
COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
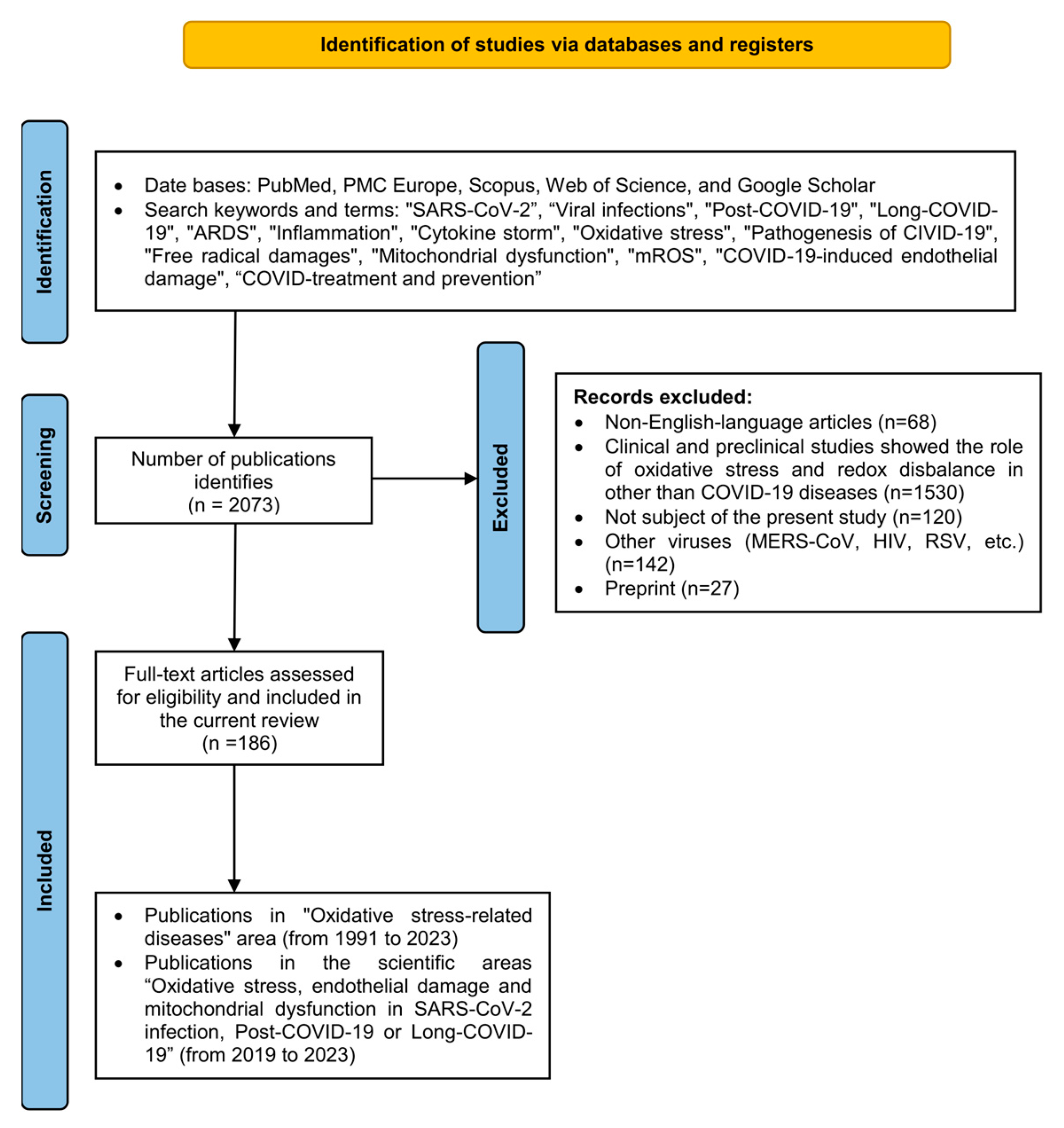
2. Literature Search, Methodology, Inclusion and Exclusion Criteria
Inclusion and Exclusion Criteria
3. Oxidative Stress, Free Radicals, and Redox State Conception
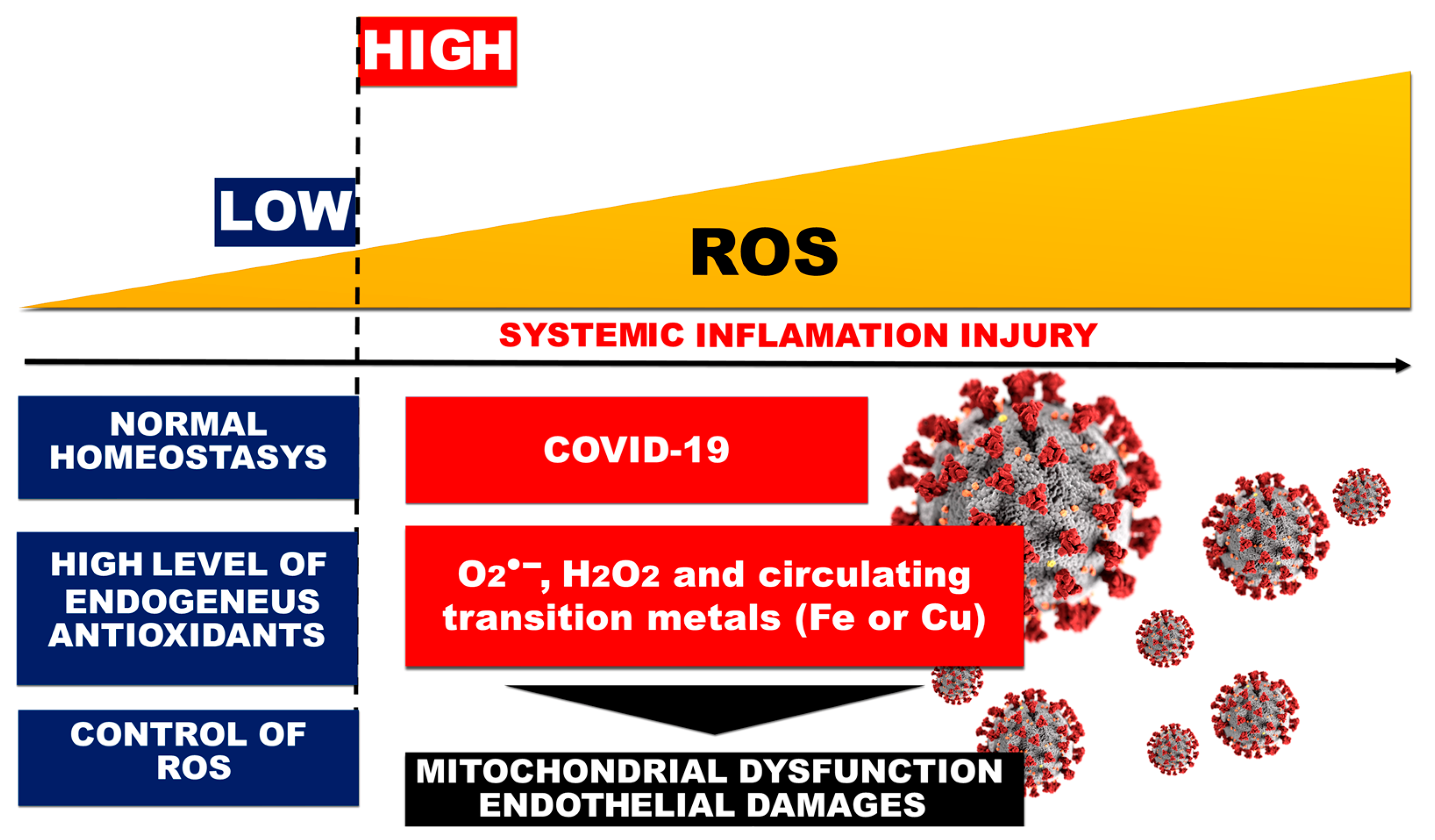
3.1. Free Radicals Reactive Oxygen Species and Reactive Nitrogen Species (RONS)
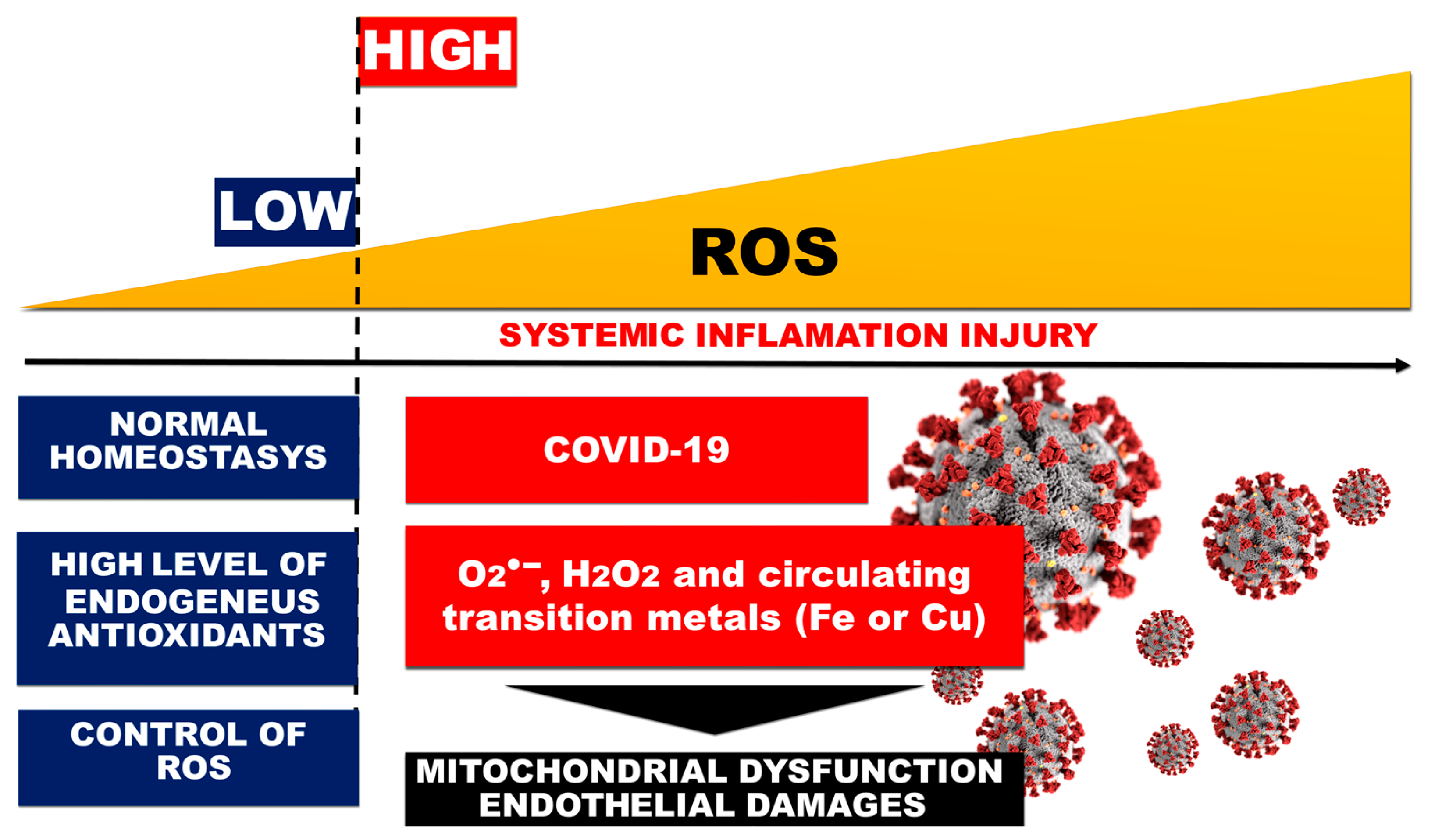
3.2. Redox State Change, Oxidative Stress, and ROS Generation in COVID-19-Related ARDS
3.3. Acute Respiratory Distress Syndrome
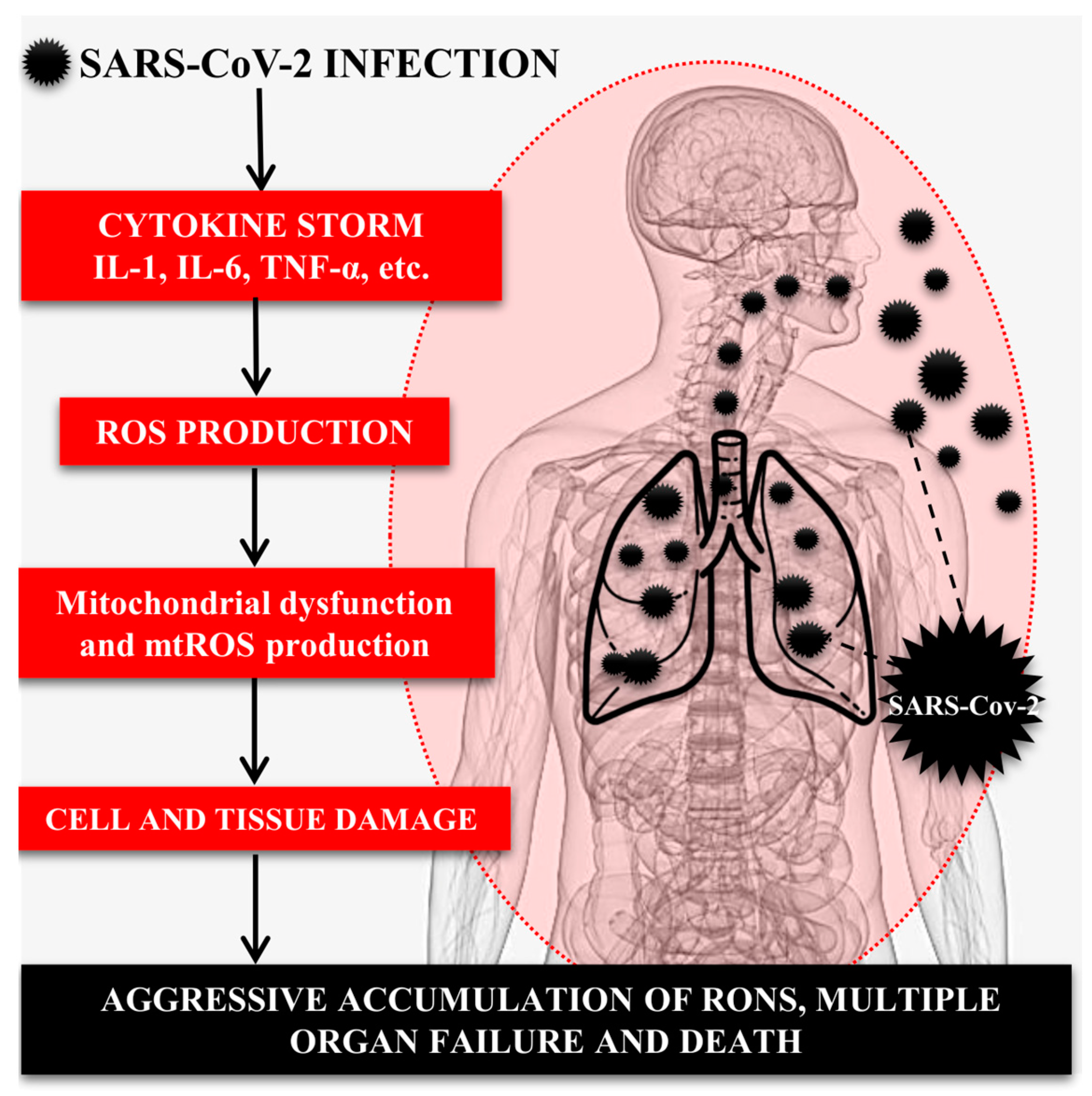
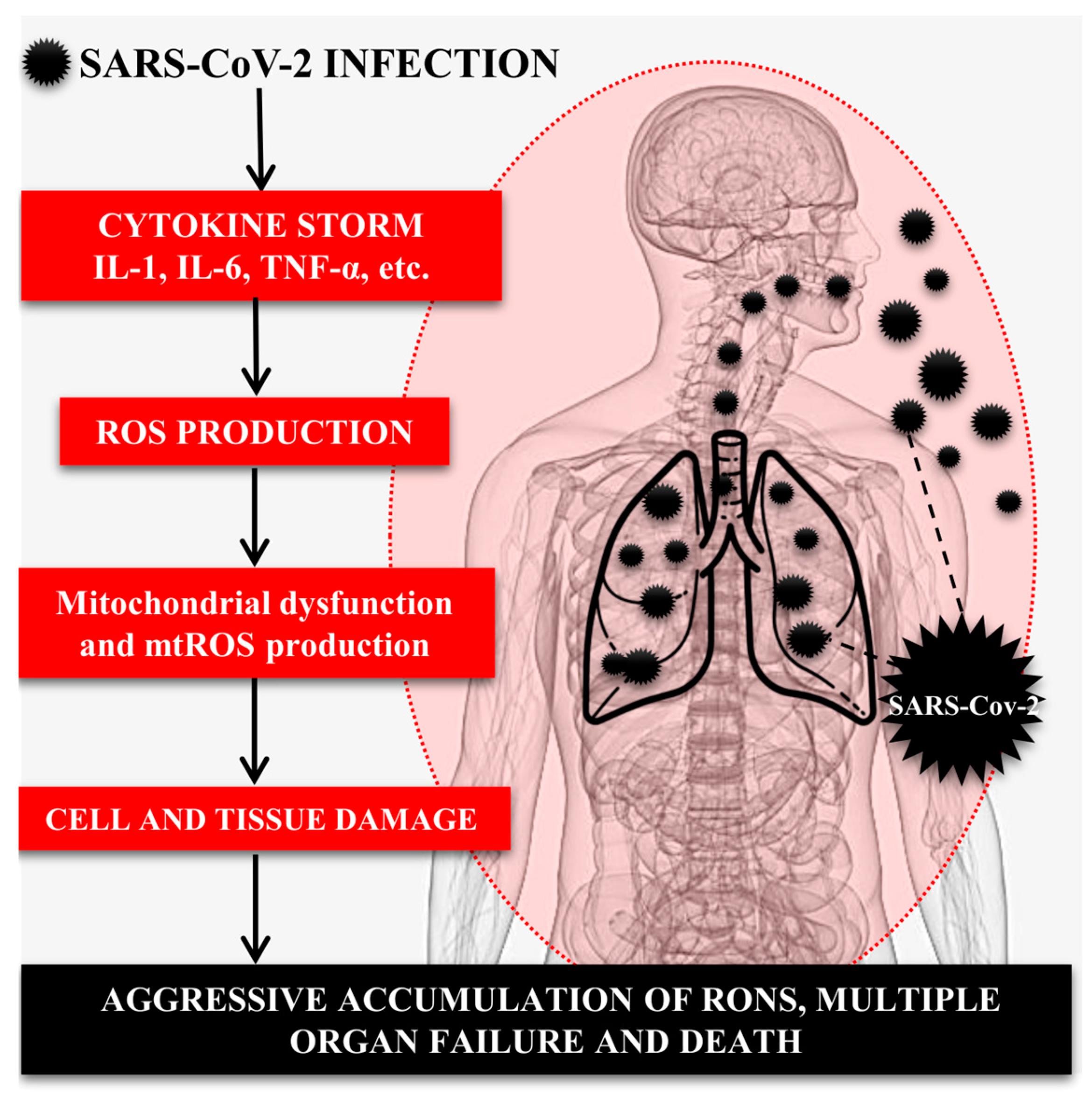
4. Role of the Immune System, Inflammatory Cytokines, and OS in SARS-CoV-2-Induced Lung Injury
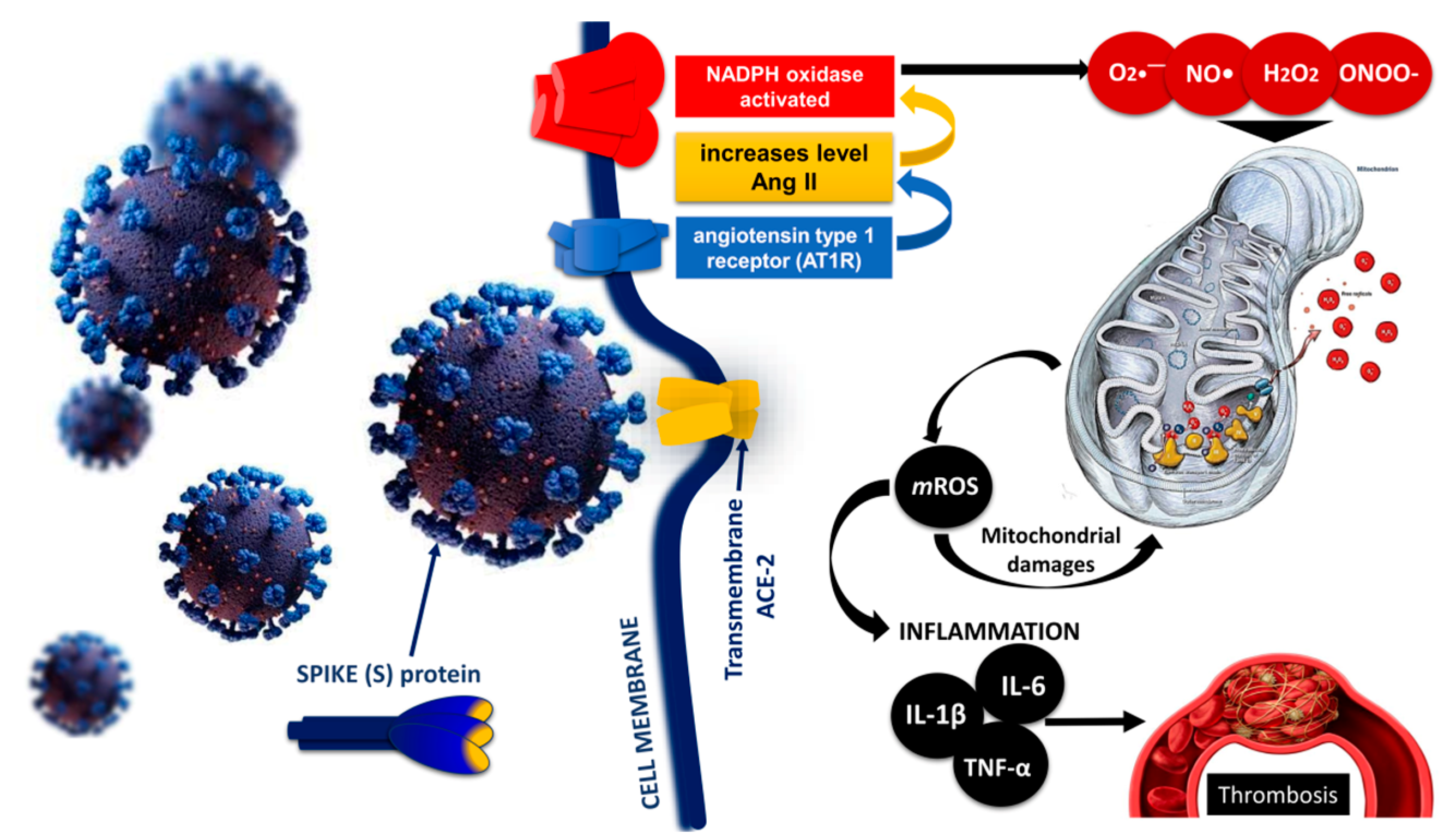
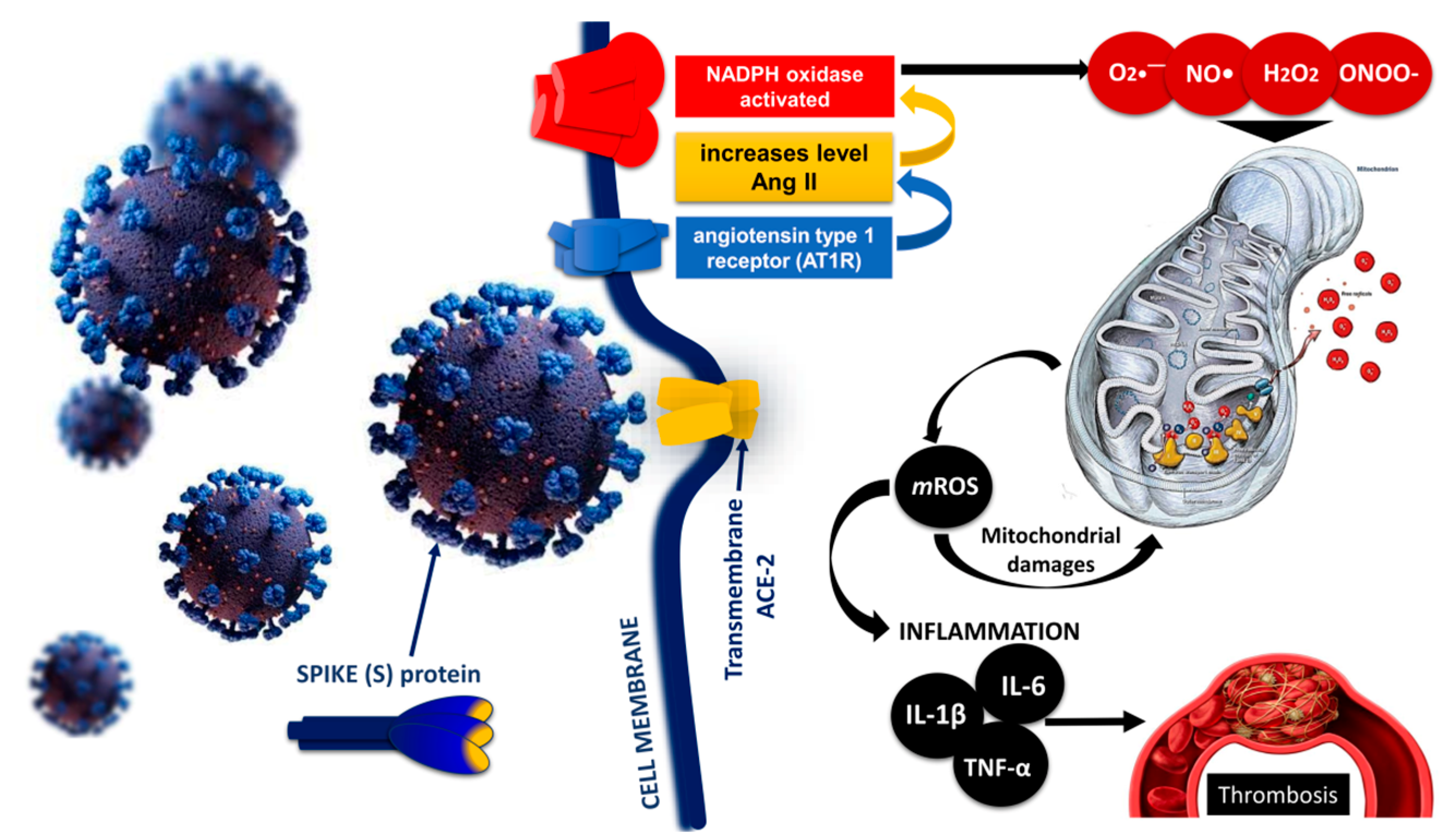
5. Role of the Mitochondria in the Pathogenesis of COVID-19
Mitochondrial Imbalance and Endothelial and Mitochondrial Dysfunction in COVID-19
6. Endothelial Injury and Thrombosis: Disseminated Intravascular Coagulopathy (DIC) and Venous Thromboembolism in COVID-19
6.1. Oxidative Stress and Endothelial Dysfunction in Liver Sinusoidal Endothelial Cells
6.2. Endothelial Damage of the Vascular Layer as a Result of Oxidative Stress in COVID-19
6.3. DIC and Thrombosis in COVID-19 Cases
7. Oxidative Stress and Endothelial Dysfunction in Post-Acute Sequelae of COVID-19 or Long-COVID-19 Syndrome
8. COVID-19: Treatment and Prevention
Endothelial Oxidative Stress Treatment and Prevention in the Context of COVID-19
