
The cGAS/STING/IFN-1 Response in Squamous Head and Neck Cancer Cells after Genotoxic Challenges and Abrogation of the ATR-Chk1 and Fanconi Anemia Axis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Clonogenic Survival
2.2. Cell Cycle and G2/M Checkpoint Abrogation
2.3. Induction of Micronuclei
2.4. cGAS/STING/IFN-1 Response
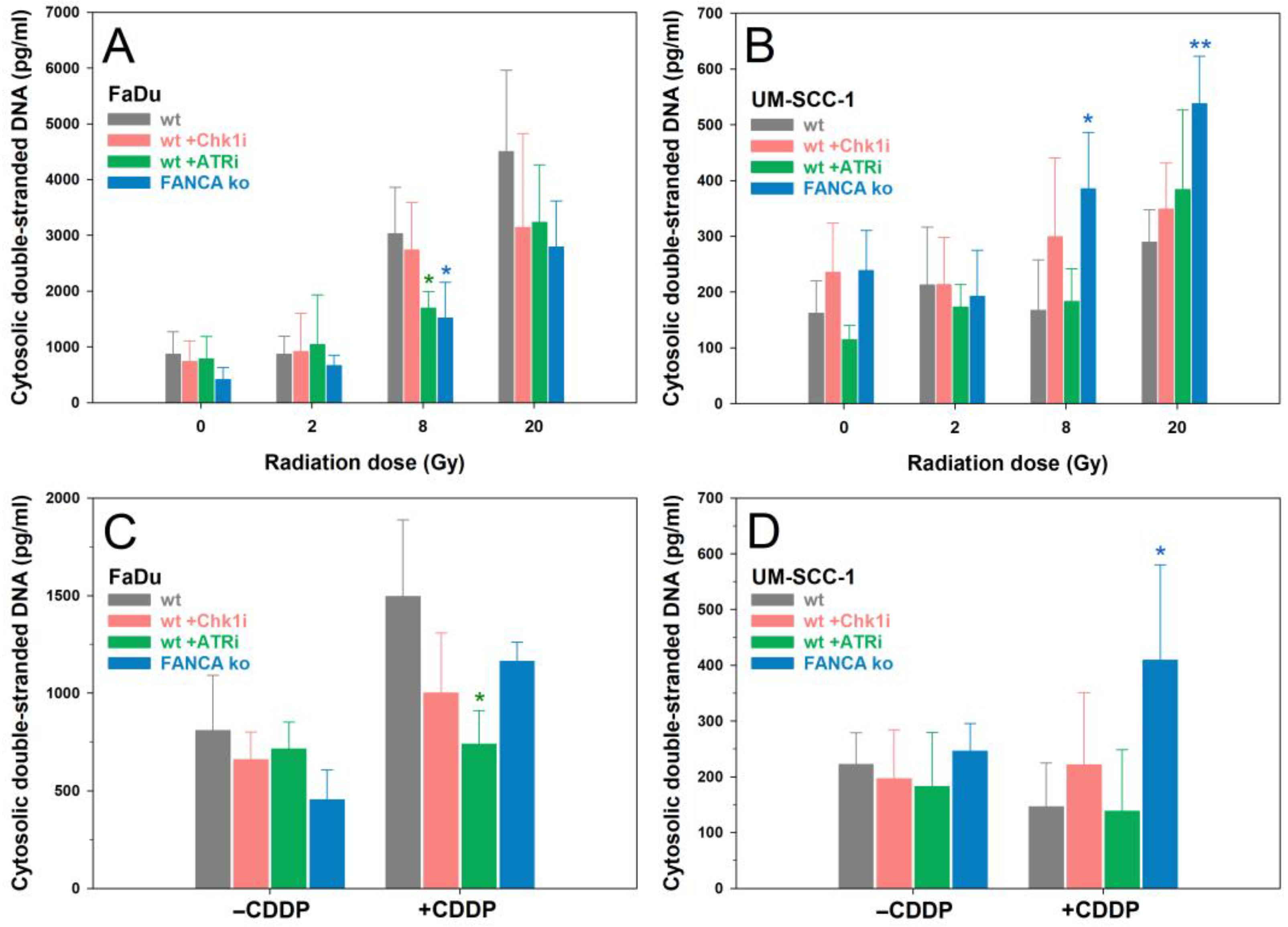
2.5. cdsDNA
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatments and Irradiation
4.3. Clonogenic Survival Assay
4.4. Cell Cycle Analysis
4.5. Cytokinesis Block Micronucleus Assay
4.6. Immunostaining
4.7. Western Blot
4.8. Quantification of Cytosolic DNA
4.9. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Belcher, R.; Hayes, K.; Fedewa, S.; Chen, A.Y. Current treatment of head and neck squamous cell cancer. J. Surg. Oncol. 2014, 110, 551–574. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Brakenhoff, R.H.; Leemans, C.R. Treatment choice for locally advanced head and neck cancers on the basis of risk factors: Biological risk factors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23 (Suppl. 10), x173–x177. [Google Scholar] [CrossRef] [PubMed]
- Porceddu, S.V.; Scotté, F.; Aapro, M.; Salmio, S.; Castro, A.; Launay-Vacher, V.; Licitra, L. Treating Patients With Locally Advanced Squamous Cell Carcinoma of the Head and Neck Unsuitable to Receive Cisplatin-Based Therapy. Front. Oncol. 2019, 9, 1522. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.G.; Cohen, E.E. Current Treatment Options for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3305–3313. [Google Scholar] [CrossRef] [PubMed]
- Adelstein, D.; Gillison, M.L.; Pfister, D.G.; Spencer, S.; Adkins, D.; Brizel, D.M.; Burtness, B.; Busse, P.M.; Caudell, J.J.; Cmelak, A.J.; et al. NCCN Guidelines Insights: Head and Neck Cancers, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 761–770. [Google Scholar] [CrossRef]
- Lee, N.Y.; Ferris, R.L.; Psyrri, A.; Haddad, R.I.; Tahara, M.; Bourhis, J.; Harrington, K.; Chang, P.M.; Lin, J.C.; Razaq, M.A.; et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet. Oncol. 2021, 22, 450–462. [Google Scholar] [CrossRef]
- Machiels, J.P.; Tao, Y.; Burtness, B.; Tahara, M.; Licitra, L.; Rischin, D.; Waldron, J.; Simon, C.; Gregoire, V.; Harrington, K.; et al. Pembrolizumab given concomitantly with chemoradiation and as maintenance therapy for locally advanced head and neck squamous cell carcinoma: KEYNOTE-412. Future Oncol. 2020, 16, 1235–1243. [Google Scholar] [CrossRef]
- Tao, Y.; Biau, J.; Sun, X.S.; Sire, C.; Martin, L.; Alfonsi, M.; Prevost, J.B.; Modesto, A.; Lafond, C.; Tourani, J.M.; et al. Pembrolizumab versus cetuximab concurrent with radiotherapy in patients with locally advanced squamous cell carcinoma of head and neck unfit for cisplatin (GORTEC 2015-01 PembroRad): A multicenter, randomized, phase II trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2023, 34, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Bauml, J.; Seiwert, T.Y.; Pfister, D.G.; Worden, F.; Liu, S.V.; Gilbert, J.; Saba, N.F.; Weiss, J.; Wirth, L.; Sukari, A.; et al. Pembrolizumab for Platinum- and Cetuximab-Refractory Head and Neck Cancer: Results from a Single-Arm, Phase II Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet. Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Aryankalayil, M.J.; Coleman, C.N.; Formenti, S.C. Emerging evidence for adapting radiotherapy to immunotherapy. Nat. Rev. Clin. Oncol. 2023, 20, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Sharon, S.; Daher-Ghanem, N.; Zaid, D.; Gough, M.J.; Kravchenko-Balasha, N. The immunogenic radiation and new players in immunotherapy and targeted therapy for head and neck cancer. Front. Oral Health 2023, 4, 1180869. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front Oncol 2019, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Vaes, R.D.W.; Hendriks, L.E.L.; Vooijs, M.; De Ruysscher, D. Biomarkers of Radiotherapy-Induced Immunogenic Cell Death. Cells 2021, 10, 930. [Google Scholar] [CrossRef]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Luo, X.; Donnelly, C.R.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Bortnik, V.; McMillan, N.A.; Idris, A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb. Pathog. 2019, 132, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Bortnik, V.; Wu, M.; Julcher, B.; Salinas, A.; Nikolic, I.; Simpson, K.J.; McMillan, N.A.; Idris, A. Loss of HPV type 16 E7 restores cGAS-STING responses in human papilloma virus-positive oropharyngeal squamous cell carcinomas cells. J. Microbiol. Immunol. Infect. Wei Mian Yu Gan Ran Za Zhi 2021, 54, 733–739. [Google Scholar] [CrossRef]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Landelouci, K.; Sinha, S.; Pépin, G. Type-I Interferon Signaling in Fanconi Anemia. Front. Cell. Infect. Microbiol. 2022, 12, 820273. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, C.V.M.; Vossen, D.M.; Borgmann, K.; Hageman, F.; Grénman, R.; Verwijs-Janssen, M.; Mout, L.; Kluin, R.J.C.; Nieuwland, M.; Severson, T.M.; et al. Fanconi anemia and homologous recombination gene variants are associated with functional DNA repair defects in vitro and poor outcome in patients with advanced head and neck squamous cell carcinoma. Oncotarget 2018, 9, 18198–18213. [Google Scholar] [CrossRef] [PubMed]
- Wreesmann, V.B.; Estilo, C.; Eisele, D.W.; Singh, B.; Wang, S.J. Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J. Oto-Rhino-Laryngol. Relat. Spec. 2007, 69, 218–225. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Chinn, S.B.; Donovan, F.X.; Chowdhury, N.I.; Kamat, A.; Adeyemo, A.A.; Thomas, J.W.; Vemulapalli, M.; Hussey, C.S.; Reid, H.H.; et al. Assessing the spectrum of germline variation in Fanconi anemia genes among patients with head and neck carcinoma before age 50. Cancer 2017, 123, 3943–3954. [Google Scholar] [CrossRef]
