
p53 in the Molecular Circuitry of Bone Marrow Failure Syndromes


{kind=link}
{kind=link}
Abstract
:1. Too Much of a Good Thing: The p53Δ31 Mutant Mouse Models a Telomere Biology Disorder
2. The Plot Thickens: Germline p53 Activation Underlies Features of Several Bone Marrow Failure Syndromes
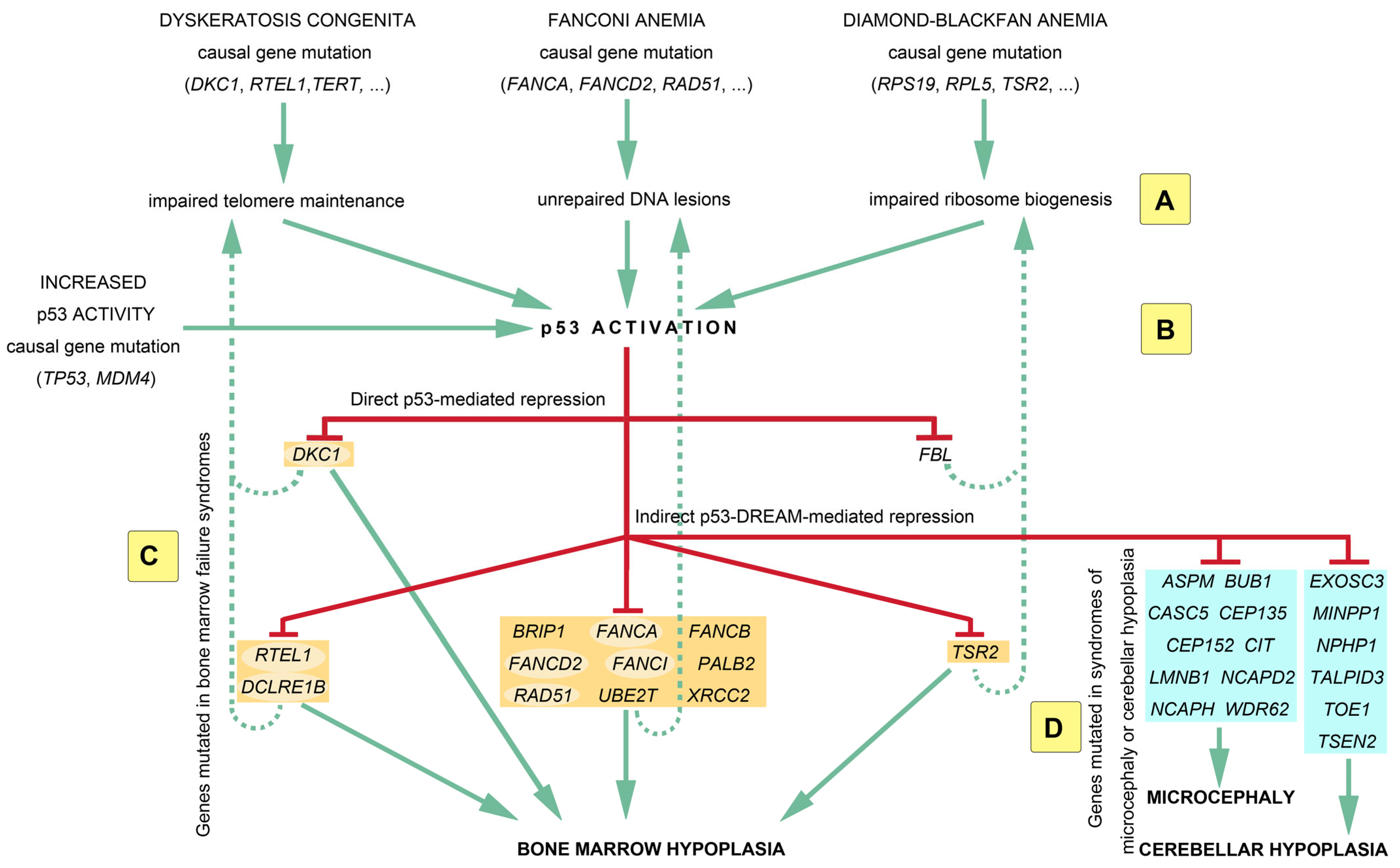
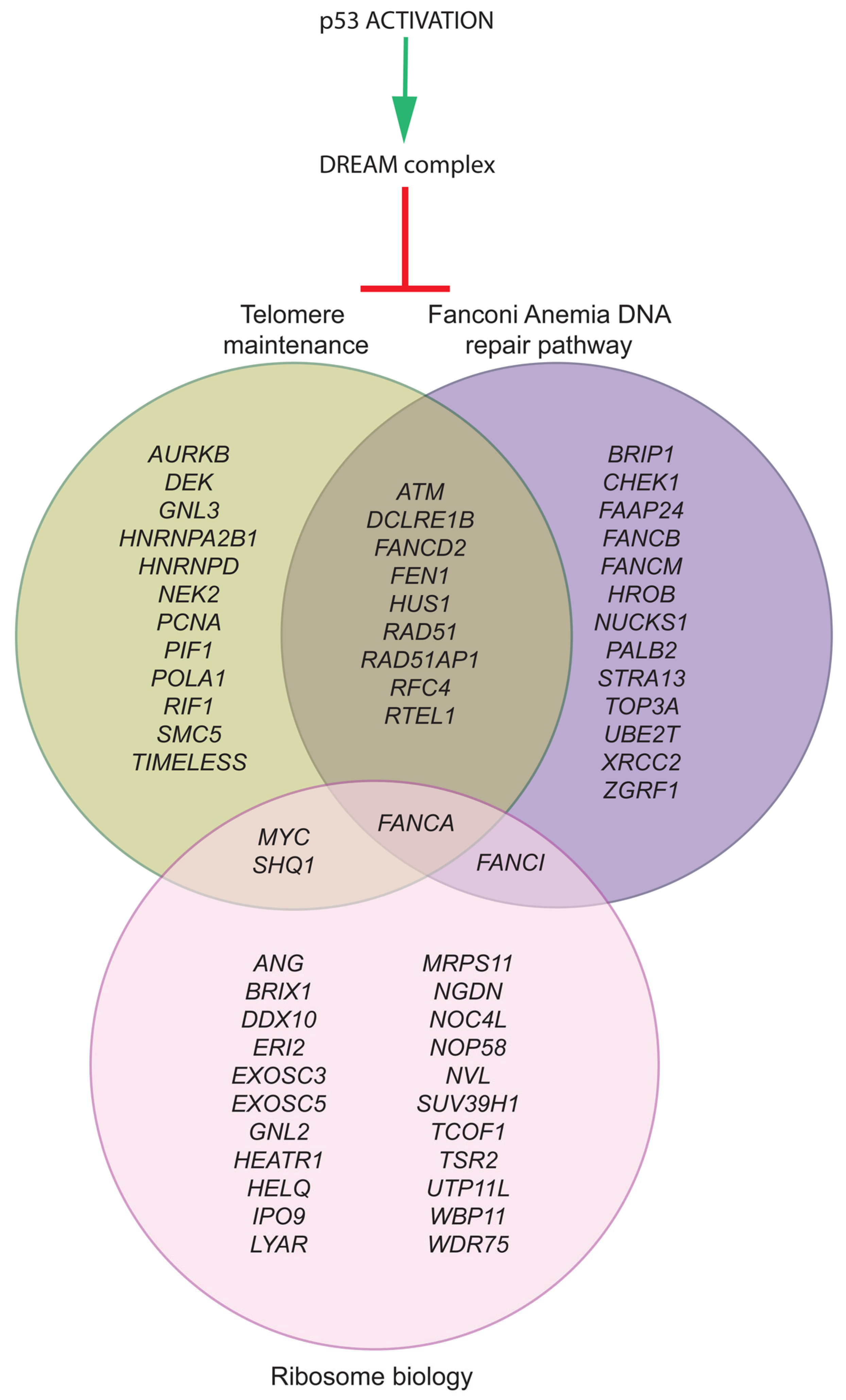
3. Food for Thought: Modeling p53 in a Circuitry of Genes Associated with Bone Marrow Failure Syndromes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hosteller, R.; Cleary, K.; Signer, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al. Mutations in the P53 Gene Occur in Diverse Human Tumour Types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ Line P53 Mutations in a Familial Syndrome of Breast Cancer, Sarcomas, and Other Neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Toledo, F.; Bardot, B. Cancer: Three Birds with One Stone. Nature 2009, 460, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Foord, O.S.; Bhattacharya, P.; Reich, Z.; Rotter, V. A DNA Binding Domain Is Contained in the C-Terminus of Wild Type P53 Protein. Nucleic Acids Res. 1991, 19, 5191–5198. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the P53 Pathway: In Vitro Hypotheses, in Vivo Veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, I.; Jaber, S.; Draskovic, I.; Bardot, B.; Fang, M.; Bouarich-Bourimi, R.; Lejour, V.; Charbonnier, L.; Soudais, C.; Bourdon, J.-C.; et al. Mutant Mice Lacking the P53 C-Terminal Domain Model Telomere Syndromes. Cell Rep. 2013, 3, 2046–2058. [Google Scholar] [CrossRef]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. P53 Is Required for Radiation-Induced Apoptosis in Mouse Thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef]
- Hamard, P.-J.; Barthelery, N.; Hogstad, B.; Mungamuri, S.K.; Tonnessen, C.A.; Carvajal, L.A.; Senturk, E.; Gillespie, V.; Aaronson, S.A.; Merad, M.; et al. The C Terminus of P53 Regulates Gene Expression by Multiple Mechanisms in a Target- and Tissue-Specific Manner in Vivo. Genes Dev. 2013, 27, 1868–1885. [Google Scholar] [CrossRef]
- McGowan, K.A.; Li, J.Z.; Park, C.Y.; Beaudry, V.; Tabor, H.K.; Sabnis, A.J.; Zhang, W.; Fuchs, H.; de Angelis, M.H.; Myers, R.M.; et al. Ribosomal Mutations Cause P53-Mediated Dark Skin and Pleiotropic Effects. Nat. Genet. 2008, 40, 963–970. [Google Scholar] [CrossRef]
- Liu, D.; Ou, L.; Clemenson, G.D.; Chao, C.; Lutske, M.E.; Zambetti, G.P.; Gage, F.H.; Xu, Y. Puma Is Required for P53-Induced Depletion of Adult Stem Cells. Nat. Cell Biol. 2010, 12, 993–998. [Google Scholar] [CrossRef]
- Mendrysa, S.M.; McElwee, M.K.; Michalowski, J.; O’Leary, K.A.; Young, K.M.; Perry, M.E. Mdm2 Is Critical for Inhibition of P53 during Lymphopoiesis and the Response to Ionizing Irradiation. Mol. Cell. Biol. 2003, 23, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Terzian, T.; Xiong, S.; Van Pelt, C.S.; Audiffred, A.; Box, N.F.; Lozano, G. The P53-Mdm2 Network in Progenitor Cell Expansion during Mouse Postnatal Development. J. Pathol. 2007, 213, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Terzian, T.; Wang, Y.; Van Pelt, C.S.; Box, N.F.; Travis, E.L.; Lozano, G. Haploinsufficiency of Mdm2 and Mdm4 in Tumorigenesis and Development. Mol. Cell. Biol. 2007, 27, 5479–5485. [Google Scholar] [CrossRef]
- Parry, E.M.; Alder, J.K.; Qi, X.; Chen, J.J.-L.; Armanios, M. Syndrome Complex of Bone Marrow Failure and Pulmonary Fibrosis Predicts Germline Defects in Telomerase. Blood 2011, 117, 5607–5611. [Google Scholar] [CrossRef] [PubMed]