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Losso, J. The potential of dietary bioactive compounds against SARS-CoV-2 and COVID-19-induced endothelial dysfunction. Molecules 2022, 27, 1623. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020, 7, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Pelle, M.C.; Zaffina, I.; Lucà, S.; Forte, V.; Trapanese, V.; Melina, M.; Giofrè, F.; Arturi, F. Endothelial Dysfunction in COVID-19: Potential Mechanisms and Possible Therapeutic Options. Life 2022, 12, 1605. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA statement. Ann. Intern. Med. 2009, 151, 264–269. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. Biomed. Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef]
- Lushchak, V.I.; Storey, K.B. Oxidative stress concept updated: Definitions, classifications, and regulatory pathways implicated. EXCLI J. 2021, 20, 956–967. [Google Scholar] [CrossRef]
- Schwarz, K.B. Oxidative stress during viral infection: A review. Free Radic. Biol. Med. 1996, 21, 641–649. [Google Scholar] [CrossRef]
- Kaul, P.; Biagioli, M.; Singh, I.; Turner, R. Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J. Infect. Dis. 2000, 181, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Forcados, G.; Muhammad, A.; Oladipo, O.; Makama, S.; Meseko, C.A. Metabolic Implications of Oxidative Stress and Inflammatory Process in SARS-CoV-2 Pathogenesis: Therapeutic Potential of Natural Antioxidants. Front. Cell Infect. Microbiol. 2021, 11, 654813. [Google Scholar] [CrossRef]
