


Anti-NMDA and Anti-AMPA Receptor Antibodies in Central Disorders: Preclinical Approaches to Assess Their Pathological Role and Translatability to Clinic



Abstract
:1. Introduction
2. NMDA Receptors and Anti-GluN Autoantibodies in Central Disorders
3. AMPA Receptors and Anti-GluA Autoantibodies in Central Disorders
4. Pathogenic Mechanisms of Neuronal Cell-Surface Autoantibodies
- (i)
- Modify the in–out receptors’ movements in plasma membranes by either stabilizing the receptor protein in the phospholipid bilayer or, alternatively, by increasing its endocytosis and subsequent association with cytosolic proteins (i.e., beta-arrestin), which would favor protein degradation [18]. In the first case, antibodies would stabilize the receptors in membranes and potentially amplify the associated signal. Differently, in the second case, they would reduce the number of receptors and accelerate their endocytosis and cytosolic degradation, causing a loss of function and a concomitant receptor depletion in the targeted cells. To note, the mechanisms of the in–out trafficking of proteins in neuronal plasma membranes control the density and therefore the signals of several receptors, including NMDA receptors [66], AMPA receptors [67] but also GABAB receptors [68] and glycinergic receptors [69]. Therefore, by interfering with the trafficking of these receptors, autoantibodies would alter the efficiency of either the excitatory or the inhibitory synaptic plasticity, exposing the brain to a detrimental condition;
- (ii)
- Behave either as agonists or antagonists, or even imitate allosteric ligands, influencing the responsiveness of the receptors toward the endogenous ligand(s) [70,71]. As a matter of fact, although the available literature is more consistent with an antagonistic profile [72], data also exist supporting an agonist-like activity in neurons [73];
- (iii)
- Disrupt the protein–protein interactions that control the strength of the synaptic connections, affecting the stability of the synapses themselves. This is the case of LGI1 antibodies which block the interaction with LGI1 with the ADAM22/23 proteins, destabilizing the synaptic active zone and leading to the decreased surface accumulation of the AMPA receptors [74].
5. Cellular and Molecular Mechanisms Mediating the Impact of Anti-NMDA Receptor Antibodies in Neurons: Preclinical Results in Animals and Transability to Human
5.1. Impact of Anti-NMDA Receptor Autoantibodies in Neurons
5.2. Impact of Commercial Anti-NMDA Receptor Antibodies in Neurons
5.3. Impact of Anti-NMDA Receptor Autoantibodies in Neurons: Future Perspectives
6. Cellular and Synaptic Mechanisms Mediating the Impact of Anti-AMPA Receptor Antibodies in Neurons: Preclinical Results in Animals and Transability to Human
6.1. Impact of Anti-AMPA Receptor Antibodies in Neurons: The Anti-GluA3 Antibodies
6.2. Impact of Anti-AMPA Receptor Antibodies in Neurons: The Anti-GluA2 Antibodies
6.3. Impact of Anti-AMPA Receptor Antibodies in Neurons: The Anti-GluA1 Antibodies
6.4. Impact of Anti-AMPA Receptor Antibodies in Neurons: Role of Complement
6.5. Impact of Anti-AMPA Receptor Autoantibodies in Neurons: Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grant, S.G.N. Synaptopathies: Diseases of the synaptome. Curr. Opin. Neurobiol. 2012, 22, 522–529. [Google Scholar] [CrossRef]
- Malenka, R.C.; Nicoll, R.A. NMDA-receptor-dependent synaptic plasticity: Multiple forms and mechanisms. Trends Neurosci. 1993, 16, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Mordelt, A.; de Witte, L.D. Microglia-mediated synaptic pruning as a key deficit in neurodevelopmental disorders: Hype or hope? Curr. Opin. Neurobiol. 2023, 79, 102674. [Google Scholar] [CrossRef] [PubMed]
- Kirschstein, T.; Köhling, R. Functional changes in neuronal circuits due to antibody-driven autoimmune response. Neurobiol. Dis. 2023, 184, 106221. [Google Scholar] [CrossRef] [PubMed]
- Khojah, O.; Makkawi, S.; Alghamdi, S. Anti-mGluR1 encephalitis: Case illustration and systematic review. Front. Neurol. 2023, 14, 1142160. [Google Scholar] [CrossRef]
- Paas, Y. The pathophysiological mechanism underlying Rasmussen’s encephalitis: A debate. Trends Neurosci. 1998, 21, 468–469. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Verde, F.; Morelli, L.; Rizzo, G.; Ricciardiello, F.; Liguori, R. Neural Surface Antibodies and Neurodegeneration: Clinical Commonalities and Pathophysiological Relationships. Biomedicines 2023, 11, 666. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef]
