
Integrated Transcriptome and Metabolome Analysis Reveals the Molecular Mechanism of Rust Resistance in Resistant (Youkang) and Susceptive (Tengjiao) Zanthoxylum armatum Cultivars

 , ,
, ,
Abstract
:1. Introduction
2. Results
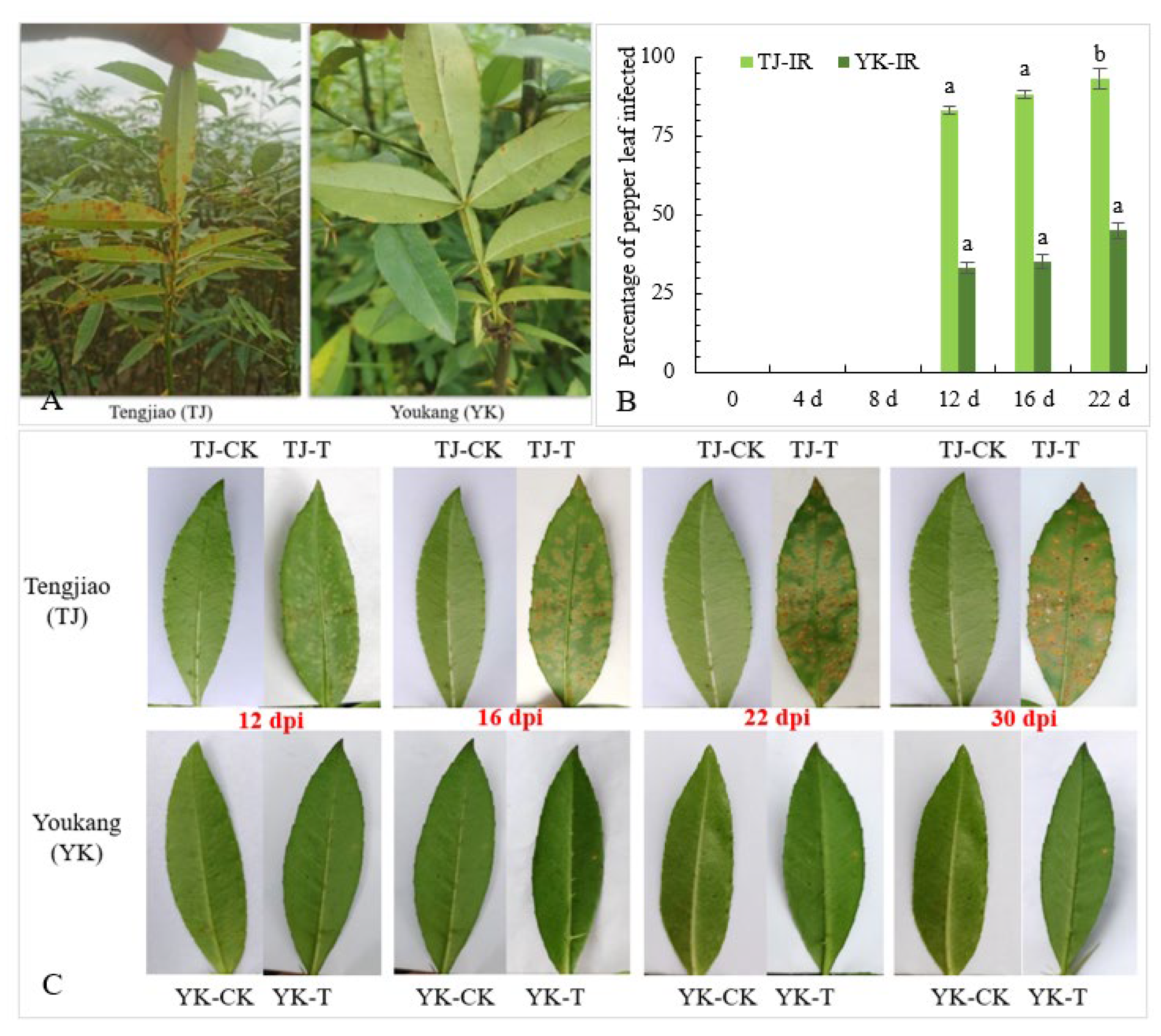
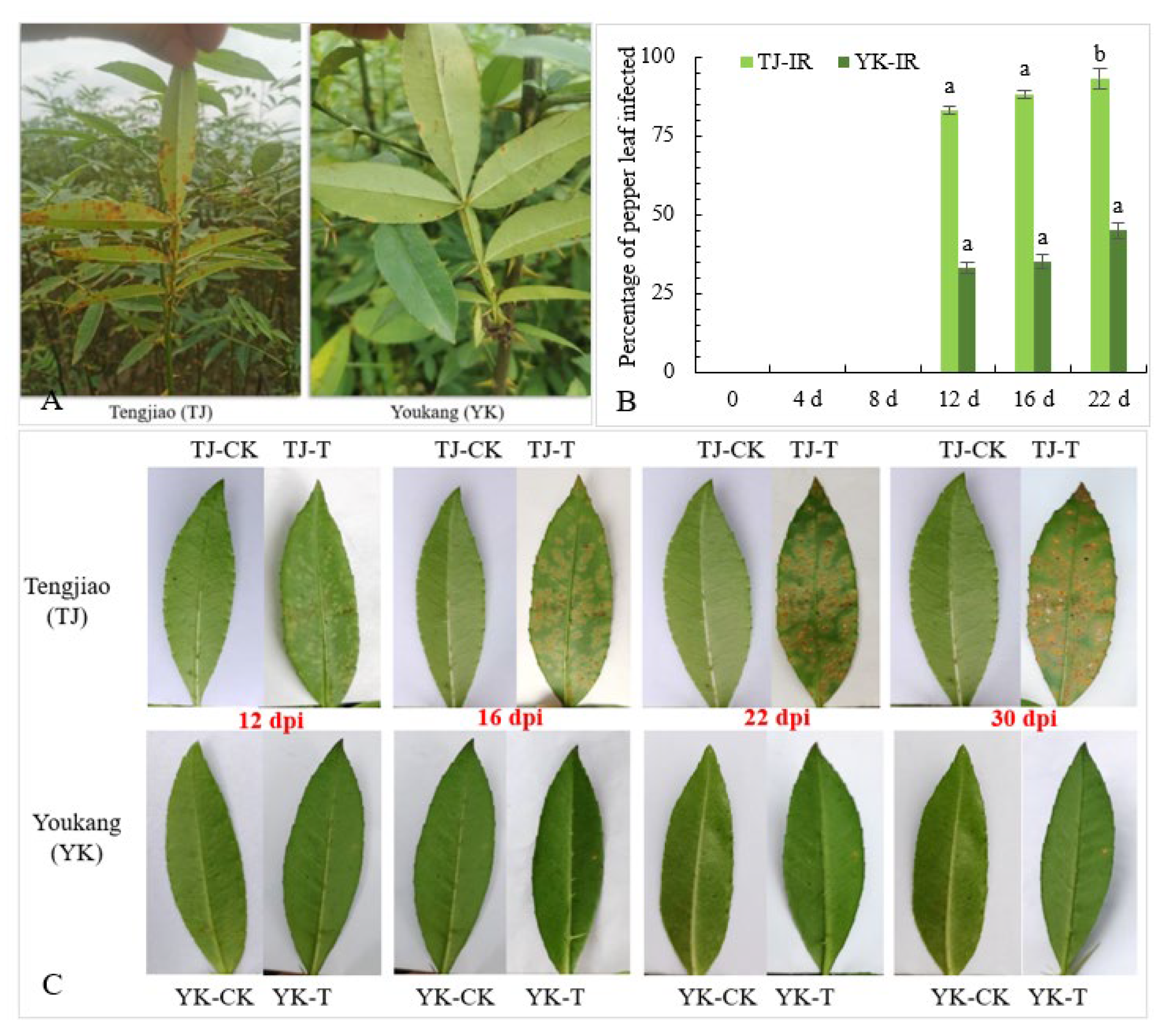