- Vollbracht, C.; Kraft, K. Oxidative Stress and Hyper-Inflammation as Major Drivers of Severe COVID-19 and Long COVID: Implications for the Benefit of High-Dose Intravenous Vitamin C. Front. Pharmacol. 2022, 13, 899198. [Google Scholar] [CrossRef]
- Chernyak, B.; Popova, E.; Prikhodko, A.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and Oxidative Stress. Biochem. Mosc. 2020, 85, 1543–1553. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.; Puri, B.; Olive, L.; Marx, W.; O’Neil, A.; Athan, E.; Carvalho, A.F.; Maes, M.; Walder, K.; et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 2020, 258, 118166. [Google Scholar] [CrossRef]
- Muhammad, Y.; Kani, Y.A.; Iliya, S.; Muhammad, J.B.; Binji, A.; El-Fulaty Ahmad, A.; Kabir, M.B.; Umar Bindawa, K.; Ahmed, A. Deficiency of antioxidants and increased oxidative stress in COVID-19 patients: A cross-sectional comparative study in Jigawa, Northwestern Nigeria. SAGE Open Med. 2021, 1, 2050312121991246. [Google Scholar] [CrossRef]
- Khan, N.; Singla, M.; Samal, S.; Lodha, R.; Medigeshi, G.R. Respiratory syncytial virus-induced oxidative stress leads to an increase in Labile Zinc Pools in Lung Epithelial Cells. Msphere 2020, 5, e00447–e004420. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J. Reactive oxygen species: Drivers of physiological and pathological processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Villamena, F.A. Molecular Basis of Oxidative Stress: Chemistry, Mechanisms, and Disease Pathogenesis; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Nedel, W.; Deutschendorf, C.; Portela, L.V.C. Sepsis-induced mitochondrial dysfunction: A narrative review. World J. Crit. Care Med. 2023, 12, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Rivera, A.