- Pittaluga, A. Presynaptic release-regulating NMDA receptors in isolated nerve terminals: A narrative review. Br. J. Pharmacol. 2021, 178, 1001–1017. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Menniti, F.S.; Traynelis, S.F. NMDA Receptors in the Central Nervous System. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1677, pp. 1–80. [Google Scholar]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Wang, Y.T. GluN2B-containing NMDARs in the mammalian brain: Pharmacology, physiology, and pathology. Front. Mol. Neurosci. 2023, 16, 1190324. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Tüzün, E.; Wu, H.-Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef]
- Zhang, Q.; Tanaka, K.; Sun, P.; Nakata, M.; Yamamoto, R.; Sakimura, K.; Matsui, M.; Kato, N. Suppression of synaptic plasticity by cerebrospinal fluid from anti-NMDA receptor encephalitis patients. Neurobiol. Dis. 2012, 45, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Moscato, E.H.; Peng, X.; Jain, A.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Acute mechanisms underlying antibody effects in anti–N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2014, 76, 108–119. [Google Scholar] [CrossRef]
- Venuti, A.; Pastori, C.; Siracusano, G.; Pennisi, R.; Riva, A.; Tommasino, M.; Sciortino, M.T.; Lopalco, L. The Abrogation of Phosphorylation Plays a Relevant Role in the CCR5 Signalosome Formation with Natural Antibodies to CCR5. Viruses 2018, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Kreye, J.; Wenke, N.K.; Chayka, M.; Leubner, J.; Murugan, R.; Maier, N.; Jurek, B.; Ly, L.-T.; Brandl, D.; Rost, B.R.; et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 2016, 139, 2641–2652. [Google Scholar] [CrossRef]
- McKeon, G.; Parker, S.; Warren, N.; Scott, J.G. The Patient Experience of Recovery Following Anti-NMDA Receptor Encephalitis: A Qualitative Content Analysis. J. Neuropsychiatry Clin. Neurosci. 2021, 33, 57–63. [Google Scholar] [CrossRef]
- Heine, J.; Kopp, U.A.; Klag, J.; Ploner, C.J.; Prüss, H.; Finke, C. Long-Term Cognitive Outcome in Anti–N-Methyl-D-Aspartate Receptor Encephalitis. Ann. Neurol. 2021, 90, 949–961. [Google Scholar] [CrossRef]
- Hirose, S.; Hara, M.; Kamei, S.; Dalmau, J.; Nakajima, H. Characteristics of clinical relapses and patient-oriented long-term outcomes of patients with anti-N-methyl-d-aspartate receptor encephalitis. J. Neurol. 2022, 269, 2486–2492. [Google Scholar] [CrossRef]
- Hunter, D.; Jamet, Z.; Groc, L. Autoimmunity and NMDA receptor in brain disorders: Where do we stand? Neurobiol. Dis. 2021, 147, 105161. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Stanic, J.; Scheggia, D.; Benussi, A.; Borroni, B.; Di Luca, M. NMDA and AMPA Receptor Autoantibodies in Brain Disorders: From Molecular Mechanisms to Clinical Features. Cells 2021, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Masdeu, J.C.; Dalmau, J.; Berman, K.F. NMDA Receptor Internalization by Autoantibodies: A Reversible Mechanism Underlying Psychosis? Trends Neurosci. 2016, 39, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Kayser, M.S.; Dalmau, J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr. Res. 2016, 176, 36–40. [Google Scholar] [CrossRef]
- Nakazawa, K.; Sapkota, K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar] [CrossRef]
- Steiner, J.; Walter, M.; Glanz, W.; Sarnyai, Z.; Bernstein, H.-G.; Vielhaber, S.; Kästner, A.; Skalej, M.; Jordan, W.; Schiltz, K.; et al. Increased Prevalence of Diverse N-Methyl-D-Aspartate Glutamate Receptor Antibodies in Patients with an Initial Diagnosis of Schizophrenia: Specific Relevance of IgG NR1a Antibodies for Distinction from N-Methyl-D-Aspartate Glutamate Receptor Encephalitis. JAMA Psychiatry 2013, 70, 271–278. [Google Scholar] [CrossRef]
- Hara, M.; Martinez-Hernandez, E.; Ariño, H.; Armangué, T.; Spatola, M.; Petit-Pedrol, M.; Saiz, A.; Rosenfeld, M.R.; Graus, F.; Dalmau, J. Clinical and pathogenic significance of IgG, IgA, and IgM antibodies against the NMDA receptor. Neurology 2018, 90, e1386–e1394. [Google Scholar] [CrossRef]
- Doss, S.; Wandinger, K.-P.; Hyman, B.T.; Panzer, J.A.; Synofzik, M.; Dickerson, B.; Mollenhauer, B.; Scherzer, C.R.; Ivinson, A.J.; Finke, C.; et al. High prevalence of NMDA receptor IgA/IgM antibodies in different dementia types. Ann. Clin. Transl. Neurol. 2014, 1, 822–832. [Google Scholar] [CrossRef]
- Gibson, L.L.; McKeever, A.; Cullen, A.E.; Nicholson, T.R.; Aarsland, D.; Zandi, M.S.; Pollak, T.A. Neuronal surface autoantibodies in dementia: A systematic review and meta-analysis. J. Neurol. 2021, 268, 2769–2779. [Google Scholar] [CrossRef]
- Busse, S.; Busse, M.; Brix, B.; Probst, C.; Genz, A.; Bogerts, B.; Stoecker, W.; Steiner, J. Seroprevalence of n-methyl-d-aspartate glutamate receptor (NMDA-R) autoantibodies in aging subjects without neuropsychiatric disorders and in dementia patients. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 545–550. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef]