- Khincha, P.P.; Savage, S.A. Genomic Characterization of the Inherited Bone Marrow Failure Syndromes. Semin. Hematol. 2013, 50, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Cole, H.N. Dyskeratosis Congenita with Pigmentation, Dystrophia Unguium, and Leucokeratosis Oris: Review of the Known Cases Reported to Date and Discussion of the Disease from Various Aspects. AMA Arch. Dermatol. 1955, 71, 451. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Tutton, S.; Azzam, G.A.; Stong, N.; Vladimirova, O.; Wiedmer, A.; Monteith, J.A.; Beishline, K.; Wang, Z.; Deng, Z.; Riethman, H.; et al. Subtelomeric P53 Binding Prevents Accumulation of DNA Damage at Human Telomeres. EMBO J. 2016, 35, 193–207. [Google Scholar] [CrossRef]
- Toledo, F. P53: A Two-Faced Regulator of Telomere Metabolism? (Comment on. DOI 10.1002/bies.201600078). Bioessays 2016, 38, 938. [Google Scholar] [CrossRef]
- Aix, E.; Gutiérrez-Gutiérrez, Ó.; Sánchez-Ferrer, C.; Aguado, T.; Flores, I. Postnatal Telomere Dysfunction Induces Cardiomyocyte Cell-Cycle Arrest through P21 Activation. J. Cell Biol. 2016, 213, 571–583. [Google Scholar] [CrossRef]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere Shortening and Tumor Formation by Mouse Cells Lacking Telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Wang, Y.; Guo, X.; Ramchandani, S.; Ma, J.; Shen, M.-F.; Garcia, D.A.; Deng, Y.; Multani, A.S.; You, M.J.; et al. Pot1b Deletion and Telomerase Haploinsufficiency in Mice Initiate an ATR-Dependent DNA Damage Response and Elicit Phenotypes Resembling Dyskeratosis Congenita. Mol. Cell. Biol. 2009, 29, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Palm, W.; Wang, R.C.; Couto, S.S.; De Lange, T. Engineered Telomere Degradation Models Dyskeratosis Congenita. Genes Dev. 2008, 22, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Holohan, B.; Wright, W.E.; Shay, J.W. Cell Biology of Disease: Telomeropathies: An Emerging Spectrum Disorder. J. Cell Biol. 2014, 205, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.; Blackburn, E.H. The Telomere Syndromes. Nat. Rev. Genet. 2012, 13, 693–704. [Google Scholar] [CrossRef]
- Jaber, S.; Toufektchan, E.; Lejour, V.; Bardot, B.; Toledo, F. P53 Downregulates the Fanconi Anaemia DNA Repair Pathway. Nat. Commun. 2016, 7, 11091. [Google Scholar] [CrossRef]
- Tummala, H.; Walne, A.; Collopy, L.; Cardoso, S.; de la Fuente, J.; Lawson, S.; Powell, J.; Cooper, N.; Foster, A.; Mohammed, S.; et al. Poly(A)-Specific Ribonuclease Deficiency Impacts Telomere Biology and Causes Dyskeratosis Congenita. J. Clin. Investig. 2015, 125, 2151–2160. [Google Scholar] [CrossRef]
- Kocak, H.; Ballew, B.J.; Bisht, K.; Eggebeen, R.; Hicks, B.D.; Suman, S.; O’Neil, A.; Giri, N.; NCI DCEG Cancer Genomics Research Laboratory; NCI DCEG Cancer Sequencing Working Group; et al. Hoyeraal-Hreidarsson Syndrome Caused by a Germline Mutation in the TEL Patch of the Telomere Protein TPP1. Genes Dev. 2014, 28, 2090–2102. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rossi, M.L.; Singh, D.K.; Dunn, C.; Ramamoorthy, M.; Croteau, D.L.; Liu, Y.; Bohr, V.A. RECQL4, the Protein Mutated in Rothmund-Thomson Syndrome, Functions in Telomere Maintenance. J. Biol. Chem. 2012, 287, 196–209. [Google Scholar] [CrossRef]
- Joksic, I.; Vujic, D.; Guc-Scekic, M.; Leskovac, A.; Petrovic, S.; Ojani, M.; Trujillo, J.P.; Surralles, J.; Zivkovic, M.; Stankovic, A.; et al. Dysfunctional Telomeres in Primary Cells from Fanconi Anemia FANCD2 Patients. Genome Integr. 2012, 3, 6. [Google Scholar] [CrossRef]