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Alfredo, C.G.; Yalith, L.A.H.; Estefani, Y.H.C.; José, P.C. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437–478. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Tanaka, T.; Yoshida, Y.; Inagi, R. Physiological and pathophysiological role of reactive oxygen species and reactive nitrogen species in the kidney. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Ruano, C.S.M.; Hegenbarth, J.C.; Vaiman, D.; Gupta, S.; McCarthy, F.P.; Méhats, C.; McCarthy, C.; Apicella, C.; Scheel, J. Computational Models on Pathological Redox Signalling Driven by Pregnancy: A Review. Antioxidants 2022, 11, 585. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.; Go, Y.; Jones, D. Redox Systems Biology of Nutrition and Oxidative Stress. J. Nutr. 2019, 149, 553–565. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Med. Cell. Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef]
- Wieczfinska, J.; Kleniewska, P.; Pawliczak, R. Oxidative Stress-Related Mechanisms in SARS-CoV-2 Infections. Oxidative Med. Cell Longev. 2022, 2022, 5589089. [Google Scholar] [CrossRef]
- Wang, X.; Tang, G.; Liu, Y.; Liu, Y.; Zhang, L.; Chen, B.; Han, Y.; Fu, Z.; Wang, L.; Hu, G.; et al. The role of IL-6 in coronavirus, especially in COVID-19. Front. Pharmacol. 2022, 13, 1033674. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.; Peniston, H.; Sanghavi, D.; Mahapatra, S. Acute Respiratory Distress Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK436002/ (accessed on 28 September 2023).