2.1. Analysis of Rust Resistance of ‘Tengjiao’ and ‘Youkang’ Chinese Pepper Leaves
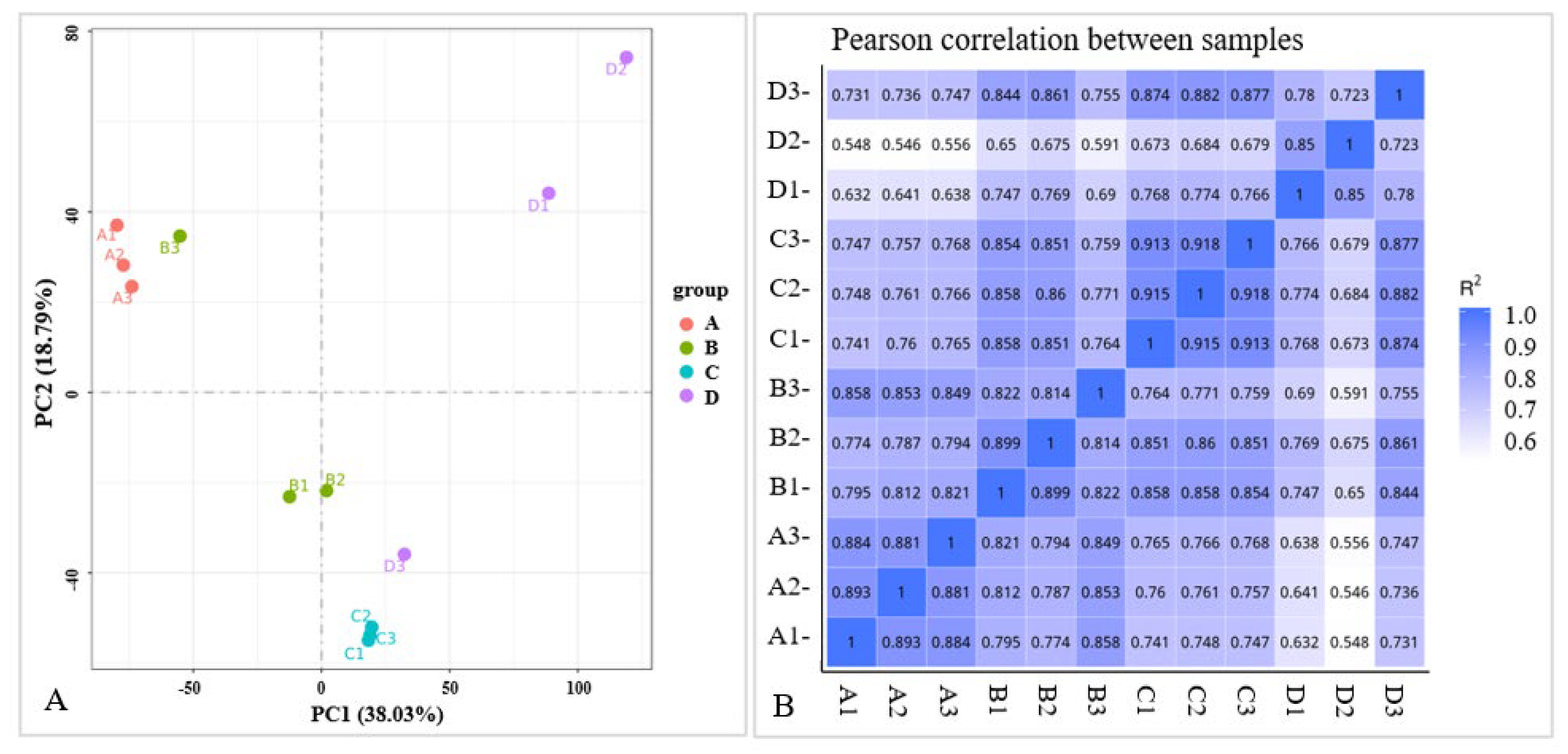