- Meetei, A.R.; Sechi, S.; Wallisch, M.; Yang, D.; Young, M.K.; Joenje, H.; Hoatlin, M.E.; Wang, W. A Multiprotein Nuclear Complex Connects Fanconi Anemia and Bloom Syndrome. Mol. Cell. Biol. 2003, 23, 3417–3426. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Yuan, F.; Rodriguez-Tello, P.; Padgaonkar, S.; Zhang, Y. Human Fanconi Anemia Complementation Group a Protein Stimulates the 5’ Flap Endonuclease Activity of FEN1. PLoS ONE 2013, 8, e82666. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Suhasini, A.N.; Brosh, R.M. Welcome the Family of FANCJ-like Helicases to the Block of Genome Stability Maintenance Proteins. Cell. Mol. Life Sci. 2009, 66, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.D.; Zacharias, N.M.; Tripathi, D.N.; Karki, M.; Bertocchio, J.-P.; Soeung, M.; He, R.; Westerman, M.E.; Gao, J.; Rao, P.; et al. Neddylation Inhibition Sensitises Renal Medullary Carcinoma Tumours to Platinum Chemotherapy. Clin. Transl. Med. 2023, 13, e1267. [Google Scholar] [CrossRef] [PubMed]
- Devany, E.; Zhang, X.; Park, J.Y.; Tian, B.; Kleiman, F.E. Positive and Negative Feedback Loops in the P53 and mRNA 3’ Processing Pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 3351–3356. [Google Scholar] [CrossRef]
- Moon, D.H.; Segal, M.; Boyraz, B.; Guinan, E.; Hofmann, I.; Cahan, P.; Tai, A.K.; Agarwal, S. Poly(A)-Specific Ribonuclease (PARN) Mediates 3’-End Maturation of the Telomerase RNA Component. Nat. Genet. 2015, 47, 1482–1488. [Google Scholar] [CrossRef]
- Boyraz, B.; Moon, D.H.; Segal, M.; Muosieyiri, M.Z.; Aykanat, A.; Tai, A.K.; Cahan, P.; Agarwal, S. Posttranscriptional Manipulation of TERC Reverses Molecular Hallmarks of Telomere Disease. J. Clin. Investig. 2016, 126, 3377–3382. [Google Scholar] [CrossRef]
- Toki, T.; Yoshida, K.; Wang, R.; Nakamura, S.; Maekawa, T.; Goi, K.; Katoh, M.C.; Mizuno, S.; Sugiyama, F.; Kanezaki, R.; et al. De Novo Mutations Activating Germline TP53 in an Inherited Bone-Marrow-Failure Syndrome. Am. J. Hum. Genet. 2018, 103, 440–447. [Google Scholar] [CrossRef]
- Fedorova, D.; Ovsyannikova, G.; Kurnikova, M.; Pavlova, A.; Konyukhova, T.; Pshonkin, A.; Smetanina, N. De Novo TP53 Germline Activating Mutations in Two Patients with the Phenotype Mimicking Diamond–Blackfan Anemia. Pediatr. Blood Cancer 2022, 69, e29558. [Google Scholar] [CrossRef]
- Kumar, R.D.; Tosur, M.; Lalani, S.R.; Mahoney, D.H.; Bertuch, A.A. The Germline P53 Activation Syndrome: A New Patient Further Refines the Clinical Phenotype. Am. J. Med. Genet. Part A 2022, 188, 2204–2208. [Google Scholar] [CrossRef]
- Toufektchan, E.; Lejour, V.; Durand, R.; Giri, N.; Draskovic, I.; Bardot, B.; Laplante, P.; Jaber, S.; Alter, B.P.; Londono-Vallejo, J.-A.; et al. Germline Mutation of MDM4, a Major P53 Regulator, in a Familial Syndrome of Defective Telomere Maintenance. Sci. Adv. 2020, 6, eaay3511. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Begemann, M.; Jakob, M.; Khurana, C.; Dey, D.; Hanafee Alali, T.; Gabriel, H.; Kricheldorf, K.; Vieri, M.; Isfort, S.; et al. Germline Variants in MDM4 Cause a Disorder of P53 Dysregulation and Insufficient Telomere Maintenance. Blood 2022, 140, 2951–2952. [Google Scholar] [CrossRef]
- Sharma, R.; Bhoopalan, S.; Han, L.; Yao, Y.; Mayberry, K.; Hansen, B.S.; Miller, S.; Khurana, C.; Erlacher, M.; Wlodarski, M.W. Germline Loss-of-Function Mutations in MDM4 Cause P53-Dependent Hematopoietic Cell Death in Patients with Variable Bone Marrow Failure Phenotypes. Blood 2022, 140, 2955–2956. [Google Scholar] [CrossRef]