- Swenson, K.E.; Swenson, E.R. Pathophysiology of Acute Respiratory Distress Syndrome and COVID-19 Lung Injury. Crit. Care Clin. 2021, 37, 749–776. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.J.A.; Ribeiro, L.R.; Gouveia, M.I.M.; Marcelino, B.D.R.; Santos, C.S.D.; Lima, K.V.B.; Lima, L.N.G.C. Hyperinflammatory Response in COVID-19: A Systematic Review. Viruses 2023, 15, 553. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, P.; Fiorillo Moreno, O.; Zarate Peñata, E.; Calderon-Villalba, A.; Pacheco Lugo, L.; Acosta Hoyos, A.; Villarreal Camacho, J.L.; Navarro Quiroz, R.; Pacheco Londoño, L.; Aroca Martinez, G.; et al. From Cell to Symptoms: The Role of SARS-CoV-2 Cytopathic Effects in the Pathogenesis of COVID-19 and Long COVID. Int. J. Mol. Sci. 2023, 24, 8290. [Google Scholar] [CrossRef]
- Gibson, P.; Qin, L.; Puah, S. COVID-19 acute respiratory distress syndrome (ARDS): Clinical features and differences from typical pre-COVID-19 ARDS. Med. J. Aust. 2020, 213, 54–56.e1. [Google Scholar] [CrossRef]
- Zhuang, Y.; Rahman, M.; Wen, Y.; Pokojovy, M.; McCaffrey, P.; Vo, A.; Walser, E.; Moen, S.; Xu, H.; Tseng, T.B. An interpretable multi-task system for clinically applicable COVID-19 diagnosis using CXR. J. X-ray Sci. Technol. 2022, 30, 847–862. [Google Scholar] [CrossRef]
- Huppert, L.; Matthay, M.; Ware, L. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef]
- Abdallat, M.; Khalil, M.; Al-Awwa, G.; Kothuru, R.; La Punzina, C. Barotrauma in COVID-19 patients. J. Lung Health Dis. 2020, 4, 8–12. [Google Scholar] [CrossRef]
- Ezeagu, R.; Olanipekun, T.; Santoshi, R.; Seneviratne, C.; Kupfer, Y. Pulmonary Barotrauma Resulting from Mechanical Ventilation in 2 Patients with a Diagnosis of COVID-19 Pneumonia. Am. J. Case Rep. 2021, 22, e927954. [Google Scholar] [CrossRef]
- Martínez Chamorro, E.; Díez Tascón, A.; Ibáñez Sanz, L.; Ossaba Vélez, S.; Borruel Nacenta, S. Radiologic diagnosis of patients with COVID-19. Radiología 2021, 63, 56–73. [Google Scholar] [CrossRef]
- Motta Junior, J.D.S.; Miggiolaro, A.F.R.D.S.; Nagashima, S.; de Paula, C.B.V.; Baena, C.P.; Scharfstein, J.; de Noronha, L. Mast Cells in Alveolar Septa of COVID-19 Patients: A Pathogenic Pathway That May Link Interstitial Edema to Immunothrombosis. Front. Immunol. 2020, 11, 574862. [Google Scholar] [CrossRef] [PubMed]
- Selickman, J.; Vrettou, C.; Mentzelopoulos, S.; Marini, J. COVID-19-Related ARDS: Key mechanistic features and treatments. J. Clin. Med. 2022, 11, 4896. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef] [PubMed]
- Tirelli, C.; De Amici, M.; Mira, S.; Nalesso, G.; Re, B.; Corsico, A.G.; Mondoni, M.; Centanni, S. Exploring the Role of Immune System and Inflammatory Cytokines in SARS-CoV-2 Induced Lung Disease: A Narrative Review. Biology 2023, 12, 177. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, F.S.; Lanzetti, M.; Nesi, R.T.; Nagato, A.C.; Silva, C.P.E.; Kennedy-Feitosa, E.; Melo, A.C.; Cattani-Cavalieri, I.; Porto, L.C.; Valenca, S.S. Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries. Antioxidants 2023, 12, 548. [Google Scholar] [CrossRef]
- Borges, L.; Gennari-Felipe, M.; Dias, B.B.; Hatanaka, E. Melatonin, Zinc, and Vitamin C: Potential Adjuvant Treatment for COVID-19 Patients. Front. Nutr. 2022, 8, 821824. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Simko, F.; Dominguez-Rodriguez, A.; Tesarik, J.; Neel, R.L.; Slominski, A.T.; Kleszczynski, K.; Martin-Gimenez, V.M.; Manucha, W.; et al. Melatonin: Highlighting its use as a potential treatment for SARS-CoV-2 infection. Cell Mol. Life Sci. 2022, 79, 143. [Google Scholar] [CrossRef]
- Jarczak, D.; Nierhaus, A. Cytokine Storm—Definition, Causes, and Implications. Int. J. Mol. Sci. 2022, 23, 11740. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 cytokine storm; what we know so far. Front. Immunol 2020, 1446. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.; Hejazi, M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T. Secondary haemophagocytic lymphohistiocytosis: Experience from the Uppsala University Hospital. Upsala J. Med. Sci. 2015, 120, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Pruett, W.; Plemmons, A.; Sanley, M. Secondary Hlh Presenting As Ards. Chest 2020, 158, A1979. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.; Brown, M. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 28. [Google Scholar] [CrossRef] [PubMed]
- Pustake, M.; Tambolkar, I.; Giri, P.; Gandhi, C. SARS, MERS and COVID-19: An overview and comparison of clinical, laboratory and radiological features. J. Fam. Med. Prim. Care 2022, 11, 10–17. [Google Scholar] [CrossRef]
- Pum, A.; Ennemoser, M.; Adage, T.; Kungl, A.J. Cytokines and chemokines in SARS-CoV-2 infections-therapeutic strategies targeting cytokine storm. Biomolecules 2021, 11, 91. [Google Scholar] [CrossRef]
- Talwar, D.; Kumar, S.; Acharya, S.; Raisinghani, N.; Madaan, S.; Hulkoti, V.; Akhilesh, A.; Khanna, S.; Shah, D.; Nimkar, S. Interleukin 6 and Its Correlation with COVID-19 in Terms of Outcomes in an Intensive Care Unit of a Rural Hospital: A Cross-sectional Study. Indian J. Crit. Care Med. 2022, 26, 39–42. [Google Scholar] [CrossRef]
- Lee, D.; Gardner, R.; Porter, D. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Lee, C.C.E.; Ali, K.; Connell, D.; Mordi, I.R.; George, J.; Lang, E.M.; Lang, C.C. COVID-19-Associated Cardiovascular Complications. Diseases 2021, 9, 47. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028456. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Yang, J.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2021, 11, 316–329. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Balansky, R.; LA Maestra, S. Antioxidants and COVID-19. J. Prev. Med. Hyg. 2021, 62, E34–E45. [Google Scholar] [CrossRef] [PubMed]
- Alwazeer, D.; Liu, F.F.; Wu, X.Y.; LeBaron, T.W. Combating Oxidative Stress and Inflammation in COVID-19 by Molecular Hydrogen Therapy: Mechanisms and Perspectives. Oxidative Med. Cell Longev. 2021, 2021, 5513868. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Amaya, M.; Voss, K. Reactive oxygen species activate NFκB (p65) and p53 and induce apoptosis in RVFV infected liver cells. Virology 2014, 449, 270–286. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, oxidative stress and innate immunity. Front. Physiol. 2018, 18, 1487. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxidative Med. Cell Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- Gonçalves, F.A.R.; Besen, B.A.M.P.; Lima, C.A.; Corá, A.P.; Pereira, A.J.R.; Perazzio, S.F.; Gouvea, C.P.; Fonseca, L.A.M.; Trindade, E.M.; Sumita, N.M.; et al. Use and misuse of biomarkers and the role of D-dimer and C-reactive protein in the management of COVID-19: A post-hoc analysis of a prospective cohort study. Clinics 2021, 76, e3547. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, M.; Hadzibegovic, A.; Kovac, M.; Jovanovic, B.; Stanisavljevic, J.; Djikic, M.; Sijan, D.; Ladjevic, N.; Palibrk, I.; Djukanovic, M.; et al. D-dimer, CRP, PCT, and IL-6 Levels at Admission to ICU Can Predict In-Hospital Mortality in Patients with COVID-19 Pneumonia. Oxidative Med. Cell Longev. 2022, 28, 8997709. [Google Scholar] [CrossRef] [PubMed]
