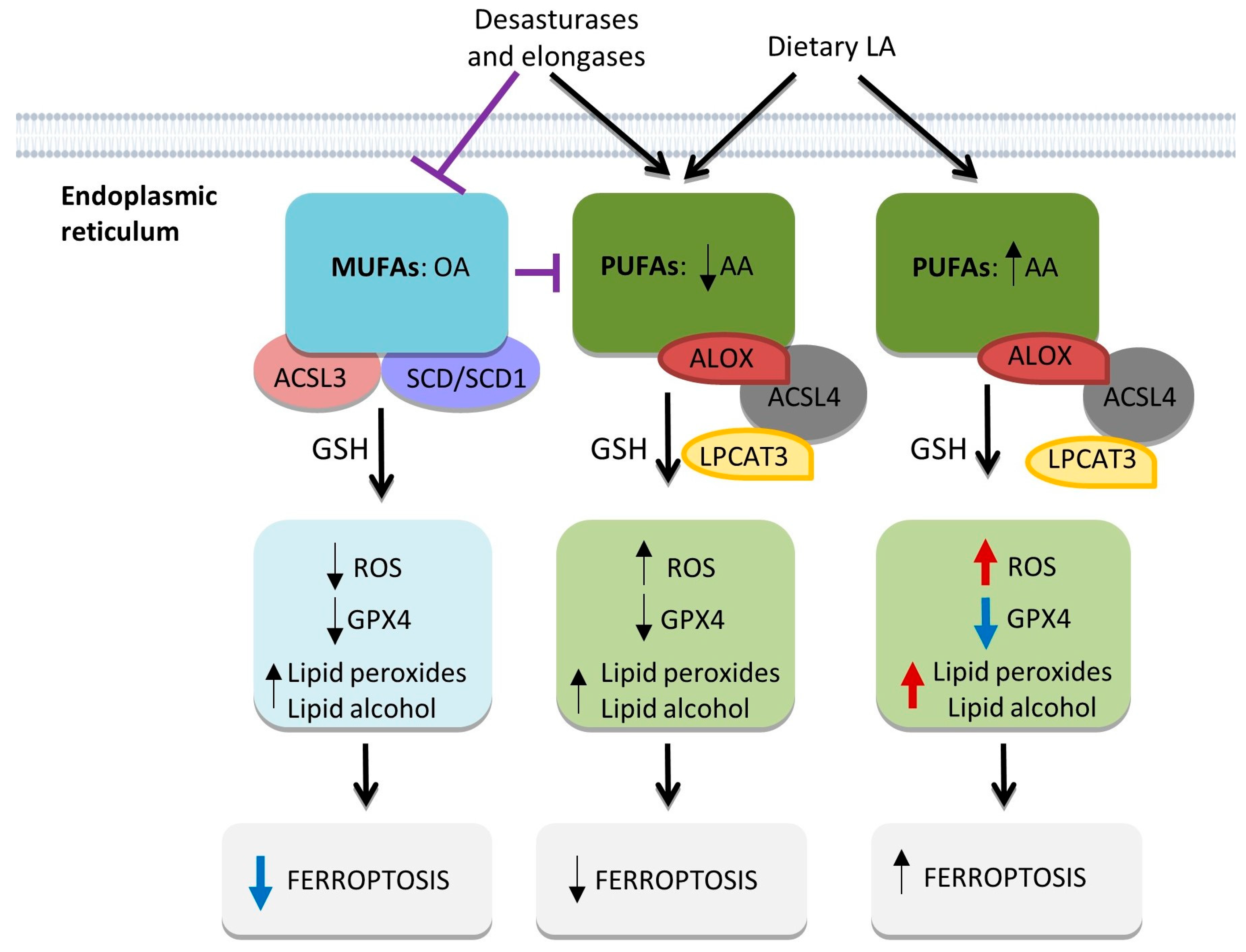
2. Reactive Oxygen Species Balance in PDAC
2.1. Generators of ROS
2.2. Scavengers of ROS
2.3. ROS Balance
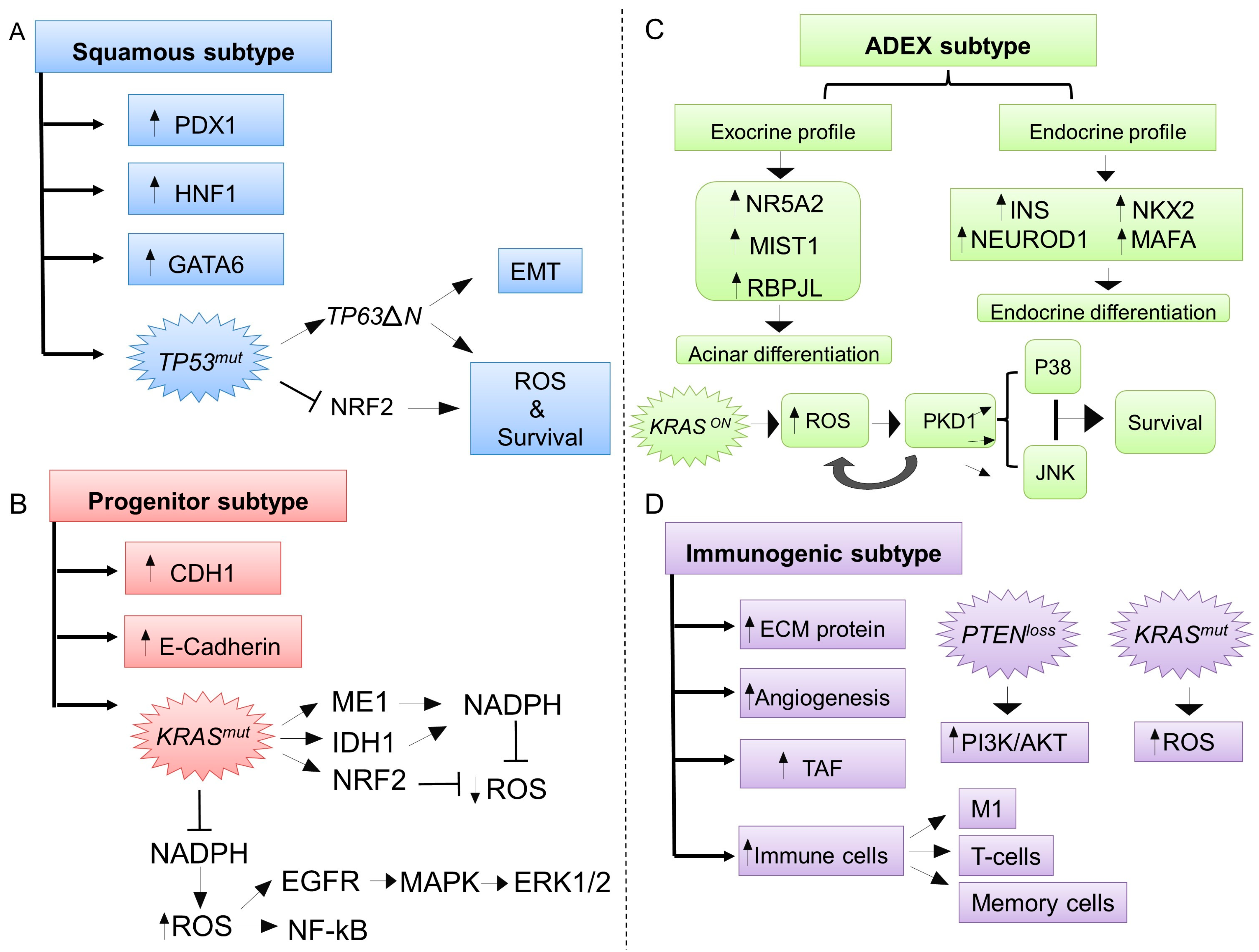
3. ROS Exhibit Specific Biological Functions According to Different Molecular Subtypes of PDAC
3.1. The Squamous Molecular Subtype and ROS
3.2. The Progenitor Molecular Subtype and ROS
3.3. The Aberrantly Differentiated Endocrine-Exocrine (ADEX, Exocrine-Like) Subtype and ROS
3.4. The Immunogenic Subtype and ROS
4. Reactive Oxygen Species and Commonly Used Chemotherapy in PDAC
5. Potential Clinical Benefits of Ferroptosis Modulation in PDAC
5.1. Targeting Oxidants
5.2. Targeting Antioxidants
5.3. Combination Approaches
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Helaly, M.; Sriwi, D.; Alkholaidi, W.S.; Almamlouk, R.; Elshaer, A.; Allaboon, R.M.; Hassan, L.H.; Khalifa, H.; Al-Alem, I. Retrograde Pancreatic Duct Stent Migration into the Biliary Tract Presenting as a Rare Early Complication of Pancreaticoduodenectomy (Whipple Procedure). Am. J. Case Rep. 2019, 20, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e15. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jansen, L.; Balavarca, Y.; Babaei, M.; van der Geest, L.; Lemmens, V.; Van Eycken, L.; De Schutter, H.; Johannesen, T.B.; Primic-Žakelj, M.; et al. Stratified Survival of Resected and Overall Pancreatic Cancer Patients in Europe and the USA in the Early Twenty-First Century: A Large, International Population-Based Study. BMC Med. 2018, 16, 125. [Google Scholar] [CrossRef]
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.-L.; Myklebust, T.Å.; Tervonen, H.; Thursfield, V.; Ransom, D.; Shack, L.; et al. Progress in Cancer Survival, Mortality, and Incidence in Seven High-Income Countries 1995-2014 (ICBP SURVMARK-2): A Population-Based Study. Lancet Oncol. 2019, 20, 1493–1505. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-Time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular Subtypes of Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Guidelines Detail. Available online: https://www.nccn.org/guidelines/guidelines-detail (accessed on 22 March 2023).