- Szaumkessel, M.; Richter, J.; Giefing, M.; Jarmuz, M.; Kiwerska, K.; Tönnies, H.; Grenman, R.; Heidemann, S.; Szyfter, K.; Siebert, R. Pyrosequencing-based DNA methylation profiling of Fanconi anemia/BRCA pathway genes in laryngeal squamous cell carcinoma. Int. J. Oncol. 2011, 39, 505–514. [Google Scholar] [CrossRef]
- van der Kamp, M.F.; Halmos, G.B.; Guryev, V.; Horvatovich, P.L.; Schuuring, E.; van der Laan, B.; van der Vegt, B.; Plaat, B.E.C.; Verhoeven, C.J. Age-specific oncogenic pathways in head and neck squamous cell carcinoma—Are elderly a different subcategory? Cell. Oncol. 2022, 45, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Webster, A.L.H.; Sanders, M.A.; Patel, K.; Dietrich, R.; Noonan, R.J.; Lach, F.P.; White, R.R.; Goldfarb, A.; Hadi, K.; Edwards, M.M.; et al. Genomic signature of Fanconi anaemia DNA repair pathway deficiency in cancer. Nature 2022, 612, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Brégnard, C.; Guerra, J.; Déjardin, S.; Passalacqua, F.; Benkirane, M.; Laguette, N. Upregulated LINE-1 Activity in the Fanconi Anemia Cancer Susceptibility Syndrome Leads to Spontaneous Pro-inflammatory Cytokine Production. EBioMedicine 2016, 8, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Alter, B.P.; Ebell, W. Cancer risks in Fanconi anemia: Findings from the German Fanconi Anemia Registry. Haematologica 2008, 93, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Auerbach, A.D.; Satagopan, J.; Giampietro, P.F.; Batish, S.D.; Huvos, A.G.; Goberdhan, A.; Shah, J.P.; Singh, B. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Busch, C.J.; Kriegs, M.; Laban, S.; Tribius, S.; Knecht, R.; Petersen, C.; Dikomey, E.; Rieckmann, T. HPV-positive HNSCC cell lines but not primary human fibroblasts are radiosensitized by the inhibition of Chk1. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 108, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Nikolaev, A.; Xing, C.; Della Manna, D.L.; Yang, E.S. CHK1/2 Inhibitor Prexasertib Suppresses NOTCH Signaling and Enhances Cytotoxicity of Cisplatin and Radiation in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2020, 19, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Zahnreich, S.; Weber, B.; Rösch, G.; Schindler, D.; Schmidberger, H. Compromised repair of radiation-induced DNA double-strand breaks in Fanconi anemia fibroblasts in G2. DNA Repair 2020, 96, 102992. [Google Scholar] [CrossRef]
- Dok, R.; Glorieux, M.; Bamps, M.; Nuyts, S. Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers. Int. J. Mol. Sci. 2021, 22, 1504. [Google Scholar] [CrossRef]
- Faulhaber, E.M.; Jost, T.; Symank, J.; Scheper, J.; Bürkel, F.; Fietkau, R.; Hecht, M.; Distel, L.V. Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells. Genes 2021, 12, 925. [Google Scholar] [CrossRef]
- van Harten, A.M.; Buijze, M.; van der Mast, R.; Rooimans, M.A.; Martens-de Kemp, S.R.; Bachas, C.; Brink, A.; Stigter-van Walsum, M.; Wolthuis, R.M.F.; Brakenhoff, R.H. Targeting the cell cycle in head and neck cancer by Chk1 inhibition: A novel concept of bimodal cell death. Oncogenesis 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Helm, A.; Fournier, C.; Durante, M. Particle radiotherapy and molecular therapies: Mechanisms and strategies towards clinical applications. Expert Rev. Mol. Med. 2022, 24, e8. [Google Scholar] [CrossRef] [PubMed]