- Smail, S.; Babaei, E.; Amin, K. Hematological, Inflammatory, Coagulation, and Oxidative/Antioxidant Biomarkers as Predictors for Severity and Mortality in COVID-19: A Prospective Cohort-Study. Int. J. Gen. Med. 2023, 16, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Mattiuzzi, C. Hemoglobin value may be decreased in patients with severe coronavirus disease 2019. Hematol. Transfus. Cell Ther. 2020, 2, 116–117. [Google Scholar] [CrossRef]
- Kang, J.; Pervaiz, S. Mitochondria: Redox metabolism and dysfunction. Biochem. Res. Int. 2012, 2012, 896751. [Google Scholar] [CrossRef]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef]
- Palma, F.; He, C.; Danes, J.; Paviani, V.; Coelho, D.R.; Gantner, B.N.; Bonini, M.G. Mitochondrial Superoxide Dismutase: What the Established, the Intriguing, and the Novel Reveal About a Key Cellular Redox Switch. Antioxid. Redox Signal. 2020, 32, 701–714. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Venditti, P. Mitochondrial Management of Reactive Oxygen Species. Antioxidants 2021, 10, 1824. [Google Scholar] [CrossRef]
- Shields, H.; Traa, A.; Van Raamsdonk, J. Beneficial and detrimental effects of reactive oxygen species on lifespan: A comprehensive review of comparative and experimental studies. Front. Cell Devel Biol. 2021, 9, 181. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Idelchik, M.; Begley, U.; Begley, T.; Melendez, J. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, F.; Ehler, J.; Bajorat, R.; Patejdl, R. Mitochondrial dysfunction in intensive care unit-acquired weakness and critical illness myopathy: A narrative review. Int. J. Mol. Sci. 2023, 24, 5516. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Jabůrek, M.; Plecitá-Hlavatá, L. Contribution of oxidative stress and impaired biogenesis of pancreatic β-Cells to Type 2 Diabetes. Antioxid. Redox Signal. 2019, 31, 722–751. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Fernández-Ayala, D.; Navas, P.; López-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef]
- Yin, F.; Cadenas, E. Mitochondria: The cellular hub of the dynamic coordinated network. Antioxid. Redox Signal. 2015, 22, 961–964. [Google Scholar] [CrossRef]
- Higashi, Y. Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease. Antioxidants 2022, 11, 1958. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.-H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, Ł.; Wojnar-Lason, K.; Kaczara, P.; Tworzydło, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef] [PubMed]
- De Michele, M.; Lorenzano, S.; Piscopo, P.; Rivabene, R.; Crestini, A.; Chistolini, A.; Stefanini, L.; Pulcinelli, F.M.; Berto, I.; Campagna, R.; et al. SARS-CoV-2 infection predicts larger infarct volume in patients with acute ischemic stroke. Front. Cardiovasc. Med. 2023, 9, 1097229. [Google Scholar] [CrossRef]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2020, 10, 1568. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Enriquez, J.; Rojas-Morales, E.; Ruiz-Garcia, M.G.; Tobón-Velasco, J.C.; Jiménez-Ortega, J.C. SARS-CoV-2 induces mitochondrial dysfunction and cell death by oxidative stress in leukocytes of COVID-19 patients. Authorea 2020. preprints. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.; Potje, S.; Fraga-Silva, T.F.C.; da Silva-Neto, J.A.; Barros, P.R.; Rodrigues, D.; Machado, M.R.; Martins, R.B.; Santos-Eichler, R.A.; Benatti, M.N.; et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vasc. Pharmacol. 2022, 142, 106946. [Google Scholar] [CrossRef]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impair. Front. Microbiol. 2022, 12, 780768. [Google Scholar] [CrossRef]
- de Las Heras, N.; Martín Giménez, V.; Ferder, L.; Ferder, L.; Manucha, W.; Lahera, V. Implications of oxidative stress and potential role of mitochondrial dysfunction in COVID-19: Therapeutic effects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef]
- Valdés-Aguayo, J.J.; Garza-Veloz, I.; Vargas-Rodríguez, J.R.; Martinez-Vazquez, M.C.; Avila-Carrasco, L.; Bernal-Silva, S.; González-Fuentes, C.; Comas-García, A.; Alvarado-Hernández, D.E.; Centeno-Ramirez, A.S.H.; et al. Peripheral Blood Mitochondrial DNA Levels Were Modulated by SARS-CoV-2 Infection Severity and Its Lessening Was Associated with Mortality Among Hospitalized Patients with COVID-19. Front. Cell Infect. Microbiol. 2021, 11, 754708. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial Dysfunction in Lung Pathogenesis. Annu. Rev. Physiol. 2017, 79, 495–515. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Tse, H.M. The role of NADPH oxidases in infectious and inflammatory diseases. Redox Biol. 2021, 48, 102159. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.; Alhoufie, S.; Hifny, A.; Schwartz, L.; Alqahtani, A.S.; Ahmed, S.B.M.; Alqahtani, A.M.; Alqahtani, S.S.; Muddathir, A.K.; Ali, H.; et al. Of mitochondrion and COVID-19. J. Enzym. Inhib. Med. Chem. 2021, 36, 1258–1267. [Google Scholar] [CrossRef]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a cellular hub in infection and inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef]
- Li, D.; Ding, X.; Xie, M.; Tian, D.; Xia, L. COVID-19-associated liver injury: From bedside to bench. J. Gastroenterol. 2021, 56, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Zapotoczny, B.; Braet, F.; Kus, E.; Ginda-Mäkelä, K.; Klejevskaja, B.; Campagna, R.; Chlopicki, S.; Szymonski, M. Actin-spectrin scaffold supports open fenestrae in liver sinusoidal endothelial cells. Traffic 2019, 20, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernández-Gea, V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 2020, 9, 929. [Google Scholar] [CrossRef]
- Romano, C.; Cozzolino, D.; Nevola, R.; Abitabile, M.; Carusone, C.; Cinone, F.; Cuomo, G.; Nappo, F.; Sellitto, A.; Umano, G.R.; et al. Liver Involvement during SARS-CoV-2 Infection Is Associated with a Worse Respiratory Outcome in COVID-19 Patients. Viruses 2023, 15, 1904. [Google Scholar] [CrossRef]
- Akkiz, H. Unraveling the Molecular and Cellular Pathogenesis of COVID-19-Associated Liver Injury. Viruses 2023, 15, 1287. [Google Scholar] [CrossRef]
- Allameh, A.; Niayesh-Mehr, R.; Aliarab, A.; Sebastiani, G.; Pantopoulos, K. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants 2023, 12, 1653. [Google Scholar] [CrossRef]
- Santoro, L.; Zaccone, V.; Falsetti, L.; Ruggieri, V.; Danese, M.; Miro, C.; Di Giorgio, A.; Nesci, A.; D’Alessandro, A.; Moroncini, G.; et al. Role of Endothelium in Cardiovascular Sequelae of Long COVID. Biomedicines 2023, 11, 2239. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Kansakar, U.; Sardu, C.; Varzideh, F.; Avvisato, R.; Wang, X.; Matarese, A.; Marfella, R.; Ziosi, M.; Gambardella, J.; et al. COVID-19 Causes Ferroptosis and Oxidative Stress in Human Endothelial Cells. Antioxidants 2023, 12, 326. [Google Scholar] [CrossRef]
- Montiel, V.; Lobysheva, I.; Gérard, L.; Vermeersch, M.; Perez-Morga, D.; Castelein, T.; Mesland, J.B.; Hantson, P.; Collienne, C.; Gruson, D.; et al. Oxidative stress-induced endothelial dysfunction and decreased vascular nitric oxide in COVID-19 patients. EBioMedicine 2022, 77, 103893. [Google Scholar] [CrossRef]
- Amponsah-Offeh, M.; Diaba-Nuhoho, P.; Speier, S.; Morawietz, H. Oxidative Stress, Antioxidants and Hypertension. Antioxidants 2023, 12, 281. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Weng, J. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Fragoso-Morales, L.G.; Correa-Basurto, J.; Rosales-Hernández, M.C. Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer’s Disease Murine Models. Antioxidants 2021, 10, 218. [Google Scholar] [CrossRef]