- Müller, P.C.; Frey, M.C.; Ruzza, C.M.; Nickel, F.; Jost, C.; Gwerder, C.; Hackert, T.; Z’graggen, K.; Kessler, U. Neoadjuvant Chemotherapy in Pancreatic Cancer: An Appraisal of the Current High-Level Evidence. Pharmacology 2021, 106, 143–153. [Google Scholar] [CrossRef]
- StatBite. U.S. Pancreatic Cancer Rates. J. Natl. Cancer Inst. 2010, 102, 1822. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Ghidini, M.; Torchio, M.; Corti, F.; Tomasello, G.; Niger, M.; Prinzi, N.; Nichetti, F.; Coinu, A.; Di Bartolomeo, M.; et al. Comparative Effectiveness of Gemcitabine plus Nab-Paclitaxel and FOLFIRINOX in the First-Line Setting of Metastatic Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 484. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Melisi, D.; Macarulla, T.; Pazo-Cid, R.; Chandana, S.R.; Fouchardiere, C.D.L.; Dean, A.P.; Kiss, I.; Lee, W.; Goetze, T.O.; et al. NAPOLI-3: A Randomized, Open-Label Phase 3 Study of Liposomal Irinotecan + 5-Fluorouracil/Leucovorin + Oxaliplatin (NALIRIFOX) versus Nab-Paclitaxel + Gemcitabine in Treatment-Naïve Patients with Metastatic Pancreatic Ductal Adenocarcinoma (mPDAC). J. Clin. Oncol. 2023, 41, LBA661. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-Derived Proline Promotes Pancreatic Ductal Adenocarcinoma Cell Survival under Nutrient Limited Conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol Uptake Disruption, in Association with Chemotherapy, Is a Promising Combined Metabolic Therapy for Pancreatic Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A Phase I Study of an Agonist CD40 Monoclonal Antibody (CP-870,893) in Combination with Gemcitabine in Patients with Advanced Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.; et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4574–4586. [Google Scholar] [CrossRef]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A Lethally Irradiated Allogeneic Granulocyte-Macrophage Colony Stimulating Factor-Secreting Tumor Vaccine for Pancreatic Adenocarcinoma. A Phase II Trial of Safety, Efficacy, and Immune Activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Munro, D.; Treberg, J.R. A Radical Shift in Perspective: Mitochondria as Regulators of Reactive Oxygen Species. J. Exp. Biol. 2017, 220, 1170–1180. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The Hallmarks of Cancer Metabolism: Still Emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Camelo, F.; Le, A. The Intricate Metabolism of Pancreatic Cancers. In The Heterogeneity of Cancer Metabolism; Le, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2021; pp. 77–88. ISBN 978-3-030-65768-0. [Google Scholar]
- Park, J.K.; Coffey, N.J.; Limoges, A.; Le, A. The Heterogeneity of Lipid Metabolism in Cancer. In The Heterogeneity of Cancer Metabolism; Le, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2021; pp. 39–56. ISBN 978-3-030-65768-0. [Google Scholar]
- Oshi, M.; Gandhi, S.; Yan, L.; Tokumaru, Y.; Wu, R.; Yamada, A.; Matsuyama, R.; Endo, I.; Takabe, K. Abundance of Reactive Oxygen Species (ROS) Is Associated with Tumor Aggressiveness, Immune Response, and Worse Survival in Breast Cancer. Breast Cancer Res. Treat. 2022, 194, 231–241. [Google Scholar] [CrossRef]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Dalla Pozza, E.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/Cannabinoid Combination Triggers Autophagy in Pancreatic Cancer Cells through a ROS-Mediated Mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef]
- Stanicka, J.; Russell, E.G.; Woolley, J.F.; Cotter, T.G. NADPH Oxidase-Generated Hydrogen Peroxide Induces DNA Damage in Mutant FLT3-Expressing Leukemia Cells*. J. Biol. Chem. 2015, 290, 9348–9361. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Qiao, Y.; Liu, Y.; Zhou, J.; Wang, X.; Zheng, H.; Xu, Z.; Zhang, J.; Zhou, Y.; Qian, L.; et al. Ent-Kaurane Diterpenoids Induce Apoptosis and Ferroptosis through Targeting Redox Resetting to Overcome Cisplatin Resistance. Redox Biol. 2021, 43, 101977. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Lim, S.-O.; Yan, M.; Hsu, J.L.; Yao, J.; Wei, Y.; Chang, S.-S.; Yamaguchi, H.; Lee, H.-H.; Ke, B.; et al. TYRO3 Induces Anti–PD-1/PD-L1 Therapy Resistance by Limiting Innate Immunity and Tumoral Ferroptosis. J. Clin. Investig. 2021, 131, e139434. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in Cancer and Cancer Immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef]
- Fiorini, C.; Cordani, M.; Gotte, G.; Picone, D.; Donadelli, M. Onconase Induces Autophagy Sensitizing Pancreatic Cancer Cells to Gemcitabine and Activates Akt/mTOR Pathway in a ROS-Dependent Manner. Biochim. Biophys. Acta 2015, 1853, 549–560. [Google Scholar] [CrossRef]
- Kong, R.; Jia, G.; Cheng, Z.; Wang, Y.; Mu, M.; Wang, S.; Pan, S.; Gao, Y.; Jiang, H.; Dong, D.; et al. Dihydroartemisinin Enhances Apo2L/TRAIL-Mediated Apoptosis in Pancreatic Cancer Cells via ROS-Mediated up-Regulation of Death Receptor 5. PLoS ONE 2012, 7, e37222. [Google Scholar] [CrossRef]