- Okude, H.; Ori, D.; Kawai, T. Signaling Through Nucleic Acid Sensors and Their Roles in Inflammatory Diseases. Front. Immunol. 2020, 11, 625833. [Google Scholar] [CrossRef]
- Feng, X.; Tubbs, A.; Zhang, C.; Tang, M.; Sridharan, S.; Wang, C.; Jiang, D.; Su, D.; Zhang, H.; Chen, Z.; et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 2020, 39, e104036. [Google Scholar] [CrossRef]
- Ock, C.Y.; Kim, S.; Keam, B.; Kim, S.; Ahn, Y.O.; Chung, E.J.; Kim, J.H.; Kim, T.M.; Kwon, S.K.; Jeon, Y.K.; et al. Changes in programmed death-ligand 1 expression during cisplatin treatment in patients with head and neck squamous cell carcinoma. Oncotarget 2017, 8, 97920–97927. [Google Scholar] [CrossRef]
- Sato, H.; Jeggo, P.A.; Shibata, A. Regulation of programmed death-ligand 1 expression in response to DNA damage in cancer cells: Implications for precision medicine. Cancer Sci. 2019, 110, 3415–3423. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, L.; Zhang, S.; Tian, X.; De La Cruz, A.; George, A.; Arnoff, T.E.; El-Deiry, W.S. The role of p53 in anti-tumor immunity and response to immunotherapy. Front. Mol. Biosci. 2023, 10, 1148389. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021, 39, 494–508.e495. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Li, J.; Montrose, D.C.; Martinez, L.A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 2023, 83, 266–280.e266. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.R.; Feng, Z.; Xiao, H.D.; Friedman, D.; Cottam, B.; Fox, B.A.; Kramer, G.; Leidner, R.S.; Bell, R.B.; Young, K.H.; et al. STING expression and response to treatment with STING ligands in premalignant and malignant disease. PLoS ONE 2017, 12, e0187532. [Google Scholar] [CrossRef] [PubMed]
- Wayne, J.; Brooks, T.; Landras, A.; Massey, A.J. Targeting DNA damage response pathways to activate the STING innate immune signaling pathway in human cancer cells. FEBS J. 2021, 288, 4507–4540. [Google Scholar] [CrossRef]
- Lee, R.H.; Kang, H.; Yom, S.S.; Smogorzewska, A.; Johnson, D.E.; Grandis, J.R. Treatment of Fanconi Anemia-Associated Head and Neck Cancer: Opportunities to Improve Outcomes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5168–5187. [Google Scholar] [CrossRef]
- Kim, M.S.; Li, S.L.; Bertolami, C.N.; Cherrick, H.M.; Park, N.H. State of p53, Rb and DCC tumor suppressor genes in human oral cancer cell lines. Anticancer Res. 1993, 13, 1405–1413. [Google Scholar]