- Hammer, C.; Stepniak, B.; Schneider, A.; Papiol, S.; Tantra, M.; Begemann, M.; Sirén, A.-L.; Pardo, L.A.; Sperling, S.; Jofrry, S.M.; et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood–brain barrier integrity. Mol. Psychiatry 2014, 19, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Janssens, N.; Lesage, A.S.J. Glutamate receptor subunit expression in primary neuronal and secondary glial cultures. J. Neurochem. 2001, 77, 1457–1474. [Google Scholar] [CrossRef] [PubMed]
- Zukin, R.; Bennett, M.V. Alternatively spliced isoforms of the NMDARI receptor subunit. Trends Neurosci. 1995, 18, 306–313. [Google Scholar] [CrossRef]
- Bettler, B.; Mulle, C. Review: Neurotransmitter Receptors. II. AMPA and kainate receptors. Neuropharmacology 1995, 34, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Song, I.; Huganir, R.L. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002, 25, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, A.; Marchi, M. Synaptosomes and Metamodulation of Receptors. Methods Mol. Biol. 2022, 2417, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Summa, M.; Di Prisco, S.; Grilli, M.; Marchi, M.; Pittaluga, A. Hippocampal AMPA autoreceptors positively coupled to NMDA autoreceptors traffic in a constitutive manner and undergo adaptative changes following enriched environment training. Neuropharmacology 2011, 61, 1282–1290. [Google Scholar] [CrossRef]
- Henley, J.M.; Barker, E.A.; Glebov, O.O. Routes, destinations and delays: Recent advances in AMPA receptor trafficking. Trends Neurosci. 2011, 34, 258–268. [Google Scholar] [CrossRef]
- Triller, A.; Choquet, D. Synaptic structure and diffusion dynamics of synaptic receptors. Biol. Cell 2003, 95, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.; Olszewski, J.; Lloyd-Smith, D. Focal seizures due to chronic localized encephalitis. Neurology 1958, 8, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Reagan, T.J.; Klass, D.W. Epilepsia Partialis Continua: A Review of 32 Cases. Arch. Neurol. 1977, 34, 266–275. [Google Scholar] [CrossRef]
- Longaretti, F.; Dunkley, C.; Varadkar, S.; Vargha-Khadem, F.; Boyd, S.G.; Cross, J.H. Evolution of the EEG in children with Rasmussen’s syndrome. Epilepsia 2012, 53, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, R.S.; Nimjee, S.M.; Oakes, W.J. Long-term follow-up in children with functional hemispherectomy for Rasmussen’s encephalitis. Child’s Nerv. Syst. 2005, 21, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.E.; Granata, T.; Franzini, A.; Freri, E.; Villani, F.; Casazza, M.; De Curtis, M.; Ragona, F.; Ferroli, P.; D’incerti, L.; et al. Hemispherotomy and functional hemispherectomy: Indications and outcome. Epilepsy Res. 2010, 89, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Vining, E.P.G.; Pillas, D.J.; Pyzik, P.L.; Avellino, A.M.; Carson, B.S.; Freeman, J.M. Hemispherectomy for intractable unihemispheric epilepsy Etiology vs outcome. Neurology 2003, 61, 887–890. [Google Scholar] [CrossRef]
- Rogers, S.W.; Andrews, P.I.; Gahring, L.C.; Whisenand, T.; Cauley, K.; Crain, B.; Hughes, T.E.; Heinemann, S.F.; McNamara, J.O. Autoantibodies to Glutamate Receptor GluR3 in Rasmussen’s Encephalitis. Science 1994, 265, 648–651. [Google Scholar] [CrossRef]
- Andrews, P.I.; Dichter, M.A.; Berkovic, S.F.; Newton, M.R.; McNamara, J.O. Plasmapheresis in Rasmussen’s encephalitis. Neurology 1996, 46, 242–246. [Google Scholar] [CrossRef]
- Benussi, A.; Alberici, A.; Buratti, E.; Ghidoni, R.; Gardoni, F.; Di Luca, M.; Padovani, A.; Borroni, B. Toward a Glutamate Hypothesis of Frontotemporal Dementia. Front. Neurosci. 2019, 13, 304. [Google Scholar] [CrossRef]
- Murley, A.G.; Rowe, J.B. Neurotransmitter deficits from frontotemporal lobar degeneration. Brain 2018, 141, 1263–1285. [Google Scholar] [CrossRef] [PubMed]
- Chessell, I.P.; Procter, A.W.; Francis, P.T.; Bowen, D.M. d-Cycloserine, a putative cognitive enhancer, facilitates activation of the N-methyl-d-aspartate receptor-ionophore complex in Alzheimer brain. Brain Res. 1991, 565, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.M.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana, I.; Alberici, A.; Bonomi, E.; Ottaviani, R.; Kumar, R.; Archetti, S.; Manes, M.; Cosseddu, M.; Buratti, E.; Padovani, A.; et al. Antinuclear antibodies in Frontotemporal Dementia: The tip’s of autoimmunity iceberg? J. Neuroimmunol. 2018, 325, 61–63. [Google Scholar] [CrossRef]
- Sjögren, M.; Wallin, A. Pathophysiological aspects of frontotemporal dementia—Emphasis on cytoskeleton proteins and autoimmunity. Mech. Ageing Dev. 2001, 122, 1923–1935. [Google Scholar] [CrossRef]
- Scheltens, N.M.E.; Tijms, B.M.; Koene, T.; Barkhof, F.; Teunissen, C.E.; Wolfsgruber, S.; Wagner, M.; Kornhuber, J.; Peters, O.; Cohn-Sheehy, B.I.; et al. Cognitive subtypes of probable Alzheimer’s disease robustly identified in four cohorts. Alzheimer’s Dement. 2017, 13, 1226–1236. [Google Scholar] [CrossRef]
- Borroni, B.; Stanic, J.; Verpelli, C.; Mellone, M.; Bonomi, E.; Alberici, A.; Bernasconi, P.; Culotta, L.; Zianni, E.; Archetti, S.; et al. Anti-AMPA GluA3 antibodies in Frontotemporal dementia: A new molecular target. Sci. Rep. 2017, 7, 6723. [Google Scholar] [CrossRef]