- Jagust, P.; Alcalá, S.; Sainz Jr, B.; Heeschen, C.; Sancho, P. Glutathione Metabolism Is Essential for Self-Renewal and Chemoresistance of Pancreatic Cancer Stem Cells. World J. Stem Cells 2020, 12, 1410–1428. [Google Scholar] [CrossRef]
- Lee, M.; Cho, T.; Jantaratnotai, N.; Wang, Y.T.; McGeer, E.; McGeer, P.L. Depletion of GSH in Glial Cells Induces Neurotoxicity: Relevance to Aging and Degenerative Neurological Diseases. FASEB J. 2010, 24, 2533–2545. [Google Scholar] [CrossRef]
- Commoner, B.; Townsend, J.; Pake, G.E. Free Radicals in Biological Materials. Nature 1954, 174, 689–691. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Chang, C.J. Chemistry and Biology of Reactive Oxygen Species in Signaling or Stress Responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial Respiratory Chain-Dependent Generation of Superoxide Anion and Its Release into the Intermembrane Space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native Rates of Superoxide Production from Multiple Sites in Isolated Mitochondria Measured Using Endogenous Reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef]
- Zweier, J.L.; Hemann, C.; Kundu, T.; Ewees, M.G.; Khaleel, S.A.; Samouilov, A.; Ilangovan, G.; El-Mahdy, M.A. Cytoglobin Has Potent Superoxide Dismutase Function. Proc. Natl. Acad. Sci. USA 2021, 118, e2105053118. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Tian, J.; Zhang, H.; Guo, Y.; Yang, J.; Zhu, C.; Song, M.; Wang, P.; Liu, Z.; Cancilla, J.; et al. Loss of Mitochondrial Aconitase Promotes Colorectal Cancer Progression via SCD1-Mediated Lipid Remodeling. Mol. Metab. 2021, 48, 101203. [Google Scholar] [CrossRef]
- Fenton, H.J.H. LXXIII.—Oxidation of Tartaric Acid in Presence of Iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Wu, Y.; Lu, J.; Antony, S.; Juhasz, A.; Liu, H.; Jiang, G.; Meitzler, J.L.; Hollingshead, M.; Haines, D.C.; Butcher, D.; et al. Activation of TLR4 Is Required for the Synergistic Induction of Dual Oxidase 2 and Dual Oxidase A2 by IFN-γ and Lipopolysaccharide in Human Pancreatic Cancer Cell Lines. J. Immunol. 2013, 190, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Furuta, S.; Mitsushita, J.; Shang, W.H.; Ito, M.; Yokoo, Y.; Yamaura, M.; Ishizone, S.; Nakayama, J.; Konagai, A.; et al. Inhibition of NADPH Oxidase 4 Activates Apoptosis via the AKT/Apoptosis Signal-Regulating Kinase 1 Pathway in Pancreatic Cancer PANC-1 Cells. Oncogene 2006, 25, 3699–3707. [Google Scholar] [CrossRef]
- Lee, J.K.; Edderkaoui, M.; Truong, P.; Ohno, I.; Jang, K.-T.; Berti, A.; Pandol, S.J.; Gukovskaya, A.S. NADPH Oxidase Promotes Pancreatic Cancer Cell Survival via Inhibiting JAK2 Dephosphorylation by Tyrosine Phosphatases. Gastroenterology 2007, 133, 1637–1648. [Google Scholar] [CrossRef]
- Landry, W.D.; Cotter, T.G. ROS Signalling, NADPH Oxidases and Cancer. Biochem. Soc. Trans. 2014, 42, 934–938. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS Signalling in the Biology of Cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, M.; Liang, B.; Xie, Z.; Zhao, Z.; Asfa, S.; Choi, H.C.; Zou, M.-H. Reduction of AMP-Activated Protein Kinase Alpha2 Increases Endoplasmic Reticulum Stress and Atherosclerosis in Vivo. Circulation 2010, 121, 792–803. [Google Scholar] [CrossRef]
- Ma, W.F.; Boudreau, H.E.; Leto, T.L. Pan-Cancer Analysis Shows TP53 Mutations Modulate the Association of NOX4 with Genetic Programs of Cancer Progression and Clinical Outcome. Antioxidants 2021, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-L.; Yang, L.; Fu, S.-W.; Lin, W.-F.; Gao, Y.-J.; Chen, H.-Y.; Ge, Z.-Z. Overexpression of NOX4 Predicts Poor Prognosis and Promotes Tumor Progression in Human Colorectal Cancer. Oncotarget 2017, 8, 33586–33600. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting Edge: Direct Interaction of TLR4 with NAD(P)H Oxidase 4 Isozyme Is Essential for Lipopolysaccharide-Induced Production of Reactive Oxygen Species and Activation of NF-Kappa B. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef]
- Dong, P.-T.; Zhan, Y.; Jusuf, S.; Hui, J.; Dagher, Z.; Mansour, M.K.; Cheng, J.-X. Photoinactivation of Catalase Sensitizes Candida Albicans and Candida Auris to ROS-Producing Agents and Immune Cells. Adv. Sci. 2022, 9, e2104384. [Google Scholar] [CrossRef] [PubMed]
- Calvani, N.E.D.; De Marco Verissimo, C.; Jewhurst, H.L.; Cwiklinski, K.; Flaus, A.; Dalton, J.P. Two Distinct Superoxidase Dismutases (SOD) Secreted by the Helminth Parasite Fasciola Hepatica Play Roles in Defence against Metabolic and Host Immune Cell-Derived Reactive Oxygen Species (ROS) during Growth and Development. Antioxidants 2022, 11, 1968. [Google Scholar] [CrossRef]
- Fernandes, A.P.; Holmgren, A. Glutaredoxins: Glutathione-Dependent Redox Enzymes with Functions Far Beyond a Simple Thioredoxin Backup System. Antioxid. Redox Signal. 2004, 6, 63–74. [Google Scholar] [CrossRef]