- Lessel, D.; Wu, D.; Trujillo, C.; Ramezani, T.; Lessel, I.; Alwasiyah, M.K.; Saha, B.; Hisama, F.M.; Rading, K.; Goebel, I.; et al. Dysfunction of the MDM2/P53 Axis Is Linked to Premature Aging. J. Clin. Investig. 2017, 127, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Bluteau, O.; Sebert, M.; Leblanc, T.; Peffault de Latour, R.; Quentin, S.; Lainey, E.; Hernandez, L.; Dalle, J.-H.; Sicre de Fontbrune, F.; Lengline, E.; et al. A Landscape of Germ Line Mutations in a Cohort of Inherited Bone Marrow Failure Patients. Blood 2018, 131, 717–732. [Google Scholar] [CrossRef]
- La Torre, M.; Merigliano, C.; Burla, R.; Mottini, C.; Zanetti, G.; Del Giudice, S.; Carcuro, M.; Virdia, I.; Bucciarelli, E.; Manni, I.; et al. Mice with Reduced Expression of the Telomere-Associated Protein Ft1 Develop P53-Sensitive Progeroid Traits. Aging Cell 2018, 17, e12730. [Google Scholar] [CrossRef]
- Ghadaouia, S.; Olivier, M.-A.; Martinez, A.; Kientega, T.; Qin, J.; Lambert-Lanteigne, P.; Cardin, G.B.; Autexier, C.; Malaquin, N.; Rodier, F. Homologous Recombination-Mediated Irreversible Genome Damage Underlies Telomere-Induced Senescence. Nucleic Acids Res. 2021, 49, 11690–11707. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.-S.; Pondarre, C.; et al. Bone Marrow Failure in Fanconi Anemia Is Triggered by an Exacerbated P53/P21 DNA Damage Response That Impairs Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; de Lange, T. P53- and ATM-Dependent Apoptosis Induced by Telomeres Lacking TRF2. Science 1999, 283, 1321–1325. [Google Scholar] [CrossRef]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; DePinho, R.A. P53 Deficiency Rescues the Adverse Effects of Telomere Loss and Cooperates with Telomere Dysfunction to Accelerate Carcinogenesis. Cell 1999, 97, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Winford-Thomas, D.; Bond, J.A.; Wyllie, F.S.; Jones, C.J. Does Telomere Shortening Drive Selection for p53 Mutation in Human Cancer? Mol. Carcinog. 1995, 12, 119–123. [Google Scholar] [CrossRef]
- Bhoopalan, S.V.; Yen, J.S.; Mayuranathan, T.; Mayberry, K.D.; Yao, Y.; Lillo Osuna, M.A.; Jang, Y.; Liyanage, J.S.S.; Blanc, L.; Ellis, S.R.; et al. An RPS19-Edited Model for Diamond-Blackfan Anemia Reveals TP53-Dependent Impairment of Hematopoietic Stem Cell Activity. JCI Insight 2023, 8, e161810. [Google Scholar] [CrossRef] [PubMed]
- Jaako, P.; Flygare, J.; Olsson, K.; Quere, R.; Ehinger, M.; Henson, A.; Ellis, S.; Schambach, A.; Baum, C.; Richter, J.; et al. Mice with Ribosomal Protein S19 Deficiency Develop Bone Marrow Failure and Symptoms like Patients with Diamond-Blackfan Anemia. Blood 2011, 118, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Fok, W.C.; de Oliveira Niero, E.L.; Dege, C.; Brenner, K.A.; Sturgeon, C.M.; Batista, L.F.Z. P53 Mediates Failure of Human Definitive Hematopoiesis in Dyskeratosis Congenita. Stem Cell Rep. 2017, 9, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Marion, W.; Boettcher, S.; Ruiz-Torres, S.; Lummertz da Rocha, E.; Lundin, V.; Morris, V.; Chou, S.; Zhao, A.M.; Kubaczka, C.; Aumais, O.; et al. An Induced Pluripotent Stem Cell Model of Fanconi Anemia Reveals Mechanisms of P53-Driven Progenitor Cell Differentiation. Blood Adv. 2020, 4, 4679–4692. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Brandt, L.T.L.; Wang, X.; Russell, H.; Mitchell, E.; Kamimae-Lanning, A.N.; Brown, J.M.; Dingler, F.A.; Garaycoechea, J.I.; Isobe, T.; et al. Genotoxic Aldehyde Stress Prematurely Ages Hematopoietic Stem Cells in a P53-Driven Manner. Mol. Cell 2023, 83, 2417–2433.e7. [Google Scholar] [CrossRef]