- Mone, A.P.; Cheney, C.; Banks, A.L.; Tridandapani, S.; Mehter, N.; Guster, S.; Lin, T.; Eisenbeis, C.F.; Young, D.C.; Byrd, J.C. Alemtuzumab induces caspase-independent cell death in human chronic lymphocytic leukemia cells through a lipid raft-dependent mechanism. Leukemia 2006, 20, 272–279. [Google Scholar] [CrossRef]
- Katsavos, S.; Coles, A. Alemtuzumab as Treatment for Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a032029. [Google Scholar] [CrossRef]
- Buscarinu, M.C.; Fornasiero, A.; Pellicciari, G.; Reniè, R.; Landi, A.C.; Bozzao, A.; Cappelletti, C.; Bernasconi, P.; Ristori, G.; Salvetti, M. Autoimmune Encephalitis and CSF Anti-GluR3 Antibodies in an MS Patient after Alemtuzumab Treatment. Brain Sci. 2019, 9, 299. [Google Scholar] [CrossRef]
- Giarola, B.; Massey, J.; Barnett, Y.; Rodrigues, M.; Sutton, I. Autoimmune encephalitis following alemtuzumab treatment of multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 28, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Negi, N.; Das, B.K. Decoding intrathecal immunoglobulins and B cells in the CNS: Their synthesis, function, and regulation. Int. Rev. Immunol. 2020, 39, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J.; Venkatesan, A. Pathogenic mechanisms in neuronal surface autoantibody-mediated encephalitis. J. Neuroimmunol. 2022, 368, 577867. [Google Scholar] [CrossRef]
- Prüss, H. Autoantibodies in neurological disease. Nat. Rev. Immunol. 2021, 21, 798–813. [Google Scholar] [CrossRef] [PubMed]
- Groc, L.; Choquet, D. Linking glutamate receptor movements and synapse function. Science 2020, 368, eaay4631. [Google Scholar] [CrossRef] [PubMed]
- He, J.-G.; Zhou, H.-Y.; Wang, F.; Chen, J.-G. Dysfunction of Glutamatergic Synaptic Transmission in Depression: Focus on AMPA Receptor Trafficking. Biol. Psychiatry Glob. Open Sci. 2023, 3, 187–196. [Google Scholar] [CrossRef]
- Benke, D. GABAB receptor trafficking and interacting proteins: Targets for the development of highly specific therapeutic strategies to treat neurological disorders? Biochem. Pharmacol. 2013, 86, 1525–1530. [Google Scholar] [CrossRef]
- Schaefer, N.; Roemer, V.; Janzen, D.; Villmann, C. Impaired Glycine Receptor Trafficking in Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 291. [Google Scholar] [CrossRef]
- Olivero, G.; Vergassola, M.; Cisani, F.; Usai, C.; Pittaluga, A. Immuno-Pharmacological Characterization of Presynaptic GluN3A-Containing NMDA Autoreceptors: Relevance to Anti-NMDA Receptor Autoimmune Diseases. Mol. Neurobiol. 2019, 56, 6142–6155. [Google Scholar] [CrossRef]
- Haubrich, J.; Font, J.; Quast, B.R.; Goupil-Lamy, A.; Scholler, P.; Nevoltris, D.; Acher, F.; Chames, P.; Rondard, P.; Prézeau, L.; et al. A Nanobody Activating Metabotropic Glutamate Receptor 4 Discriminates between Homo-and Heterodimers. Proc. Natl. Acad. Sci. USA 2021, 18, e2105848118. [Google Scholar] [CrossRef]
- Lancaster, E.; Lai, M.; Peng, X.; Hughes, E.; Constantinescu, R.; Raizer, J.; Friedman, D.; Skeen, M.B.; Grisold, W.; Kimura, A.; et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: Case series and characterisation of the antigen. Lancet Neurol. 2010, 9, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Twyman, R.E.; Gahring, L.C.; Spiess, J.; Rogers, S.W. Glutamate receptor antibodies activate a subset of receptors and reveal an agonist binding site. Neuron 1995, 14, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, T.; Fukata, Y.; Yamasaki, M.; Miyazaki, T.; Yokoi, N.; Takashima, H.; Watanabe, M.; Watanabe, O.; Fukata, M. Autoantibodies to Epilepsy-Related LGI1 in Limbic Encephalitis Neutralize LGI1-ADAM22 Interaction and Reduce Synaptic AMPA Receptors. J. Neurosci. 2013, 33, 18161–18174. [Google Scholar] [CrossRef] [PubMed]
- Körtvelyessy, P.; Bauer, J.; Stoppel, C.M.; Brück, W.; Gerth, I.; Vielhaber, S.; Wiedemann, F.R.; Heinze, H.J.; Bartels, C.; Bien, C.G. Complement-associated neuronal loss in a patient with CASPR2 antibody–associated encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e75. [Google Scholar] [CrossRef]
- Merega, E.; Di Prisco, S.; Lanfranco, M.; Severi, P.; Pittaluga, A. Complement selectively elicits glutamate release from nerve endings in different regions of mammal central nervous system. J. Neurochem. 2014, 129, 473–483. [Google Scholar] [CrossRef]