- Jones, D.P. Redox Potential of GSH/GSSG Couple: Assay and Biological Significance. Methods Enzym. 2002, 348, 93–112. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Brigelius-Flohé, R.; Aumann, K.D.; Roveri, A.; Schomburg, D.; Flohé, L. Diversity of Glutathione Peroxidases. Methods Enzym. 1995, 252, 38–53. [Google Scholar] [CrossRef]
- Zhang, W.; Gong, M.; Zhang, W.; Mo, J.; Zhang, S.; Zhu, Z.; Wang, X.; Zhang, B.; Qian, W.; Wu, Z.; et al. Thiostrepton Induces Ferroptosis in Pancreatic Cancer Cells through STAT3/GPX4 Signalling. Cell Death Dis. 2022, 13, 630. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.-L. Nrf2 Inhibition Reverses Resistance to GPX4 Inhibitor-Induced Ferroptosis in Head and Neck Cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, T.; Lai, J.; Zeng, D.; Chen, W.; Zhang, X.; Zhu, X.; Zhang, G.; Hu, Z. Silencing TRPM2 Enhanced Erastin- and RSL3-Induced Ferroptosis in Gastric Cancer Cells through Destabilizing HIF-1α and Nrf2 Proteins. Cytotechnology 2022, 74, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.G. Superoxide Dismutase in Redox Biology: The Roles of Superoxide and Hydrogen Peroxide. Anti-Cancer Agents Med. Chem. Anti-Cancer Agents 2011, 11, 341–346. [Google Scholar] [CrossRef]
- Nie, S.; Shi, Z.; Shi, M.; Li, H.; Qian, X.; Peng, C.; Ding, X.; Zhang, S.; Lv, Y.; Wang, L.; et al. PPARγ/SOD2 Protects Against Mitochondrial ROS-Dependent Apoptosis via Inhibiting ATG4D-Mediated Mitophagy to Promote Pancreatic Cancer Proliferation. Front. Cell Dev. Biol. 2022, 9, 745554. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Chen, Y.; Xue, F.; Liu, K.; Zhu, B.; Gao, J.; Yin, J.; Zhang, C.; Li, G. Contribution of Ferroptosis and GPX4′s Dual Functions to Osteoarthritis Progression. eBioMedicine 2022, 76, 103847. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a Biomarker and Contributor of Ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Ye, Z.; Zhuo, Q.; Hu, Q.; Xu, X.; Liu, M.; Zhang, Z.; Xu, W.; Liu, W.; Fan, G.; Qin, Y.; et al. FBW7-NRA41-SCD1 Axis Synchronously Regulates Apoptosis and Ferroptosis in Pancreatic Cancer Cells. Redox Biol. 2021, 38, 101807. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bajari, R.; Andric, D.; Gerthoffert, F.; Lepsa, A.; Nahal-Bose, H.; Stein, L.D.; Ferretti, V. The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 2019, 37, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Majewski, I.J.; Bernards, R. Taming the Dragon: Genomic Biomarkers to Individualize the Treatment of Cancer. Nat. Med. 2011, 17, 304–312. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Singhi, A.D.; Wood, L.D. Early Detection of Pancreatic Cancer Using DNA-Based Molecular Approaches. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 457–468. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Martin-Galan, M.; Garcia-Foncillas, J. The Match between Molecular Subtypes, Histology and Microenvironment of Pancreatic Cancer and Its Relevance for Chemoresistance. Cancers 2021, 13, 322. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal Transition Is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Jiang, Y.-Y.; Jiang, Y.; Li, C.-Q.; Zhang, Y.; Dakle, P.; Kaur, H.; Deng, J.-W.; Lin, R.Y.-T.; Han, L.; Xie, J.-J.; et al. TP63, SOX2, and KLF5 Establish a Core Regulatory Circuitry That Controls Epigenetic and Transcription Patterns in Esophageal Squamous Cell Carcinoma Cell Lines. Gastroenterology 2020, 159, 1311–1327.e19. [Google Scholar] [CrossRef]
- Zhao, T.; Ye, S.; Tang, Z.; Guo, L.; Ma, Z.; Zhang, Y.; Yang, C.; Peng, J.; Chen, J. Loss-of-Function of P53 Isoform Δ113p53 Accelerates Brain Aging in Zebrafish. Cell Death Dis. 2021, 12, 151. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The Antioxidant Function of the P53 Tumor Suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Cano, C.E.; Gommeaux, J.; Pietri, S.; Culcasi, M.; Garcia, S.; Seux, M.; Barelier, S.; Vasseur, S.; Spoto, R.P.; Pébusque, M.-J.; et al. Tumor Protein 53-Induced Nuclear Protein 1 Is a Major Mediator of P53 Antioxidant Function. Cancer Res. 2009, 69, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Seillier, M.; Pouyet, L.; N’Guessan, P.; Nollet, M.; Capo, F.; Guillaumond, F.; Peyta, L.; Dumas, J.-F.; Varrault, A.; Bertrand, G.; et al. Defects in Mitophagy Promote Redox-Driven Metabolic Syndrome in the Absence of TP53INP1. EMBO Mol. Med. 2015, 7, 802–818. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins Activate Nrf2 by Promoting P62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct Interaction between Nrf2 and P21(Cip1/WAF1) Upregulates the Nrf2-Mediated Antioxidant Response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H Attenuates the Expression of Phase 2 Detoxifying Enzymes and Promotes the Survival of Cells with High Levels of Reactive Oxygen Species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Storz, P. KRas, ROS and the Initiation of Pancreatic Cancer. Small GTPases 2017, 8, 38–42. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Döppler, H.; Liou, G.-Y.; Storz, P. Downregulation of TRAF2 Mediates NIK-Induced Pancreatic Cancer Cell Proliferation and Tumorigenicity. PLoS ONE 2013, 8, e53676. [Google Scholar] [CrossRef] [PubMed]