- Li, X.; Wilson, A.F.; Du, W.; Pang, Q. Cell-Cycle-Specific Function of P53 in Fanconi Anemia Hematopoietic Stem and Progenitor Cell Proliferation. Stem Cell Rep. 2018, 10, 339–346. [Google Scholar] [CrossRef]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for Ribosomal Protein Genes Causes Selective Activation of P53 in Human Erythroid Progenitor Cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef]
- Ghemlas, I.; Li, H.; Zlateska, B.; Klaassen, R.; Fernandez, C.V.; Yanofsky, R.A.; Wu, J.; Pastore, Y.; Silva, M.; Lipton, J.H.; et al. Improving Diagnostic Precision, Care and Syndrome Definitions Using Comprehensive next-Generation Sequencing for the Inherited Bone Marrow Failure Syndromes. J. Med. Genet. 2015, 52, 575–584. [Google Scholar] [CrossRef]
- Steier, W.; Van Voolen, G.A.; Selmanowitz, V.J. Dyskeratosis Congenita: Relationship to Fanconi’s Anemia. Blood 1972, 39, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Leteurtre, F.; Rocha, V.; Guardiola, P.; Berger, R.; Daniel, M.-T.; Noguera, M.H.; Maarek, O.; Roux, G.L.E.; de la Salmonière, P.; et al. Abnormal Telomere Metabolism in Fanconi’s Anaemia Correlates with Genomic Instability and the Probability of Developing Severe Aplastic Anaemia. Br. J. Haematol. 2003, 120, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Leteurtre, F.; Li, X.; Guardiola, P.; Le Roux, G.; Sergère, J.C.; Richard, P.; Carosella, E.D.; Gluckman, E. Accelerated Telomere Shortening and Telomerase Activation in Fanconi’s Anaemia. Br. J. Haematol. 1999, 105, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Little, J.B. Suppression of Cytotoxic Effect of Mitomycin-C by Superoxide Dismutase in Fanconi’s Anemia and Dyskeratosis Congenita Fibroblasts. Carcinogenesis 1983, 4, 795–799. [Google Scholar] [CrossRef]
- Ballew, B.J.; Joseph, V.; De, S.; Sarek, G.; Vannier, J.-B.; Stracker, T.; Schrader, K.A.; Small, T.N.; O’Reilly, R.; Manschreck, C.; et al. A Recessive Founder Mutation in Regulator of Telomere Elongation Helicase 1, RTEL1, Underlies Severe Immunodeficiency and Features of Hoyeraal Hreidarsson Syndrome. PLoS Genet. 2013, 9, e1003695. [Google Scholar] [CrossRef]
- Castillo Duque de Estrada, N.M.; Thoms, M.; Flemming, D.; Hammaren, H.M.; Buschauer, R.; Ameismeier, M.; Baßler, J.; Beck, M.; Beckmann, R.; Hurt, E. Structure of Nascent 5S RNPs at the Crossroad between Ribosome Assembly and MDM2-P53 Pathways. Nat. Struct. Mol. Biol. 2023, 30, 1119–1131. [Google Scholar] [CrossRef]
- Eastham, M.J.; Pelava, A.; Wells, G.R.; Lee, J.K.; Lawrence, I.R.; Stewart, J.; Deichner, M.; Hertle, R.; Watkins, N.J.; Schneider, C. The Induction of P53 Correlates with Defects in the Production, but Not the Levels, of the Small Ribosomal Subunit and Stalled Large Ribosomal Subunit Biogenesis. Nucleic Acids Res. 2023, 51, 9397–9414. [Google Scholar] [CrossRef]
- Golomb, L.; Volarevic, S.; Oren, M. P53 and Ribosome Biogenesis Stress: The Essentials. FEBS Lett. 2014, 588, 2571–2579. [Google Scholar] [CrossRef]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the Tumor Suppressor P53 upon Impairment of Ribosome Biogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 817–830. [Google Scholar] [CrossRef]
- Franklin, D.A.; Liu, S.; Jin, A.; Cui, P.; Guo, Z.; Arend, K.C.; Moorman, N.J.; He, S.; Wang, G.G.; Wan, Y.Y.; et al. Ribosomal Protein RPL11 Haploinsufficiency Causes Anemia in Mice via Activation of the RP-MDM2-P53 Pathway. J. Biol. Chem. 2023, 299, 102739. [Google Scholar] [CrossRef]