- Merega, E.; Di Prisco, S.; Severi, P.; Kalfas, F.; Pittaluga, A. Antibody/receptor protein immunocomplex in human and mouse cortical nerve endings amplifies complement-induced glutamate release. Neurosci. Lett. 2015, 600, 50–55. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Wright, S.K.; Vincent, A. In vivo Mechanisms of Antibody-Mediated Neurological Disorders: Animal Models and Potential Implications. Front. Neurol. 2019, 10, 1394. [Google Scholar] [CrossRef]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System. Physiol. Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rey, J.; Rossi, E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; et al. Articles Anti-NMDA-Receptor Encephalitis: Case Series and Analysis of the Effects of Antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Jézéquel, J.; Johansson, E.M.; Dupuis, J.P.; Rogemond, V.; Gréa, H.; Kellermayer, B.; Hamdani, N.; Le Guen, E.; Rabu, C.; Lepleux, M.; et al. Dynamic disorganization of synaptic NMDA receptors triggered by autoantibodies from psychotic patients. Nat. Commun. 2017, 8, 1791. [Google Scholar] [CrossRef]
- Mikasova, L.; De Rossi, P.; Bouchet, D.; Georges, F.; Rogemond, V.; Didelot, A.; Meissirel, C.; Honnorat, J.; Groc, L. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012, 135, 1606–1621. [Google Scholar] [CrossRef] [PubMed]
- Ladépêche, L.; Planagumà, J.; Thakur, S.; Suárez, I.; Hara, M.; Borbely, J.S.; Sandoval, A.; Laparra-Cuervo, L.; Dalmau, J.; Lakadamyali, M. NMDA Receptor Autoantibodies in Autoimmune Encephalitis Cause a Subunit-Specific Nanoscale Redistribution of NMDA Receptors. Cell Rep. 2018, 23, 3759–3768. [Google Scholar] [CrossRef] [PubMed]
- Planagumà, J.; Haselmann, H.; Mannara, F.; Petit-Pedrol, M.; Grünewald, B.; Aguilar, E.; Röpke, L.; Martín-García, E.; Titulaer, M.J.; Jercog, P.; et al. Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann. Neurol. 2016, 80, 388–400. [Google Scholar] [CrossRef]
- Malviya, M.; Barman, S.; Golombeck, K.S.; Planagumà, J.; Mannara, F.; Strutz-Seebohm, N.; Wrzos, C.; Demir, F.; Baksmeier, C.; Steckel, J.; et al. NMDAR encephalitis: Passive transfer from man to mouse by a recombinant antibody. Ann. Clin. Transl. Neurol. 2017, 4, 768–783. [Google Scholar] [CrossRef] [PubMed]
- Planagumà, J.; Leypoldt, F.; Mannara, F.; Gutiérrez-Cuesta, J.; Martín-García, E.; Aguilar, E.; Titulaer, M.J.; Petit-Pedrol, M.; Jain, A.; Balice-Gordon, R.; et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015, 138, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Mannara, F.; Radosevic, M.; Planagumà, J.; Soto, D.; Aguilar, E.; García-Serra, A.; Maudes, E.; Pedreño, M.; Paul, S.; Doherty, J.; et al. Allosteric modulation of NMDA receptors prevents the antibody effects of patients with anti-NMDAR encephalitis. Brain 2020, 143, 2709–2720. [Google Scholar] [CrossRef]
- Gu, J.; Jin, T.; Li, Z.; Chen, H.; Xia, H.; Xu, X.; Gui, Y. Exosomes expressing neuronal autoantigens induced immune response in antibody-positive autoimmune encephalitis. Mol. Immunol. 2021, 131, 164–170. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Spruce, L.A.; Dalmau, J.; Seeholzer, S.H.; Lynch, D.R. Anti-NMDA Receptor Encephalitis Antibody Binding Is Dependent on Amino Acid Identity of a Small Region within the GluN1 Amino Terminal Domain. J. Neurosci. 2012, 32, 11082–11094. [Google Scholar] [CrossRef]
- Olivero, G.; Cisani, F.; Marimpietri, D.; Di Paolo, D.; Gagliani, M.C.; Podestà, M.; Cortese, K.; Pittaluga, A. The Depolarization-Evoked, Ca2+-Dependent Release of Exosomes From Mouse Cortical Nerve Endings: New Insights Into Synaptic Transmission. Front. Pharmacol. 2021, 12, 670158. [Google Scholar] [CrossRef]
- Luccini, E.; Musante, V.; Neri, E.; Raiteri, M.; Pittaluga, A. N-methyl-D-aspartate autoreceptors respond to low and high agonist concentrations by facilitating, respectively, exocytosis and carrier-mediated release of glutamate in rat hippocampus. J. Neurosci. Res. 2007, 85, 3657–3665. [Google Scholar] [CrossRef]
- Musante, V.; Summa, M.; Cunha, R.A.; Raiteri, M.; Pittaluga, A. Pre-synaptic glycine GlyT1 transporter—NMDA receptor interaction: Relevance to NMDA autoreceptor activation in the presence of Mg2+ ions. J. Neurochem. 2011, 117, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Javitt, D. From Revolution to Evolution: The Glutamate Hypothesis of Schizophrenia and its Implication for Treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.O.; Bustillo, J.R. Glutamatergic dysfunction in Schizophrenia. Transl. Psychiatry 2022, 12, 500. [Google Scholar] [CrossRef] [PubMed]
- Olivero, G.; Grilli, M.; Marchi, M.; Pittaluga, A. Metamodulation of presynaptic NMDA receptors: New perspectives for pharmacological interventions. Neuropharmacology 2023, 234, 109570. [Google Scholar] [CrossRef]
- Missale, C.; Fiorentini, C.; Busi, C.; Collo, G.; Spano, P.F. The NMDA/D1 Receptor Complex as a New Target in Drug Development. Curr. Top. Med. Chem. 2006, 6, 801–808. [Google Scholar] [CrossRef]
- Dean, C.A.; Metzbower, S.R.; Dessain, S.K.; Blanpied, T.A.; Benavides, D.R. Regulation of NMDA Receptor Signaling at Single Synapses by Human Anti-NMDA Receptor Antibodies. Front. Mol. Neurosci. 2022, 15, 940005. [Google Scholar] [CrossRef]