- Vaziri-Gohar, A.; Zarei, M.; Brody, J.R.; Winter, J.M. Metabolic Dependencies in Pancreatic Cancer. Front. Oncol. 2018, 8, 617. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- von Figura, G.; Morris, J.P.; Wright, C.V.E.; Hebrok, M. Nr5a2 Maintains Acinar Cell Differentiation and Constrains Oncogenic Kras-Mediated Pancreatic Neoplastic Initiation. Gut 2014, 63, 656–664. [Google Scholar] [CrossRef]
- Hale, M.A.; Swift, G.H.; Hoang, C.Q.; Deering, T.G.; Masui, T.; Lee, Y.-K.; Xue, J.; MacDonald, R.J. The Nuclear Hormone Receptor Family Member NR5A2 Controls Aspects of Multipotent Progenitor Cell Formation and Acinar Differentiation during Pancreatic Organogenesis. Development 2014, 141, 3123–3133. [Google Scholar] [CrossRef]
- Cowell, C.F.; Döppler, H.; Yan, I.K.; Hausser, A.; Umezawa, Y.; Storz, P. Mitochondrial Diacylglycerol Initiates Protein-Kinase D1-Mediated ROS Signaling. J. Cell Sci. 2009, 122, 919–928. [Google Scholar] [CrossRef]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic Cancer Stroma: An Update on Therapeutic Targeting Strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Garrido-Martin, E.M.; Mellows, T.W.P.; Clarke, J.; Ganesan, A.-P.; Wood, O.; Cazaly, A.; Seumois, G.; Chee, S.J.; Alzetani, A.; King, E.V.; et al. M1hot Tumor-Associated Macrophages Boost Tissue-Resident Memory T Cells Infiltration and Survival in Human Lung Cancer. J. Immunother. Cancer 2020, 8, e000778. [Google Scholar] [CrossRef]
- Tanaka, S. Molecular Pathogenesis and Targeted Therapy of Pancreatic Cancer. Ann. Surg. Oncol. 2016, 23, 197–205. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative Treatment of β-Elemene and Cetuximab Is Sensitive to KRAS Mutant Colorectal Cancer Cells by Inducing Ferroptosis and Inhibiting Epithelial-Mesenchymal Transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab Promotes RSL3-Induced Ferroptosis by Suppressing the Nrf2/HO-1 Signalling Pathway in KRAS Mutant Colorectal Cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a P53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a Novel P53 Target Gene Regulating Energy Metabolism and Antioxidant Function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the System xC-/Glutathione Axis Selectively Targets Cancers with Mutant-P53 Accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.-D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Richard, D.; Kefi, K.; Barbe, U.; Bausero, P.; Visioli, F. Polyunsaturated Fatty Acids as Antioxidants. Pharmacol. Res. 2008, 57, 451–455. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Illés, E.; Patra, S.G.; Marks, V.; Mizrahi, A.; Meyerstein, D. The FeII(Citrate) Fenton Reaction under Physiological Conditions. J. Inorg. Biochem. 2020, 206, 111018. [Google Scholar] [CrossRef]
- Lopes, T.J.S.; Luganskaja, T.; Vujić Spasić, M.; Hentze, M.W.; Muckenthaler, M.U.; Schümann, K.; Reich, J.G. Systems Analysis of Iron Metabolism: The Network of Iron Pools and Fluxes. BMC Syst. Biol. 2010, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J.; Hider, R.C.; Wilson, M.T. Iron Chelators Can Protect against Oxidative Stress through Ferryl Heme Reduction. Free Radic. Biol. Med. 2008, 44, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The Glutathione Peroxidase Gpx4 Prevents Lipid Peroxidation and Ferroptosis to Sustain Treg Cell Activation and Suppression of Antitumor Immunity. Cell Rep. 2021, 35, 109235. [Google Scholar] [CrossRef] [PubMed]
- Bekkali, N.L.H.; Oppong, K.W. Pancreatic Ductal Adenocarcinoma Epidemiology and Risk Assessment: Could We Prevent? Possibility for an Early Diagnosis. Endosc. Ultrasound 2017, 6, S58–S61. [Google Scholar] [CrossRef]
- Tian, N.; Wu, D.; Zhu, L.; Zeng, M.; Li, J.; Wang, X. A Predictive Model for Recurrence after Upfront Surgery in Patients with Resectable Pancreatic Ductal Adenocarcinoma (PDAC) by Using Preoperative Clinical Data and CT Characteristics. BMC Med. Imaging 2022, 22, 116. [Google Scholar] [CrossRef]
- Groot, V.P.; Gemenetzis, G.; Blair, A.B.; Rivero-Soto, R.J.; Yu, J.; Javed, A.A.; Burkhart, R.A.; Rinkes, I.H.M.B.; Molenaar, I.Q.; Cameron, J.L.; et al. Defining and Predicting Early Recurrence in 957 Patients With Resected Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2019, 269, 1154–1162. [Google Scholar] [CrossRef]
- Maisonneuve, P. Epidemiology and Burden of Pancreatic Cancer. La Presse Médicale 2019, 48, e113–e123. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Gocho, T.; Aguilar, M.; Wu, M.; Zhuang, Z.-N.; Fu, J.; Yanaga, K.; Huang, P.; Chiao, P.J. Mechanisms of Overcoming Intrinsic Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma through the Redox Modulation. Mol. Cancer Ther. 2015, 14, 788–798. [Google Scholar] [CrossRef]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M. Regulation of NADPH Oxidase Subunit P22(Phox) by NF-kB in Human Aortic Smooth Muscle Cells. Arch. Physiol. Biochem. 2007, 113, 163–172. [Google Scholar] [CrossRef]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual Roles of Nrf2 in Cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef]