- Froldi, G.; Dorigo, P. Endothelial dysfunction in Coronavirus disease 2019 (COVID-19): Gender and age influences. Med. Hypotheses 2020, 144, 110015. [Google Scholar] [CrossRef] [PubMed]
- Miggiolaro, A.F.R.S.; da Silva, F.P.G.; Wiedmer, D.B.; Godoy, T.M.; Borges, N.H.; Piper, G.W.; Oricil, A.G.G.; Klein, C.K.; Hlatchuk, E.C.; Dagostini, J.C.H.; et al. COVID-19 and Pulmonary Angiogenesis: The Possible Role of Hypoxia and Hyperinflammation in the Overexpression of Proteins Involved in Alveolar Vascular Dysfunction. Viruses 2023, 15, 706. [Google Scholar] [CrossRef] [PubMed]
- Giordo, R.; Paliogiannis, P.; Mangoni, A.; Pintus, G. SARS-CoV-2 and endothelial cell interaction in COVID-19: Molecular perspectives. Vasc. Biol. 2021, 3, R15–R23. [Google Scholar] [CrossRef] [PubMed]
- De Pablo-Moreno, J.A.; Serrano, L.; Revuelta, L.J.; Sánchez, M.J.; Liras, A. The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. Int. J. Mol. Sci. 2022, 23, 8283. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Angelini, D.E.; Kaatz, S.; Rosovsky, R.P.; Zon, R.L.; Pillai, S.; Robertson, W.E.; Elavalakanar, P.; Patell, R.; Khorana, A. COVID-19 and venous thromboembolism: A narrative review. Res. Pract. Thromb. Haemost. 2022, 6, e12666. [Google Scholar] [CrossRef]
- Sutanto, H.; Soegiarto, G. Risk of Thrombosis during and after a SARS-CoV-2 Infection: Pathogenesis, Diagnostic Approach, and Management. Hematol. Rep. 2023, 15, 225–243. [Google Scholar] [CrossRef]
- Niculae, C.; Hristea, A.; Moroti, R. Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review. Biomedicines 2023, 11, 929. [Google Scholar] [CrossRef]
- Maruhashi, T.; Higashi, Y. Pathophysiological Association of Endothelial Dysfunction with Fatal Outcome in COVID-19. Int. J. Mol. Sci. 2021, 22, 5131. [Google Scholar] [CrossRef]
- Gross, P.L.; Chan, N.C. Thromboembolism in Older Adults. Front. Med. 2021, 7, 470016. [Google Scholar] [CrossRef]
- Yamada, S.; Asakura, H. Therapeutic strategies for disseminated intravascular coagulation associated with aortic aneurysm. Int. J. Mol. Sci. 2022, 23, 1296. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Iba, T. COVID-19 coagulopathy: Is it disseminated intravascular coagulation? Intern. Emerg. Med. 2021, 16, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cheng, Z.; Luo, L.; Zhu, Y.; Lin, W.; Ming, Z.; Chen, W.; Hu, Y. Incidence and impact of disseminated intravascular coagulation in COVID-19 a systematic review and meta-analysis. Thromb. Res. 2021, 201, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, C.; Jourdi, G.; Adjambri, E.; Walborn, A.; Patel, P.; Fareed, J.; Elalamy, I.; Hoppensteadt, D.; Gerotziafas, G.T. Disseminated Intravascular Coagulation: An Update on Pathogenesis, Diagnosis, and Therapeutic Strategies. Clin. Appl. Thromb. Hemost. 2018, 9, 8S–28S. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.; Levy, J. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Schwartz, R.A. Disseminated intravascular coagulation: A devastating systemic disorder of special concern with COVID-19. Dermatol. Ther. 2020, 33, 14053. [Google Scholar] [CrossRef]
- Page, E.M.; Ariëns, R.A.S. Mechanisms of thrombosis and cardiovascular complications in COVID-19. Thromb. Res. 2021, 200, 1–8. [Google Scholar] [CrossRef]
- Milovanovic, L.; Hessey, E.; Sebastianski, M.; Keto-Lambert, D.; Vandermeer, B.; Bagshaw, S.M.; Rewa, O. Epidemiology, clinical characteristics and treatment of critically ill patients with COVID-19): A protocol for a living systematic review. BMJ Open 2021, 11, e042008. [Google Scholar] [CrossRef]
- Chang, R.; Mamun, A.; Dominic, A.; Le, N.T. SARS-CoV-2 Mediated Endothelial Dysfunction: The Potential Role of Chronic Oxidative Stress. Front. Physiol. 2021, 11, 605908. [Google Scholar] [CrossRef]
- Pincemail, J.; Cavalier, E.; Charlier, C.; Cheramy–Bien, J.-P.; Brevers, E.; Courtois, A.; Fadeur, M.; Meziane, S.; Goff, C.L.; Misset, B.; et al. Oxidative Stress Status in COVID-19 Patients Hospitalized in Intensive Care Unit for Severe Pneumonia. A Pilot Study. Antioxidants 2021, 10, 257. [Google Scholar] [CrossRef]
- Soetedjo, N.N.M.; Iryaningrum, M.R.; Damara, F.A.; Permadhi, I.; Sutanto, L.B.; Hartono, H.; Rasyid, H. Prognostic properties of hypoalbuminemia in COVID-19 patients: A systematic review and diagnostic meta-analysis. Clin. Nutr. ESPEN 2021, 45, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Lambadiari, V.; Korakas, E.; Oikonomou, E.; Bletsa, E.; Kountouri, A.; Goliopoulou, A.; Ikonomidis, I.; Siasos, G. COVID-19, Endothelium and the Cardiometabolic Patient: A Possible Role for Capillary Leak Syndrome. Biomedicines 2022, 10, 2379. [Google Scholar] [CrossRef] [PubMed]
- Behzadifard, M.; Soleimani, M. NETosis and SARS-CoV-2 infection related thrombosis: A narrative review. Thromb. J. 2022, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Ogawa, H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int. J. Hematol. 2021, 113, 45–57. [Google Scholar] [CrossRef]
- Fischer, A.; Badier, N.; Zhang, L.; Elbéji, A.; Wilmes, P.; Oustric, P.; Benoy, C.; Ollert, M.; Fagherazzi, G. Long COVID Classification: Findings from a Clustering Analysis in the Predi-COVID Cohort Study. Int. J. Environ. Res. Public Health 2022, 19, 16018. [Google Scholar] [CrossRef] [PubMed]
- Haffke, M.; Freitag, H.; Rudolf, G.; Seifert, M.; Doehner, W.; Scherbakov, N.; Hanitsch, L.; Wittke, K.; Bauer, S.; Konietschke, F.; et al. Endothelial dysfunction and altered endothelial biomarkers in patients with post-COVID-19 syndrome and chronic fatigue syndrome (ME/CFS). J. Transl. Med. 2022, 138, 2022. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Maltezou, H.C.; Pavli, A.; Tsakris, A. Post-COVID Syndrome: An Insight on Its Pathogenesis. Vaccines 2021, 9, 497. [Google Scholar] [CrossRef]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Tsilingiris, D.; Vallianou, N.G.; Karampela, I.; Christodoulatos, G.S.; Papavasileiou, G.; Petropoulou, D.; Magkos, F.; Dalamaga, M. Laboratory Findings and Biomarkers in Long COVID: What Do We Know So Far? Insights into Epidemiology, Pathogenesis, Therapeutic Perspectives and Challenges. Int. J. Mol. Sci. 2023, 24, 10458. [Google Scholar] [CrossRef]
- Stufano, A.; Isgrò, C.; Palese, L.L.; Caretta, P.; De Maria, L.; Lovreglio, P.; Sardanelli, A.M. Oxidative Damage and Post-COVID Syndrome: A Cross-Sectional Study in a Cohort of Italian Workers. Int. J. Mol. Sci. 2023, 24, 7445. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, A.; Pranata, R.; Lim, M.A.; Akbara, M.R.; Martha, J.W. Endotheliopathy marked by high von Willebrand factor (vWF) antigen in COVID-19 is associated with poor outcome: A systematic review and meta-analysis. Int. J. Infect. Dis. 2022, 117, 267–273. [Google Scholar] [CrossRef]
- Chakrabortty, R.; Rahman, S.; Jahan, R.; Dip, A.H.; Rahman, M.M. Nintedanib in the management of pulmonary fibrosis after COVID-19: A case report. BIRDEM Med. J. 2021, 11, 148–152. [Google Scholar] [CrossRef]
- Alam, M.S.; Czajkowsky, D.M. SARS-CoV-2 infection and oxidative stress: Pathophysiological insight into thrombosis and therapeutic opportunities. Cytokine Growth Factor Rev. 2022, 63, 44–57. [Google Scholar] [CrossRef]