- Malina, K.C.-K.; Ganor, Y.; Levite, M.; Teichberg, V.I. Autoantibodies Against an Extracellular Peptide of the GluR3 Subtype of AMPA Receptors Activate Both Homomeric and Heteromeric AMPA Receptor Channels. Neurochem. Res. 2006, 31, 1181–1190. [Google Scholar] [CrossRef]
- Carlson, N.G.; Gahring, L.C.; Rogers, S.W. Identification of the amino acids on a neuronal glutamate receptor recognized by an autoantibody from a patient with paraneoplastic syndrome. J. Neurosci. Res. 2001, 63, 480–485. [Google Scholar] [CrossRef]
- Whitney, K.D.; McNamara, J.O. GluR3 Autoantibodies Destroy Neural Cells in a Complement-Dependent Manner Modulated by Complement Regulatory Proteins. J. Neurosci. 2000, 20, 7307–7316. [Google Scholar] [CrossRef]
- Lai, M.; Hughes, E.G.; Peng, X.; Zhou, L.; Gleichman, A.J.; Shu, H.; Matà, S.; Kremens, D.; Vitaliani, R.; Geschwind, M.D.; et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann. Neurol. 2009, 65, 424–434. [Google Scholar] [CrossRef]
- Peng, X.; Hughes, E.G.; Moscato, E.H.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Cellular plasticity induced by anti–α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann. Neurol. 2015, 77, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Palese, F.; Bonomi, E.; Nuzzo, T.; Benussi, A.; Mellone, M.; Zianni, E.; Cisani, F.; Casamassa, A.; Alberici, A.; Scheggia, D.; et al. Anti-GluA3 antibodies in frontotemporal dementia: Effects on glutamatergic neurotransmission and synaptic failure. Neurobiol. Aging 2020, 86, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Cisani, F.; Olivero, G.; Usai, C.; Van Camp, G.; Maccari, S.; Morley-Fletcher, S.; Pittaluga, A.M. Antibodies Against the NH2-Terminus of the GluA Subunits Affect the AMPA-Evoked Releasing Activity: The Role of Complement. Front. Immunol. 2021, 12, 586521. [Google Scholar] [CrossRef]
- Levite, M.; Fleidervish, I.A.; Schwarz, A.; Pelled, D.; Futerman, A.H. Autoantibodies to the Glutamate Receptor Kill Neurons via Activation of the Receptor Ion Channel. J. Autoimmun. 1999, 13, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Ganor, Y.; Goldberg-Stern, H.; Cohen, R.; Teichberg, V.; Levite, M. Glutamate receptor antibodies directed against AMPA receptors subunit 3 peptide B (GluR3B) can be produced in DBA/2J mice, lower seizure threshold and induce abnormal behavior. Psychoneuroendocrinology 2014, 42, 106–117. [Google Scholar] [CrossRef]
- Carlson, N.G.; Gahring, L.C.; Twyman, R.E.; Rogers, S.W. Identification of Amino Acids in the Glutamate Receptor, GluR3, Important for Antibody-Binding and Receptor-Specific Activation*. J. Biol. Chem. 1997, 272, 11295–11301. [Google Scholar] [CrossRef]
- Day, C.; Silva, J.-P.; Munro, R.; Baker, T.S.; Wolff, C.; Bithell, A.; Stephens, G.J. Anti-AMPA Receptor Autoantibodies Reduce Excitatory Currents in Rat Hippocampal Neurons. Pharmaceuticals 2023, 16, 77. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 2013, 15, 11–27. [Google Scholar] [CrossRef]
- Jain, A.; Balice-Gordon, R. Cellular, synaptic, and circuit effects of antibodies in autoimmune CNS synaptopathies. Handb. Clin. Neurol. 2016, 133, 77–93. [Google Scholar] [CrossRef]
- Gahring, L.C.; Rogers, S.W.; Twyman, R.E. Autoantibodies to glutamate receptor subunit GluR2 in nonfamilial olivopontocerebellar degeneration. Neurology 1997, 48, 494–500. [Google Scholar] [CrossRef]
- He, X.-P.; Patel, M.; Whitney, K.D.; Janumpalli, S.; Tenner, A.; McNamara, J.O. Glutamate Receptor GluR3 Antibodies and Death of Cortical Cells. Neuron 1998, 20, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P.; Dean, Y.D.; McGreal, E.P.; VanBeek, J.; Morgan, B.P. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology 2000, 49, 171–186. [Google Scholar] [CrossRef] [PubMed]
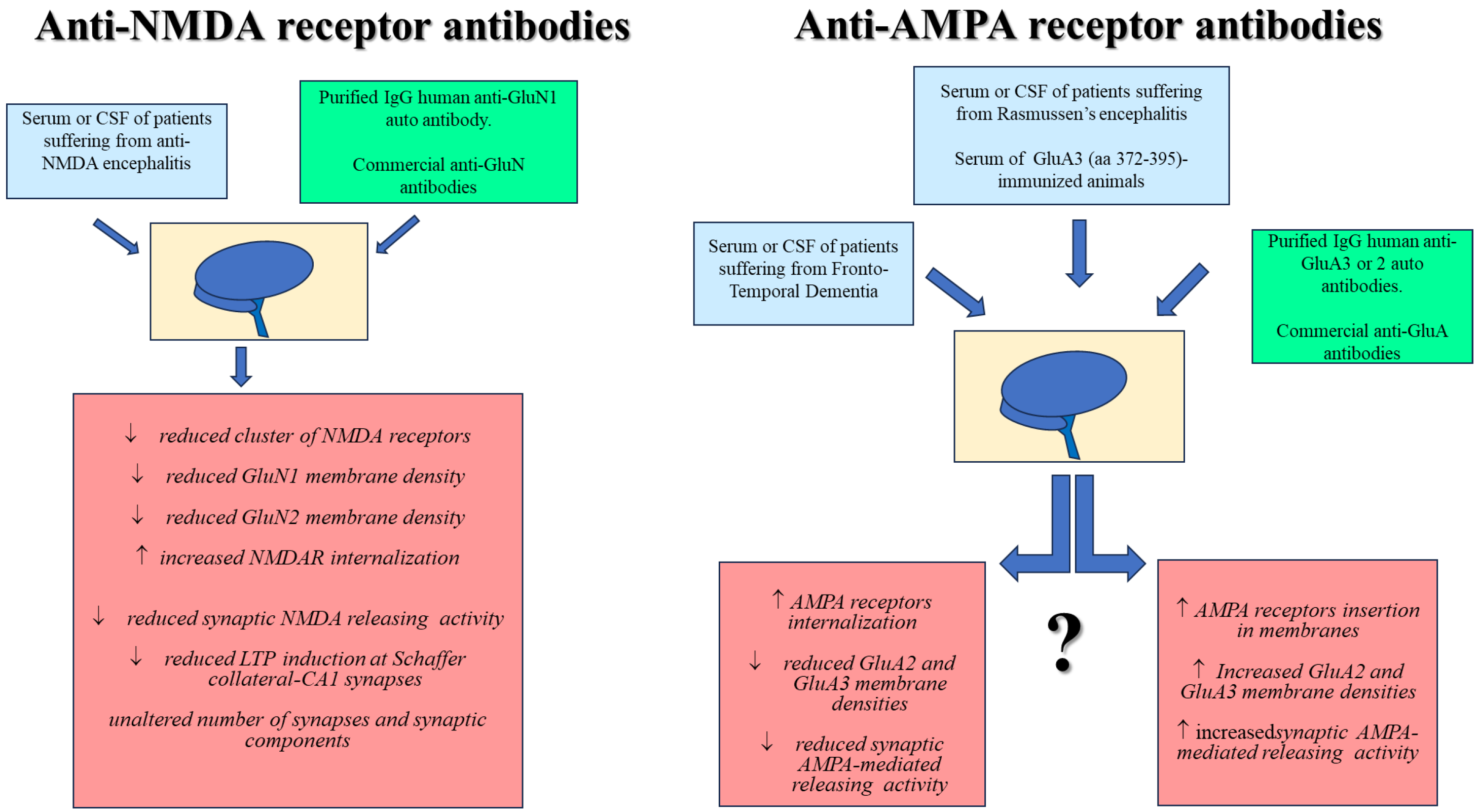

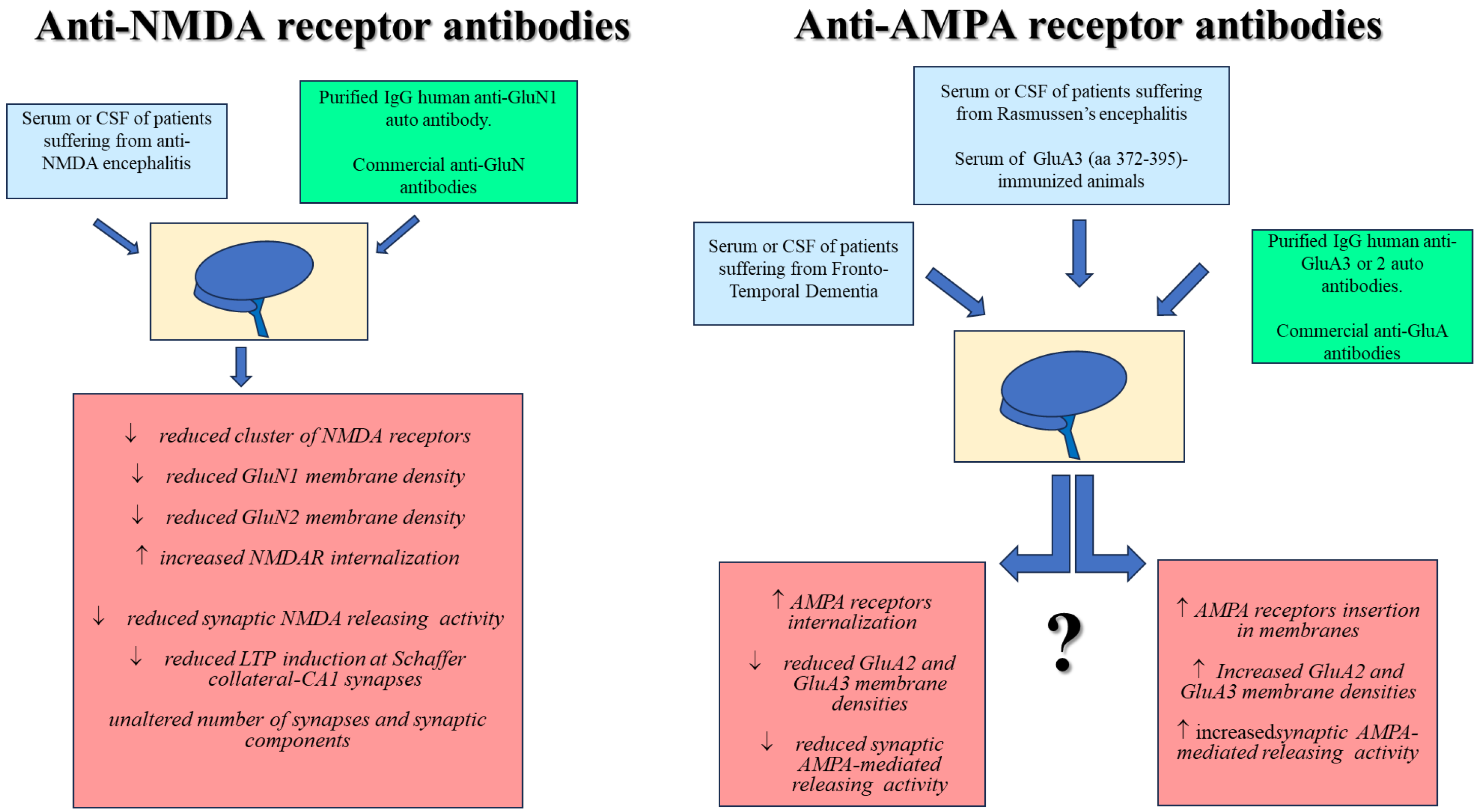{kind=link}
| Source | Targeted GluN Subunit | Experimental Paradigm | Effects | Reference |
|---|---|---|---|---|
| Serum and CSF of patients suffering from encephalitis Purified IgG human anti-GluN1 auto-antibody | GluN1 | HEK293 cells transfected with rodent (or human) GluN1 or GluN2 (A, B, C, or D) subunits. Cultured rat hippocampal neurons | Immunolabeling of NMDA receptor clusters in postsynaptic dendrites ↓ Reduced cell-surface fraction of NMDA receptors | [80] |
| CSF from patients suffering from anti-NMDA receptor encephalitis Purified IgG human anti-GluN1 autoantibody | GluN1 | Cultured rat hippocampal neurons | ↓ Reduced cluster of NMDA receptors ↓ Reduced GluN1 density ↓ Reduced GluN2 density ↓ Reduced synaptic NMDA currents Unaltered number of synapses and synaptic components | [15] |