- Eskandari, M.R.; Moghaddam, F.; Shahraki, J.; Pourahmad, J. A Comparison of Cardiomyocyte Cytotoxic Mechanisms for 5-Fluorouracil and Its pro-Drug Capecitabine. Xenobiotica 2015, 45, 79–87. [Google Scholar] [CrossRef]
- Liu, M.-P.; Liao, M.; Dai, C.; Chen, J.-F.; Yang, C.-J.; Liu, M.; Chen, Z.-G.; Yao, M.-C. Sanguisorba Officinalis L Synergistically Enhanced 5-Fluorouracil Cytotoxicity in Colorectal Cancer Cells by Promoting a Reactive Oxygen Species-Mediated, Mitochondria-Caspase-Dependent Apoptotic Pathway. Sci. Rep. 2016, 6, 34245. [Google Scholar] [CrossRef]
- Zhu, Q.; Guo, Y.; Chen, S.; Fu, D.; Li, Y.; Li, Z.; Ni, C. Irinotecan Induces Autophagy-Dependent Apoptosis and Positively Regulates ROS-Related JNK- and P38-MAPK Pathways in Gastric Cancer Cells. OncoTargets Ther. 2020, 13, 2807–2817. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, C.; He, Z.; Zhu, F.; Wang, M.; He, R.; Zhao, C.; Shi, X.; Zhou, M.; Pan, S.; et al. Inhibition of PI3K/AKT Signaling via ROS Regulation Is Involved in Rhein-Induced Apoptosis and Enhancement of Oxaliplatin Sensitivity in Pancreatic Cancer Cells. Int. J. Biol. Sci. 2021, 17, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhao, B.; Chang, H.; Xiao, M.; Wu, Y.; Liu, Y. Paclitaxel Suppresses Proliferation and Induces Apoptosis through Regulation of ROS and the AKT/MAPK Signaling Pathway in Canine Mammary Gland Tumor Cells. Mol. Med. Rep. 2018, 17, 8289–8299. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Rui, D.; Yan, Y.; Xu, S.; Niu, Q.; Feng, G.; Wang, Y.; Li, S.; Jing, M. Oxidative Damage Induced by Arsenic in Mice or Rats: A Systematic Review and Meta-Analysis. Biol. Trace Elem. Res. 2017, 176, 154–175. [Google Scholar] [CrossRef]
- Li, X.; Ding, X.; Adrian, T.E. Arsenic Trioxide Induces Apoptosis in Pancreatic Cancer Cells via Changes in Cell Cycle, Caspase Activation, and GADD Expression. Pancreas 2003, 27, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H.; Seol, J.G.; Kim, E.S.; Hyun, J.M.; Jung, C.W.; Lee, C.C.; Kim, B.K.; Lee, Y.Y. Arsenic Trioxide-Mediated Growth Inhibition in MC/CAR Myeloma Cells via Cell Cycle Arrest in Association with Induction of Cyclin-Dependent Kinase Inhibitor, P21, and Apoptosis. Cancer Res. 2000, 60, 3065–3071. [Google Scholar] [PubMed]
- Loh, S.N. Arsenic and an Old Place: Rescuing P53 Mutants in Cancer. Cancer Cell 2021, 39, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-F.; Lu, Y.; Wu, Q.; Lou, Y.-J.; Yang, M.; Xu, J.-Y.; Sun, C.-H.; Mao, L.-P.; Xu, G.-X.; Li, L.; et al. Oral Arsenic and Retinoic Acid for High-Risk Acute Promyelocytic Leukemia. J. Hematol. Oncol. 2022, 15, 148. [Google Scholar] [CrossRef]
- Kindler, H.L.; Aklilu, M.; Nattam, S.; Vokes, E.E. Arsenic Trioxide in Patients with Adenocarcinoma of the Pancreas Refractory to Gemcitabine: A Phase II Trial of the University of Chicago Phase II Consortium. Am. J. Clin. Oncol. 2008, 31, 553–556. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C Selectively Kills KRAS and BRAF Mutant Colorectal Cancer Cells by Targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kasukabe, T.; Kumakura, S. Piperlongumine Rapidly Induces the Death of Human Pancreatic Cancer Cells Mainly through the Induction of Ferroptosis. Int. J. Oncol. 2018, 52, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of Artesunate as a Specific Activator of Ferroptosis in Pancreatic Cancer Cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e9. [Google Scholar] [CrossRef]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic Depletion of L-Cyst(e)Ine with Cyst(e)Inase Increases Reactive Oxygen Species and Suppresses Tumor Growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Z.; Cheng, J.; Pan, H.; Lin, T.; Shen, X.; Chen, W.; Chen, Q.; Gu, C.; Mao, Q.; et al. Platelet-Vesicles-Encapsulated RSL-3 Enable Anti-Angiogenesis and Induce Ferroptosis to Inhibit Pancreatic Cancer Progress. Front. Endocrinol. 2022, 13, 865655. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Song, G.; Bae, H. Laminarin Attenuates ROS-Mediated Cell Migration and Invasiveness through Mitochondrial Dysfunction in Pancreatic Cancer Cells. Antioxidants 2022, 11, 1714. [Google Scholar] [CrossRef] [PubMed]
- Abdel Hadi, N.; Reyes-Castellanos, G.; Carrier, A. Targeting Redox Metabolism in Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 1534. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Zhyvoloup, A.; Baković, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G.; et al. Protein CoAlation and Antioxidant Function of Coenzyme A in Prokaryotic Cells. Biochem. J. 2018, 475, 1909–1937. [Google Scholar] [CrossRef]