- Marcel, V.; Ghayad, S.E.; Belin, S.; Therizols, G.; Morel, A.-P.; Solano-Gonzàlez, E.; Vendrell, J.A.; Hacot, S.; Mertani, H.C.; Albaret, M.A.; et al. P53 Acts as a Safeguard of Translational Control by Regulating Fibrillarin and rRNA Methylation in Cancer. Cancer Cell 2013, 24, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, U.; Ray, A.; Sehgal, P.B. Repression of the Interleukin 6 Gene Promoter by P53 and the Retinoblastoma Susceptibility Gene Product. Proc. Natl. Acad. Sci. USA 1991, 88, 7605–7609. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, D.; Mechta, F.; Yaniv, M.; Oren, M. Wild-Type P53 Can down-Modulate the Activity of Various Promoters. Proc. Natl. Acad. Sci. USA 1991, 88, 9979–9983. [Google Scholar] [CrossRef] [PubMed]
- Peuget, S.; Selivanova, G. P53-Dependent Repression: DREAM or Reality? Cancers 2021, 13, 4850. [Google Scholar] [CrossRef]
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Benson, E.K.; Mungamuri, S.K.; Attie, O.; Kracikova, M.; Sachidanandam, R.; Manfredi, J.J.; Aaronson, S.A. P53-Dependent Gene Repression through P21 Is Mediated by Recruitment of E2F4 Repression Complexes. Oncogene 2014, 33, 3959–3969. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Wintsche, A.; Müller, G.A.; Engeland, K. Polo-like Kinase 4 Transcription Is Activated via CRE and NRF1 Elements, Repressed by DREAM through CDE/CHR Sites and Deregulated by HPV E7 Protein. Nucleic Acids Res. 2014, 42, 163–180. [Google Scholar] [CrossRef]
- Fischer, M.; Steiner, L.; Engeland, K. The Transcription Factor P53: Not a Repressor, Solely an Activator. Cell Cycle 2014, 13, 3037–3058. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The P53-P21-DREAM-CDE/CHR Pathway Regulates G2/M Cell Cycle Genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F Target Gene Analyses Identifies Cell Cycle Gene Regulatory Networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Engeland, K. Cell Cycle Arrest through Indirect Transcriptional Repression by P53: I Have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: P53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Schade, A.E.; Branigan, T.B.; Müller, G.A.; DeCaprio, J.A. Coordinating Gene Expression during the Cell Cycle. Trends Biochem. Sci. 2022, 47, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Naughtin, M.; Podsypanina, K.; Lejour, V.; Wilson, L.; Gurard-Levin, Z.A.; Orsi, G.A.; Simeonova, I.; Toufektchan, E.; Attardi, L.D.; et al. Essential Role for Centromeric Factors Following P53 Loss and Oncogenic Transformation. Genes Dev. 2017, 31, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.J.; Hess, J.L. The PAF Complex Synergizes with MLL Fusion Proteins at HOX Loci to Promote Leukemogenesis. Cancer Cell 2010, 17, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Thorsteinsdottir, U.; Mamo, A.; Kroon, E.; Jerome, L.; Bijl, J.; Lawrence, H.J.; Humphries, K.; Sauvageau, G. Overexpression of the Myeloid Leukemia-Associated Hoxa9 Gene in Bone Marrow Cells Induces Stem Cell Expansion. Blood 2002, 99, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.J.; Helgason, C.D.; Sauvageau, G.; Fong, S.; Izon, D.J.; Humphries, R.K.; Largman, C. Mice Bearing a Targeted Interruption of the Homeobox Gene HOXA9 Have Defects in Myeloid, Erythroid, and Lymphoid Hematopoiesis. Blood 1997, 89, 1922–1930. [Google Scholar] [CrossRef]
- Rakotopare, J.; Lejour, V.; Duval, C.; Eldawra, E.; Escoffier, H.; Toledo, F. A Systematic Approach Identifies P53-DREAM Pathway Target Genes Associated with Blood or Brain Abnormalities. Dis. Models Mech. 2023, 16, dmm.050376. [Google Scholar] [CrossRef]