| CSF from patients suffering from anti-NMDA receptor encephalitis CSF from non-encephalitis patients (control CSF) Commercial anti-GluN1 and anti-GluN2B antibodies | GluN1 | Transfected HEK293 cells expressing GluN1/GlUn2A or GluN1/GluN2B heteromers Cultured rat hippocampal neurons C57JBl mouse hippocampal slices | Anti-NMDA autoantibody-induced staining of NMDA receptors confirmed with commercial anti-GluN1 and anti-GluN2B Staining of native NMDARs in rat hippocampal neurons ↓ Reduced LTP induction at Schaffer collateral-CA1 synapses in mouse hippocampal slices. | [16] |
| CSF from patients suffering from anti-NMDAreceptor encephalitis Commercial anti-GluN1 and anti-GluN2B antibodies raised against the COOH terminus | GluN1 | Transfected HEK293 cells expressing GluN1/GlUn2A or GluN1/GluN2B heteromers Whole rat brain lysate and rat cortical, hippocampal, and cerebellar lysates | Patients’ CSF immunoprecipitates GluN1 proteins from whole rat brain lysate and from cortical, hippocampal, and cerebellar lysates Patients’ CSF labels NMDA receptor in transfected cells In transfected HEK cells, the short exposure (2 min) to glutamate/glycine plus patient’s CSF prolongs the open duration of the NMDA receptor-associated channel. Differently, the prolonged exposure (>24 h) decreases NMDA receptors | [89] |
| CSF from patients suffering from anti-NMDA receptor encephalitis Purified IgG human anti-GluN1 antibody | GluN1 GluN2A GluN2B | Cultured rat hippocampal neurons | ↓ GluN2-containing NMDA receptors density ↓ LTP induction at Schaffer collateral-CA1 synapses in mouse hippocampal slices ↑ Patient’s CSF increases the mobile fraction of GluN2A-NMDA receptors but decreases the GluN2B-NMDA receptors. Patient’s IgG disrupts GluN2A-NMDA receptor/EPHB2R interaction | [82] |
| CSF from patients suffering from anti-NMDA receptor encephalitis Commercial anti-GluN1 antibody | GluN1 | Cultured rat hippocampal neurons | Patient and commercial anti-GluN1 antibody have comparable distribution and immunostainings ↓ Surface NMDA receptors in both excitatory and inhibitory hippocampal neurons ↑ NMDA receptor internalization ↓ NMDA receptor-mediated current amplitude | [17] |
| Recombinant monoclonal GluN1 antibody from cerebrospinal fluid memory B-cells | GluN1 | Transfected HEK293 cells expressing GluN1/GluN2A or GluN1/GluN2B heteromers Cultured mouse hippocampal neurons Mouse brain slices | Staining of native GluN1-containing NMDA receptors in transfected HEK293 cells, in primary mouse hippocampal neurons and in mouse brain ↓ Synaptic NMDA receptor currents elicited by recombinant monoclonal GluN1 antibody | [19] |
| Commercial anti-GluN1, GluN2A/B, GluN3A antibodies | GluN1, GluN2B | Mouse hippocampal synaptosomes | ↓ NMDA receptors cluster ↓ GluN1 density ↓ GluN2 density ↓ NMDA-mediated glutamate exocytosis | [70] |
| GluN1 antibodies derived from patients with anti-NMDAR encephalitis | GluN1 | Staining of mature hippocampal primary neuron in culture Miniature spontaneous calcium transients (mSCaTs) mediated via NMDARs at synaptic spines are not altered in pathogenic GluN1 antibody-exposed conditions. Calcium does not accumulate in neuronal spines following brief exposure to pathogenic GluN1 antibodies | [97] |
| Source | Targeted GluA Subunit | Experimental Paradigm | Effects | Reference |
|---|---|---|---|---|
| Serum of rabbits immunized with a specific amino acid sequence of the GluR3 protein (the Glu3B peptide, aa 372–395) Purified IgG anti-GluA3 autoantibodies Serum from patients with active Rasmussen’s | GluA3 | Cultured fetal mouse cortical neurons | ↑ Currents in neurons, prevented by CNQX The GluR3B peptide specifically blocks the antisera- and IgG-evoked currents The anti-GluR3B antibody mimics the serum from immunized rabbit and the purified IgG. | [73] |
| Murine antibody recognizing a specific sequence of the GluR3 protein (the Glu3B peptide, aa 372–395) | GluA3 | Mouse coronal slices of somatosensory cortex Primary hippocampal cultures | Immunostaining of neurons ↑ CNQX-sensitive currents in neurons GluR3B peptide-induced neuronal death reduced by CNQX | [105] |
| Anti-GluA3 IgG isolated from GluA3 (aa 246–455)-immunized rabbits | GluA3 | Rat mixed cortical neuronal and glial cultures Whole cell recording in cultured rat cortical neurons | ↑ Release of LDH from mixed cultures in a GIKY52466 or CNQX-independent manner The plasma from immunized animals does not evoke currents in cultured neurons | [112] |
| Purified rabbit antibody recognizing a specific amino acid sequence of the GluR3 protein (the Glu3B peptide, aa 372–395) Monoclonal mouse anti-GluA3B | GluA3 | Rat brain homogenates Xenopus laevi oocytes expressing GluA3 homomeric or GluA2/A3 heteromeric AMPARs | Purified rabbit anti-GluA3 antibody elicits whole cell currents that were sensitive to CNQX | [98] |
| Murine anti-autoantibody recognizing the amino acid sequence of the GluR3 protein (the Glu3B peptide, aa 372–395) | GluA3 | DBA/2J mice immunized with the GluR3B peptide that express circulating anti-GluA3B autoantibodies | Immunized DBA/2J mice were more susceptible to pentetrazol-induced seizures | [106] |