- Pekkola-Heino, K.; Servomaa, K.; Kiuru, A.; Grenman, R. Increased radiosensitivity is associated with p53 mutations in cell lines derived from oral cavity carcinoma. Acta Oto-Laryngol. 1996, 116, 341–344. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Tang, W.; Webster, A.L.H.; Whiteaker, J.R.; Chandler, C.M.; Errazquin, R.; Roohollahi, K.; Fritzke, M.; Hoskins, E.E.; Jonlin, E.; et al. Fanconi anemia-isogenic head and neck cancer cell line pairs—A basic and translational science resource. Int. J. Cancer 2023, 153, 183–196. [Google Scholar] [CrossRef]
- Paret, C.; Russo, A.; Otto, H.; Mayer, A.; Zahnreich, S.; Wagner, W.; Samuel, D.; Scharnhorst, D.; Solomon, D.A.; Dhall, G.; et al. Personalized therapy: CNS HGNET-BCOR responsiveness to arsenic trioxide combined with radiotherapy. Oncotarget 2017, 8, 114210–114225. [Google Scholar] [CrossRef]
- Zahnreich, S.; Poplawski, A.; Hartel, C.; Eckhard, L.S.; Galetzka, D.; Hankeln, T.; Löbrich, M.; Marron, M.; Mirsch, J.; Ritter, S.; et al. Spontaneous and Radiation-Induced Chromosome Aberrations in Primary Fibroblasts of Patients with Pediatric First and Second Neoplasms. Front. Oncol. 2020, 10, 1338. [Google Scholar] [CrossRef]
- Zahnreich, S.; Gebrekidan, S.; Multhoff, G.; Vaupel, P.; Schmidberger, H.; Mayer, A. Oxygen Deprivation Modulates EGFR and PD-L1 in Squamous Cell Carcinomas of the Head and Neck. Front. Oncol. 2021, 11, 623964. [Google Scholar] [CrossRef]
- Zahnreich, S.; Yusifli, K.; Poplawski, A.; Eckhard, L.S.; Mirsch, J.; Hankeln, T.; Galetzka, D.; Marron, M.; Scholz-Kreisel, P.; Spix, C.; et al. Replication stress drives chromosomal instability in fibroblasts of childhood cancer survivors with second primary neoplasms. DNA Repair 2022, 122, 103435. [Google Scholar] [CrossRef]
- Gagnon, K.T.; Li, L.; Janowski, B.A.; Corey, D.R. Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat. Protoc. 2014, 9, 2045–2060. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahnreich, S.; El Guerzyfy, S.; Kaufmann, J.; Schmidberger, H. The cGAS/STING/IFN-1 Response in Squamous Head and Neck Cancer Cells after Genotoxic Challenges and Abrogation of the ATR-Chk1 and Fanconi Anemia Axis. Int. J. Mol. Sci. 2023, 24, 14900. https://doi.org/10.3390/ijms241914900
Zahnreich S, El Guerzyfy S, Kaufmann J, Schmidberger H. The cGAS/STING/IFN-1 Response in Squamous Head and Neck Cancer Cells after Genotoxic Challenges and Abrogation of the ATR-Chk1 and Fanconi Anemia Axis. International Journal of Molecular Sciences. 2023; 24(19):14900. https://doi.org/10.3390/ijms241914900
Chicago/Turabian StyleZahnreich, Sebastian, Soumia El Guerzyfy, Justus Kaufmann, and Heinz Schmidberger. 2023. "The cGAS/STING/IFN-1 Response in Squamous Head and Neck Cancer Cells after Genotoxic Challenges and Abrogation of the ATR-Chk1 and Fanconi Anemia Axis" International Journal of Molecular Sciences 24, no. 19: 14900. https://doi.org/10.3390/ijms241914900
APA StyleZahnreich, S., El Guerzyfy, S., Kaufmann, J., & Schmidberger, H. (2023). The cGAS/STING/IFN-1 Response in Squamous Head and Neck Cancer Cells after Genotoxic Challenges and Abrogation of the ATR-Chk1 and Fanconi Anemia Axis. International Journal of Molecular Sciences, 24(19), 14900. https://doi.org/10.3390/ijms241914900