- Fischer, M.; Schwarz, R.; Riege, K.; DeCaprio, J.A.; Hoffmann, S. TargetGeneReg 2.0: A Comprehensive Web-Atlas for P53, P63, and Cell Cycle-Dependent Gene Regulation. NAR Cancer 2022, 4, zcac009. [Google Scholar] [CrossRef]
- Zou, Z.; Ohta, T.; Miura, F.; Oki, S. ChIP-Atlas 2021 Update: A Data-Mining Suite for Exploring Epigenomic Landscapes by Fully Integrating ChIP-Seq, ATAC-Seq and Bisulfite-Seq Data. Nucleic Acids Res. 2022, 50, W175–W182. [Google Scholar] [CrossRef]
- Resnick-Silverman, L.; Zhou, R.; Campbell, M.J.; Leibling, I.; Parsons, R.; Manfredi, J.J. In Vivo RNA-Seq and ChIP-Seq Analyses Show an Obligatory Role for the C Terminus of P53 in Conferring Tissue-Specific Radiation Sensitivity. Cell Rep. 2023, 42, 112216. [Google Scholar] [CrossRef] [PubMed]
- Tung, L.T.; Wang, H.; Belle, J.I.; Petrov, J.C.; Langlais, D.; Nijnik, A. P53-Dependent Induction of P2X7 on Hematopoietic Stem and Progenitor Cells Regulates Hematopoietic Response to Genotoxic Stress. Cell Death Dis. 2021, 12, 923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Hammack, C.; Ogden, S.C.; Cheng, Y.; Lee, E.M.; Wen, Z.; Qian, X.; Nguyen, H.N.; Li, Y.; Yao, B.; et al. Molecular Signatures Associated with ZIKV Exposure in Human Cortical Neural Progenitors. Nucleic Acids Res. 2016, 44, 8610–8620. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Mages, C.F.; Wintsche, A.; Bernhart, S.H.; Müller, G.A. The DREAM Complex through Its Subunit Lin37 Cooperates with Rb to Initiate Quiescence. Elife 2017, 6, e26876. [Google Scholar] [CrossRef]
- Armanios, M. The Role of Telomeres in Human Disease. Annu. Rev. Genom. Hum. Genet. 2022, 23, 363–381. [Google Scholar] [CrossRef]
- Garus, A.; Autexier, C. Dyskerin: An Essential Pseudouridine Synthase with Multifaceted Roles in Ribosome Biogenesis, Splicing, and Telomere Maintenance. RNA 2021, 27, 1441–1458. [Google Scholar] [CrossRef]
- Wang, S.; Ding, B.; Cui, M.; Yan, W.; Xia, Q.; Meng, D.; Shen, S.; Xie, S.; Jin, H.; Zhang, X. Fanconi Anemia Pathway Genes Advance Cervical Cancer via Immune Regulation and Cell Adhesion. Front. Cell Dev. Biol. 2021, 9, 734794. [Google Scholar] [CrossRef]
- Uringa, E.-J.; Lisaingo, K.; Pickett, H.A.; Brind’Amour, J.; Rohde, J.-H.; Zelensky, A.; Essers, J.; Lansdorp, P.M. RTEL1 Contributes to DNA Replication and Repair and Telomere Maintenance. Mol. Biol. Cell 2012, 23, 2782–2792. [Google Scholar] [CrossRef]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-Phenotype Associations in Fanconi Anemia: A Literature Review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef]
- Sondalle, S.B.; Longerich, S.; Ogawa, L.M.; Sung, P.; Baserga, S.J. Fanconi Anemia Protein FANCI Functions in Ribosome Biogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Badie, S.; Escandell, J.M.; Bouwman, P.; Carlos, A.R.; Thanasoula, M.; Gallardo, M.M.; Suram, A.; Jaco, I.; Benitez, J.; Herbig, U.; et al. BRCA2 Acts as a RAD51 Loader to Facilitate Telomere Replication and Capping. Nat. Struct. Mol. Biol. 2010, 17, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Gueiderikh, A.; Maczkowiak-Chartois, F.; Rouvet, G.; Souquère-Besse, S.; Apcher, S.; Diaz, J.-J.; Rosselli, F. Fanconi Anemia A Protein Participates in Nucleolar Homeostasis Maintenance and Ribosome Biogenesis. Sci. Adv. 2021, 7, eabb5414. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zhang, F.; Barrett, B.; Ren, K.; Andreassen, P.R. A Role for Monoubiquitinated FANCD2 at Telomeres in ALT Cells. Nucleic Acids Res. 2009, 37, 1740–1754. [Google Scholar] [CrossRef]