- Trujillo, M.; Clippe, A.; Manta, B.; Ferrer-Sueta, G.; Smeets, A.; Declercq, J.-P.; Knoops, B.; Radi, R. Pre-Steady State Kinetic Characterization of Human Peroxiredoxin 5: Taking Advantage of Trp84 Fluorescence Increase upon Oxidation. Arch. Biochem. Biophys. 2007, 467, 95–106. [Google Scholar] [CrossRef]
- Zhang, Y.; Ikeno, Y.; Qi, W.; Chaudhuri, A.; Li, Y.; Bokov, A.; Thorpe, S.R.; Baynes, J.W.; Epstein, C.; Richardson, A.; et al. Mice Deficient in Both Mn Superoxide Dismutase and Glutathione Peroxidase-1 Have Increased Oxidative Damage and a Greater Incidence of Pathology but No Reduction in Longevity. J. Gerontol. Ser. A 2009, 64A, 1212–1220. [Google Scholar] [CrossRef]
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor Suppressor Function of the Plasma Glutathione Peroxidase Gpx3 in Colitis-Associated Carcinoma. Cancer Res. 2013, 73, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182.e4. [Google Scholar] [CrossRef]
- Ke, D.Y.J.; El-Sahli, S.; Wang, L. The Potential of Natural Products in the Treatment of Triple-Negative Breast Cancer. Curr. Cancer Drug Targets 2022, 22, 388–403. [Google Scholar] [CrossRef]
- Auyeung, K.K.-W.; Ko, J.K.-S. Novel Herbal Flavonoids Promote Apoptosis but Differentially Induce Cell Cycle Arrest in Human Colon Cancer Cell. Investig. New Drugs 2010, 28, 1–13. [Google Scholar] [CrossRef]
- Husain, K.; Centeno, B.A.; Coppola, D.; Trevino, J.; Sebti, S.M.; Malafa, M.P. δ-Tocotrienol, a Natural Form of Vitamin E, Inhibits Pancreatic Cancer Stem-like Cells and Prevents Pancreatic Cancer Metastasis. Oncotarget 2017, 8, 31554–31567. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.-L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 Transcription Factor by the Alkaloid Trigonelline Renders Pancreatic Cancer Cells More Susceptible to Apoptosis through Decreased Proteasomal Gene Expression and Proteasome Activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhu, Y.; Yu, H.; Liu, X.; Jiao, B.; Lu, X. Libertellenone H, a Natural Pimarane Diterpenoid, Inhibits Thioredoxin System and Induces ROS-Mediated Apoptosis in Human Pancreatic Cancer Cells. Molecules 2021, 26, 315. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, M.; Zhang, R.; Jin, Z. Inhibition of Cancer Cell Growth in Gemcitabine-Resistant Pancreatic Carcinoma by Mangiferin Phytochemical Involves Induction of Autophagy, Endogenous ROS Production, Cell Cycle Disruption, Mitochondrial Mediated Apoptosis and Suppression of Cancer Cell Migration and Invasion. J. BUON 2019, 24, 1581–1586. [Google Scholar]
- du Plessis-Stoman, D.; du Preez, J.; van de Venter, M. Combination Treatment with Oxaliplatin and Mangiferin Causes Increased Apoptosis and Downregulation of NFκB in Cancer Cell Lines. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 177–184. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Ma, J.; Li, X.-Y.; Wu, Y.; Shi, H.; Chen, Y.; Lu, G.; Shen, H.-M.; Lu, G.-D.; Zhou, J. Quercetin Induces P53-Independent Cancer Cell Death through Lysosome Activation by the Transcription Factor EB and Reactive Oxygen Species-Dependent Ferroptosis. Br. J. Pharmacol. 2021, 178, 1133–1148. [Google Scholar] [CrossRef]
- Kasukabe, T.; Honma, Y.; Okabe-Kado, J.; Higuchi, Y.; Kato, N.; Kumakura, S. Combined Treatment with Cotylenin A and Phenethyl Isothiocyanate Induces Strong Antitumor Activity Mainly through the Induction of Ferroptotic Cell Death in Human Pancreatic Cancer Cells. Oncol. Rep. 2016, 36, 968–976. [Google Scholar] [CrossRef]
- Kasukabe, T.; Okabe-Kado, J.; Kato, N.; Honma, Y.; Kumakura, S. Cotylenin A and Arsenic Trioxide Cooperatively Suppress Cell Proliferation and Cell Invasion Activity in Human Breast Cancer Cells. Int. J. Oncol. 2015, 46, 841–848. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-Stress-Mediated AMPK Activation Inhibits Ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Blazquez, C.; Lacalle-Gonzalez, C.; Sanz-Criado, L.; Ochieng’ Otieno, M.; Garcia-Foncillas, J.; Martinez-Useros, J. Iron-Dependent Cell Death: A New Treatment Approach against Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 14979. https://doi.org/10.3390/ijms241914979
Lopez-Blazquez C, Lacalle-Gonzalez C, Sanz-Criado L, Ochieng’ Otieno M, Garcia-Foncillas J, Martinez-Useros J. Iron-Dependent Cell Death: A New Treatment Approach against Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2023; 24(19):14979. https://doi.org/10.3390/ijms241914979
Chicago/Turabian StyleLopez-Blazquez, Carlos, Carlos Lacalle-Gonzalez, Lara Sanz-Criado, Michael Ochieng’ Otieno, Jesus Garcia-Foncillas, and Javier Martinez-Useros. 2023. "Iron-Dependent Cell Death: A New Treatment Approach against Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 24, no. 19: 14979. https://doi.org/10.3390/ijms241914979
APA StyleLopez-Blazquez, C., Lacalle-Gonzalez, C., Sanz-Criado, L., Ochieng’ Otieno, M., Garcia-Foncillas, J., & Martinez-Useros, J. (2023). Iron-Dependent Cell Death: A New Treatment Approach against Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 24(19), 14979. https://doi.org/10.3390/ijms241914979