- Nunn, A.; Guy, G.; Brysch, W.; Bell, J. Understanding Long COVID; Mitochondrial Health and Adaptation-Old Pathways, New Problems. Biomedicines 2022, 10, 3113. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Nassar, A.; Ibrahim, I.M.; Amin, F.G.; Magdy, M.; Elgharib, A.M.; Azzam, E.B.; Nasser, F.; Yousry, K.; Shamkh, I.M.; Mahdy, S.M.; et al. A Review of Human Coronaviruses’ Receptors: The Host-Cell Targets for the Crown Bearing Viruses. Molecules 2021, 26, 6455. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; Robertson, D.L.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Hassan, S.T.S. Shedding Light on the Effect of Natural Anti-Herpesvirus Alkaloids on SARS-CoV-2: A Treatment Option for COVID-19. Viruses 2020, 12, 476. [Google Scholar] [CrossRef]
- Yong, S.J. Long COVID or post-COVID-19 syndrome: Putative pathophysiology, risk factors, and treatments. Infect. Dis. 2021, 53, 737–754. [Google Scholar] [CrossRef]
- Akanchise, T.; Angelova, A. Potential of Nano-Antioxidants and Nanomedicine for Recovery from Neurological Disorders Linked to Long COVID Syndrome. Antioxidants 2023, 12, 393. [Google Scholar] [CrossRef]
- Fratta Pasini, A.M.; Stranieri, C.; Cominacini, L.; Mozzini, C. Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-CoV-2 Complications. Antioxidants 2021, 10, 272. [Google Scholar] [CrossRef] [PubMed]
- Farahani, M.; Niknam, Z.; Mohammadi Amirabad, L.; Amiri-Dashatan, N.; Koushki, M.; Nemati, M.; Danesh Pouya, F.; Rezaei-Tavirani, M.; Rasmi, Y.; Tayebi, L. Molecular pathways involved in COVID-19 and potential pathway-based therapeutic targets. Biomed. Pharmacother. 2022, 145, 112420. [Google Scholar] [CrossRef] [PubMed]
- Nejat, R.; Torshizi, M.F.; Najafi, D.J. S Protein, ACE2 and Host Cell Proteases in SARS-CoV-2 Cell Entry and Infectivity; Is Soluble ACE2 a Two Blade Sword? A Narrative Review. Vaccines 2023, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhang, W.; Krummenacher, C.; Chen, Y.; Zheng, Q.; Zhao, Q.; Zeng, M.S.; Xia, N.; Zeng, Y.X.; Xu, M.; et al. Targeting herpesvirus entry complex and fusogen glycoproteins with prophylactic and therapeutic agents. Trends Microbiol. 2023, 31, 788–804. [Google Scholar] [CrossRef]
- Navarro-Bielsa, A.; Gracia-Cazaña, T.; Aldea-Manrique, B.; Abadías-Granado, I.; Ballano, A.; Bernad, I.; Gilaberte, Y. COVID-19 infection and vaccines: Potential triggers of Herpesviridae reactivation. An. Bras. Dermatol. 2023, 98, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.T.S.; Šudomová, M.; Mazurakova, A.; Kubatka, P. Insights into Antiviral Properties and Molecular Mechanisms of Non-Flavonoid Polyphenols against Human Herpesviruses. Int. J. Mol. Sci. 2022, 23, 13891. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Saidara, E.; Maes, M. Persistent SARS-CoV-2 Infection, EBV, HHV-6 and Other Factors May Contribute to Inflammation and Autoimmunity in Long COVID. Viruses 2023, 15, 400. [Google Scholar] [CrossRef]
- Dursun, R.; Temiz, S.A. The clinics of HHV-6 infection in COVID-19 pandemic: Pityriasis rosea and Kawasaki disease. Dermatol. Ther. 2020, 33, e13730. [Google Scholar] [CrossRef]
- Hosomi, S.; Nishida, Y.; Fujiwara, Y. The Impact of Human Herpesviruses in Clinical Practice of Inflammatory Bowel Disease in the Era of COVID-19. Microorganisms 2021, 9, 1870. [Google Scholar] [CrossRef]
- Maia, C.M.F.; Marques, N.P.; de Lucena, E.H.G.; de Rezende, L.F.; Martelli, D.R.B.; Martelli-Júnior, H. Increased number of Herpes Zoster cases in Brazil related to the COVID-19 pandemic. Int. J. Infect. Dis. 2021, 104, 732–733. [Google Scholar] [CrossRef]
- Silva, M.J.A.; Ribeiro, L.R.; Lima, K.V.B.; Lima, L.N.G.C. Adaptive immunity to SARS-CoV-2 infection: A systematic review. Front. Immunol. 2022, 13, 1001198. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. Natural products may interfere with SARS-CoV-2 attachment to the host cell. J. Biomol. Struct. Dyn. 2021, 39, 3194–3203. [Google Scholar] [CrossRef]
- González-Maldonado, P.; Alvarenga, N.; Burgos-Edwards, A.; Flores-Giubi, M.E.; Barúa, J.E.; Romero-Rodríguez, M.C.; Soto-Rifo, R.; Valiente-Echeverría, F.; Langjahr, P.; Cantero-González, G.; et al. Screening of Natural Products Inhibitors of SARS-CoV-2 Entry. Molecules 2022, 27, 1743. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Baglivo, M.; Baronio, M.; Natalini, G.; Beccari, T.; Chiurazzi, P.; Fulcheri, E.; Petralia, P.P.; Michelini, S.; Fiorentini, G.; Miggiano, G.A.; et al. Natural small molecules as inhibitors of coronavirus lipid-dependent attachment to host cells: A possible strategy for reducing SARS-CoV-2 infectivity? Acta Biomed. 2020, 91, 161–164. [Google Scholar] [CrossRef]
- Prasansuklab, A.; Theerasri, A.; Rangsinth, P.; Sillapachaiyaporn, C.; Chuchawankul, S.; Tencomnao, T. Anti-COVID-19 drug candidates: A review on potential biological activities of natural products in the management of new coronavirus infection. J. Tradit. Complement. Med. 2021, 11, 144–157. [Google Scholar] [CrossRef]
- Bizzoca, M.E.; Leuci, S.; Mignogna, M.D.; Muzio, E.L.; Caponio, V.C.A.; Muzio, L.L. Natural compounds may contribute in preventing SARS-CoV-2 infection: A narrative review. Food Sci. Hum. Wellness 2022, 11, 1134–1142. [Google Scholar] [CrossRef]
- Kiani, A.K.; Dhuli, K.; Anpilogov, K.; Bressan, S.; Dautaj, A.; Dundar, M.; Ergoren, M.C.; Bertelli, M. Natural compounds as inhibitors of SARS-CoV-2 endocytosis: A promising approach against COVID-19. Acta Biomed. Medica Atenei Parm. 2020, 91, e2020008. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. Turning the Tide: Natural Products and Natural-Product-Inspired Chemicals as Potential Counters to SARS-CoV-2 Infection. Front. Pharmacol. 2020, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.T.; Wong, A.Y.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.; Huang, X.; et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgieva, E.; Ananiev, J.; Yovchev, Y.; Arabadzhiev, G.; Abrashev, H.; Abrasheva, D.; Atanasov, V.; Kostandieva, R.; Mitev, M.; Petkova-Parlapanska, K.; et al. COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction. Int. J. Mol. Sci. 2023, 24, 14876. https://doi.org/10.3390/ijms241914876
Georgieva E, Ananiev J, Yovchev Y, Arabadzhiev G, Abrashev H, Abrasheva D, Atanasov V, Kostandieva R, Mitev M, Petkova-Parlapanska K, et al. COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction. International Journal of Molecular Sciences. 2023; 24(19):14876. https://doi.org/10.3390/ijms241914876
Chicago/Turabian StyleGeorgieva, Ekaterina, Julian Ananiev, Yovcho Yovchev, Georgi Arabadzhiev, Hristo Abrashev, Despina Abrasheva, Vasil Atanasov, Rositsa Kostandieva, Mitko Mitev, Kamelia Petkova-Parlapanska, and et al. 2023. "COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction" International Journal of Molecular Sciences 24, no. 19: 14876. https://doi.org/10.3390/ijms241914876
APA StyleGeorgieva, E., Ananiev, J., Yovchev, Y., Arabadzhiev, G., Abrashev, H., Abrasheva, D., Atanasov, V., Kostandieva, R., Mitev, M., Petkova-Parlapanska, K., Karamalakova, Y., Koleva-Korkelia, I., Tsoneva, V., & Nikolova, G. (2023). COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction. International Journal of Molecular Sciences, 24(19), 14876. https://doi.org/10.3390/ijms241914876