- Savage, S.A. Dyskeratosis Congenita and Telomere Biology Disorders. Hematology 2022, 2022, 637–648. [Google Scholar] [CrossRef]
- Vlachos, A.; Muir, E. How I Treat Diamond-Blackfan Anemia. Blood 2010, 116, 3715–3723. [Google Scholar] [CrossRef]
- Keller, M.D.; Ganesh, J.; Heltzer, M.; Paessler, M.; Bergqvist, A.G.C.; Baluarte, H.J.; Watkins, D.; Rosenblatt, D.S.; Orange, J.S. Severe Combined Immunodeficiency Resulting from Mutations in MTHFD1. Pediatrics 2013, 131, e629–e634. [Google Scholar] [CrossRef]
- Vial, Y.; Lainey, E.; Leblanc, T.; Baudouin, V.; Dourthe, M.E.; Gressens, P.; Verloes, A.; Cavé, H.; Drunat, S. De Novo NUF2 Variant in a Novel Inherited Bone Marrow Failure Syndrome Including Microcephaly and Renal Hypoplasia. Br. J. Haematol. 2022, 199, 739–743. [Google Scholar] [CrossRef]
- Barroso-González, J.; García-Expósito, L.; Hoang, S.M.; Lynskey, M.L.; Roncaioli, J.L.; Ghosh, A.; Wallace, C.T.; de Vitis, M.; Modesti, M.; Bernstein, K.A.; et al. RAD51AP1 Is an Essential Mediator of Alternative Lengthening of Telomeres. Mol. Cell 2019, 76, 11–26.e7. [Google Scholar] [CrossRef]
- Sarkar, J.; Liu, Y. Fanconi Anemia Proteins in Telomere Maintenance. DNA Repair 2016, 43, 107–112. [Google Scholar] [CrossRef]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a Regulator of Ribosome Biogenesis and Protein Synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Brannvoll, A.; Xue, X.; Kwon, Y.; Kompocholi, S.; Simonsen, A.K.W.; Viswalingam, K.S.; Gonzalez, L.; Hickson, I.D.; Oestergaard, V.H.; Mankouri, H.W.; et al. The ZGRF1 Helicase Promotes Recombinational Repair of Replication-Blocking DNA Damage in Human Cells. Cell Rep. 2020, 32, 107849. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Elfaki, M.; Alkuraya, F.S. Exome Sequencing Reveals a Novel Fanconi Group Defined by XRCC2 Mutation. J. Med. Genet. 2012, 49, 184–186. [Google Scholar] [CrossRef]
- Leman, A.R.; Dheekollu, J.; Deng, Z.; Lee, S.W.; Das, M.M.; Lieberman, P.M.; Noguchi, E. Timeless Preserves Telomere Length by Promoting Efficient DNA Replication through Human Telomeres. Cell Cycle 2012, 11, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Ivanauskiene, K.; Delbarre, E.; McGhie, J.D.; Küntziger, T.; Wong, L.H.; Collas, P. The PML-Associated Protein DEK Regulates the Balance of H3.3 Loading on Chromatin and Is Important for Telomere Integrity. Genome Res. 2014, 24, 1584–1594. [Google Scholar] [CrossRef]
- Grozdanov, P.N.; Roy, S.; Kittur, N.; Meier, U.T. SHQ1 Is Required Prior to NAF1 for Assembly of H/ACA Small Nucleolar and Telomerase RNPs. RNA 2009, 15, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakotopare, J.; Toledo, F. p53 in the Molecular Circuitry of Bone Marrow Failure Syndromes. Int. J. Mol. Sci. 2023, 24, 14940. https://doi.org/10.3390/ijms241914940
Rakotopare J, Toledo F. p53 in the Molecular Circuitry of Bone Marrow Failure Syndromes. International Journal of Molecular Sciences. 2023; 24(19):14940. https://doi.org/10.3390/ijms241914940
Chicago/Turabian StyleRakotopare, Jeanne, and Franck Toledo. 2023. "p53 in the Molecular Circuitry of Bone Marrow Failure Syndromes" International Journal of Molecular Sciences 24, no. 19: 14940. https://doi.org/10.3390/ijms241914940
APA StyleRakotopare, J., & Toledo, F. (2023). p53 in the Molecular Circuitry of Bone Marrow Failure Syndromes. International Journal of Molecular Sciences, 24(19), 14940. https://doi.org/10.3390/ijms241914940