| Serum and CSF containing anti-GluA3 autoantibodies from patients suffering from FTD | GluA3 | Rat hippocampal neuronal primary cultures differentiated neurons from human-induced pluripotent stem cells (hIPSC) | ↓ GluA3 subunit synaptic localization of AMPA receptor in hippocampal neuronal primary cultures ↓ Dendritic spines AMPA receptor in cultured hippocampal neurons ↓ GluA3 subunit fraction in the postsynaptic fraction of cultured hIPSC | [58] |
| Serum of patients suffering from FTD-Tau neuropathology with a high titer of anti-GluA3 autoantibody | GluA3 | Mouse cortical synaptosomes | The serum does not evoke glutamate exocytosis ↓ AMPA-evoked glutamate release | [103] |
| Commercial anti-GluA3 antibody | GluA3 | Mouse cortical synaptosomes | ↑ GluA2 and GluA3 subunits insertion in synaptosomal plasma membranes The antibody does not evoke glutamate exocytosis ↑ AMPA-evoked glutamate release | [104] |
| IgG Anti-GluA3 antibody from rabbit immunized with the GluR3B peptide | GluA3 | Mouse whole brain lysates Primary hippocampal neurons | Anti-GluA3 immunostaining in the whole brain lysate ↓ excitatory postsynaptic currents (EPSCs) in primary hippocampal neurons | [108] |
| Serum of a patient suffering from progressive sporadic olivopontocerebellar atrophy Purified IgM anti-GluA2 autoantibodies | GluA2 | Cultured fetal mouse cortical neurons | ↑ Currents in neurons in a CNQX-sensitive manner Currents were blocked by a synthetic peptide corresponding to the specific epitope region of GluR2 (aa 369–393) | [111] |
| Purified IgM anti-GluA1 and GluA2 autoantibodies from the CSF of patients suffering from anti-AMPA receptor encephalitis Commercial anti-GluA1 and GluA2 antibody | GluA2 | Primary rat hippocampal neuron and astrocyte cocultures | ↓ Surface AMPA receptor protein and synaptic localization Unmodified glutamatergic synapse density and cell viability ↑ Internalization of AMPA receptor clusters ↓ Miniature excitatory postsynaptic currents (mEPSCs) ↓ Miniature inhibitory postsynaptic currents (mIPSCs) ↑Intrinsic neuronal excitability after 48 h of treatment with patient CSF Commercial anti-GluA2 antibody does not cause receptor internalization | [102] |
| Anti-GluA2 autoantibodies purified from patients suffering from limbic encephalitis | GluA2 | Human embryonic kidney 293 (HEK293) cells | ↓ GluA2-containing AMPA receptor clusters number at synapses with a smaller decrease in overall AMPA receptor cluster density | [101] |
| Commercial anti-GluA2 antibody | GluA2 | Mouse cortical synaptosomes | ↑ Insertion of GluA2 and GluA3 subunits in synaptosomal plasma membranes The anti-GluA2 antibody does not evoke glutamate exocytosis ↑ AMPA-evoked glutamate release | [104] |
| Purified IgM anti-GluA1 autoantibodies from the CSF of anti-AMPA receptor encephalitis patients Commercial anti-GluA1 antibody | GluA1 | Primary rat hippocampal neuron and astrocyte cocultures | ↓ Localization of synaptic surface AMPA receptor protein The anti-GluA1 antibody does not modify the glutamatergic synapse density and the cell viability ↑ Internalization of AMPA receptor clusters ↓ Miniature excitatory postsynaptic currents (mEPSCs) ↓ Miniature inhibitory postsynaptic currents (mIPSCs) ↑ Intrinsic neuronal excitability after 48 h of treatment with patient CSF Commercial anti-GluA1 antibody does not cause receptor internalization | [102] |
| Commercial anti-GluA1 antibody | GluA1 | Mouse cortical synaptosomes | The anti-GluA1 antibody does not evoke glutamate exocytosis The anti-GluA1 antibody does not modify the AMPA-evoked glutamate release | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivero, G.; Roggeri, A.; Pittaluga, A. Anti-NMDA and Anti-AMPA Receptor Antibodies in Central Disorders: Preclinical Approaches to Assess Their Pathological Role and Translatability to Clinic. Int. J. Mol. Sci. 2023, 24, 14905. https://doi.org/10.3390/ijms241914905
Olivero G, Roggeri A, Pittaluga A. Anti-NMDA and Anti-AMPA Receptor Antibodies in Central Disorders: Preclinical Approaches to Assess Their Pathological Role and Translatability to Clinic. International Journal of Molecular Sciences. 2023; 24(19):14905. https://doi.org/10.3390/ijms241914905
Chicago/Turabian StyleOlivero, Guendalina, Alessandra Roggeri, and Anna Pittaluga. 2023. "Anti-NMDA and Anti-AMPA Receptor Antibodies in Central Disorders: Preclinical Approaches to Assess Their Pathological Role and Translatability to Clinic" International Journal of Molecular Sciences 24, no. 19: 14905. https://doi.org/10.3390/ijms241914905
APA StyleOlivero, G., Roggeri, A., & Pittaluga, A. (2023). Anti-NMDA and Anti-AMPA Receptor Antibodies in Central Disorders: Preclinical Approaches to Assess Their Pathological Role and Translatability to Clinic. International Journal of Molecular Sciences, 24(19), 14905. https://doi.org/10.3390/ijms241914905