2.2. Transcriptome Data Analysis
2.2.1. RNA Quality Determination and Sequencing Results
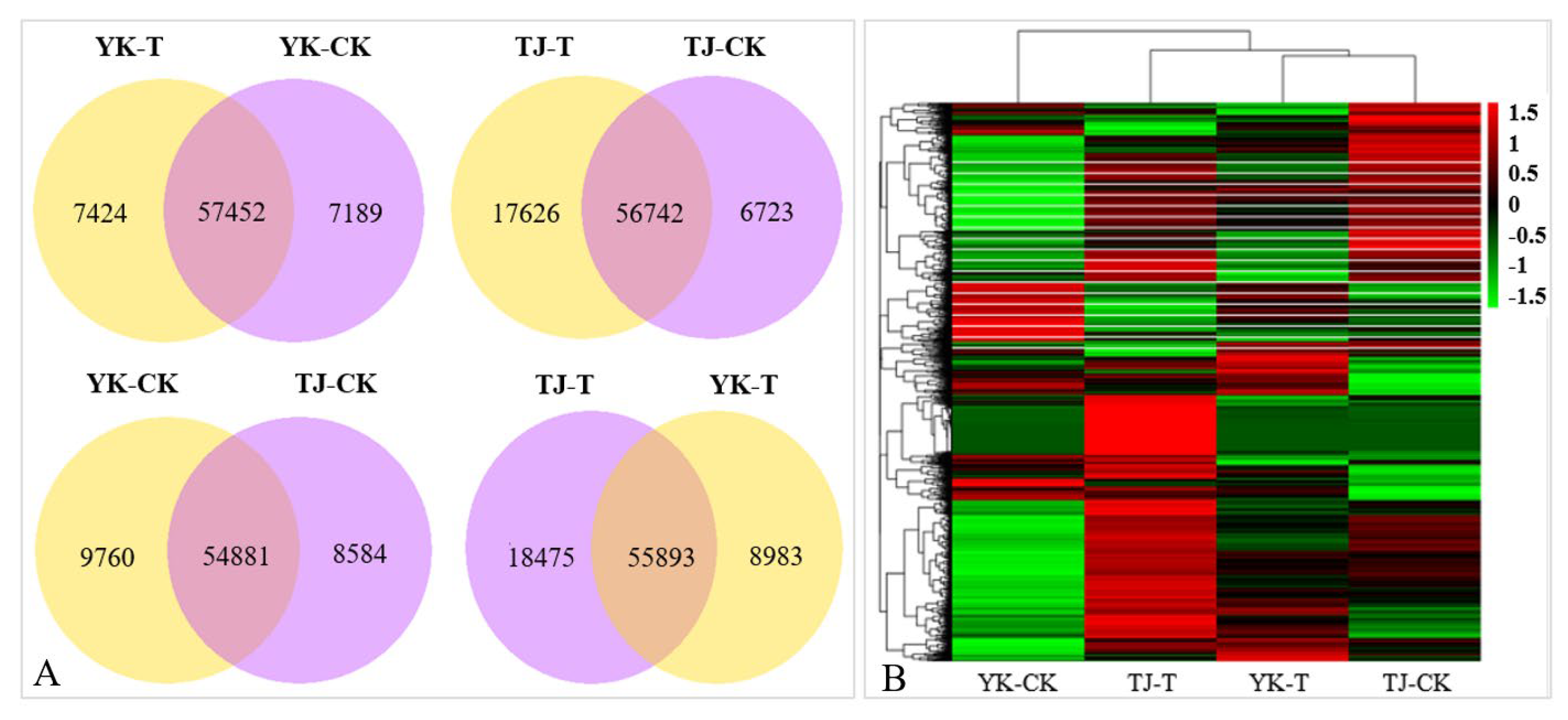
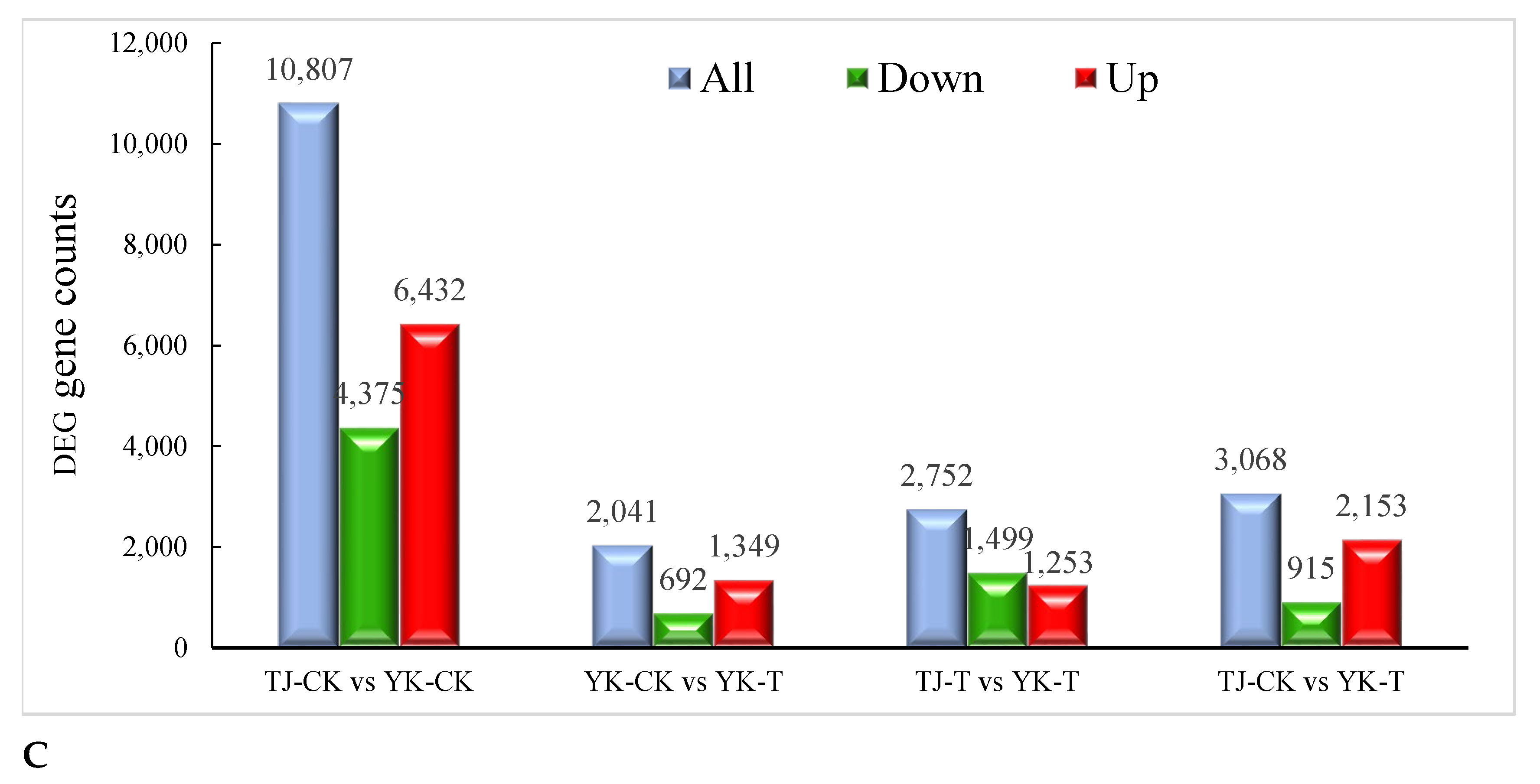
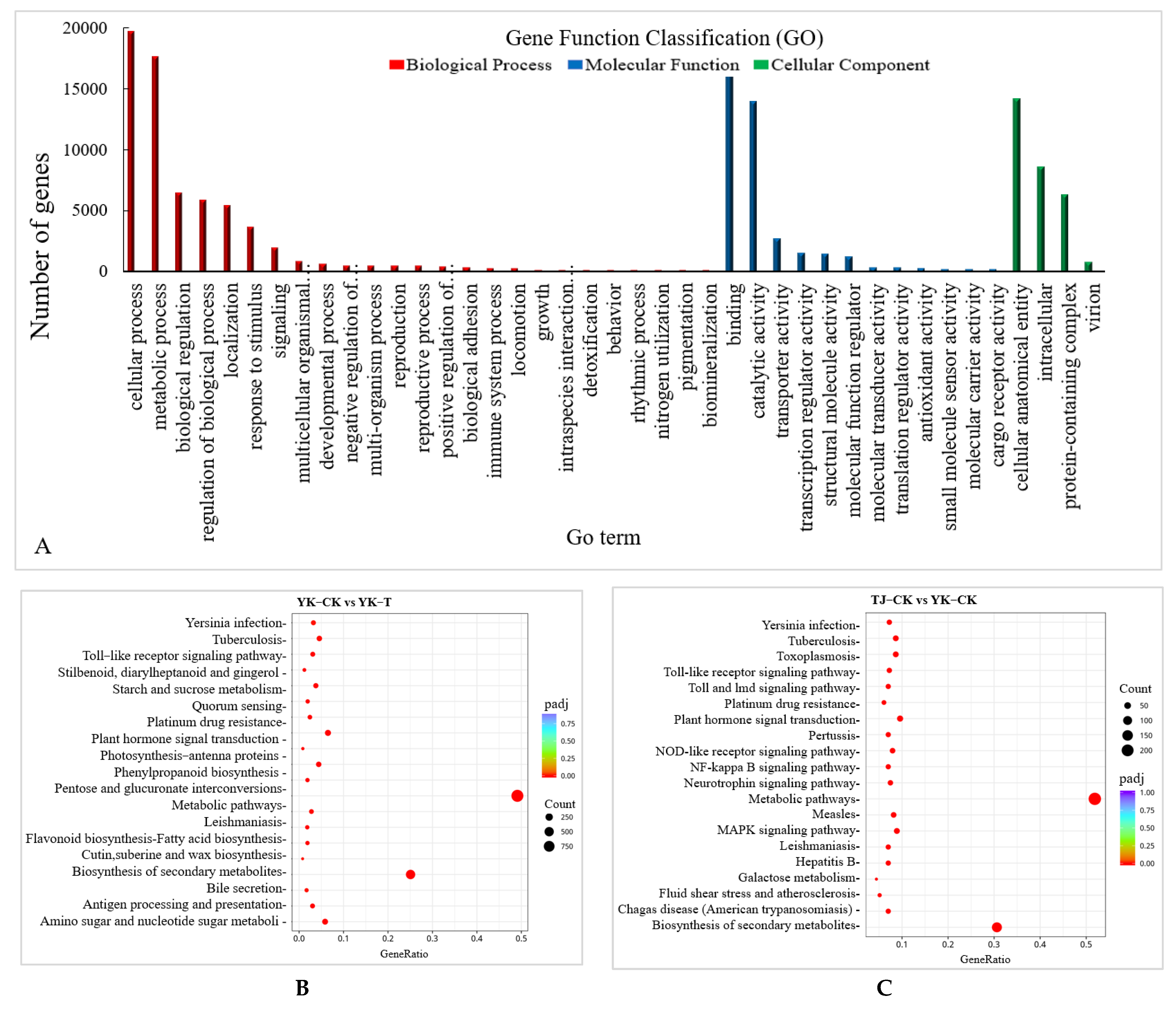
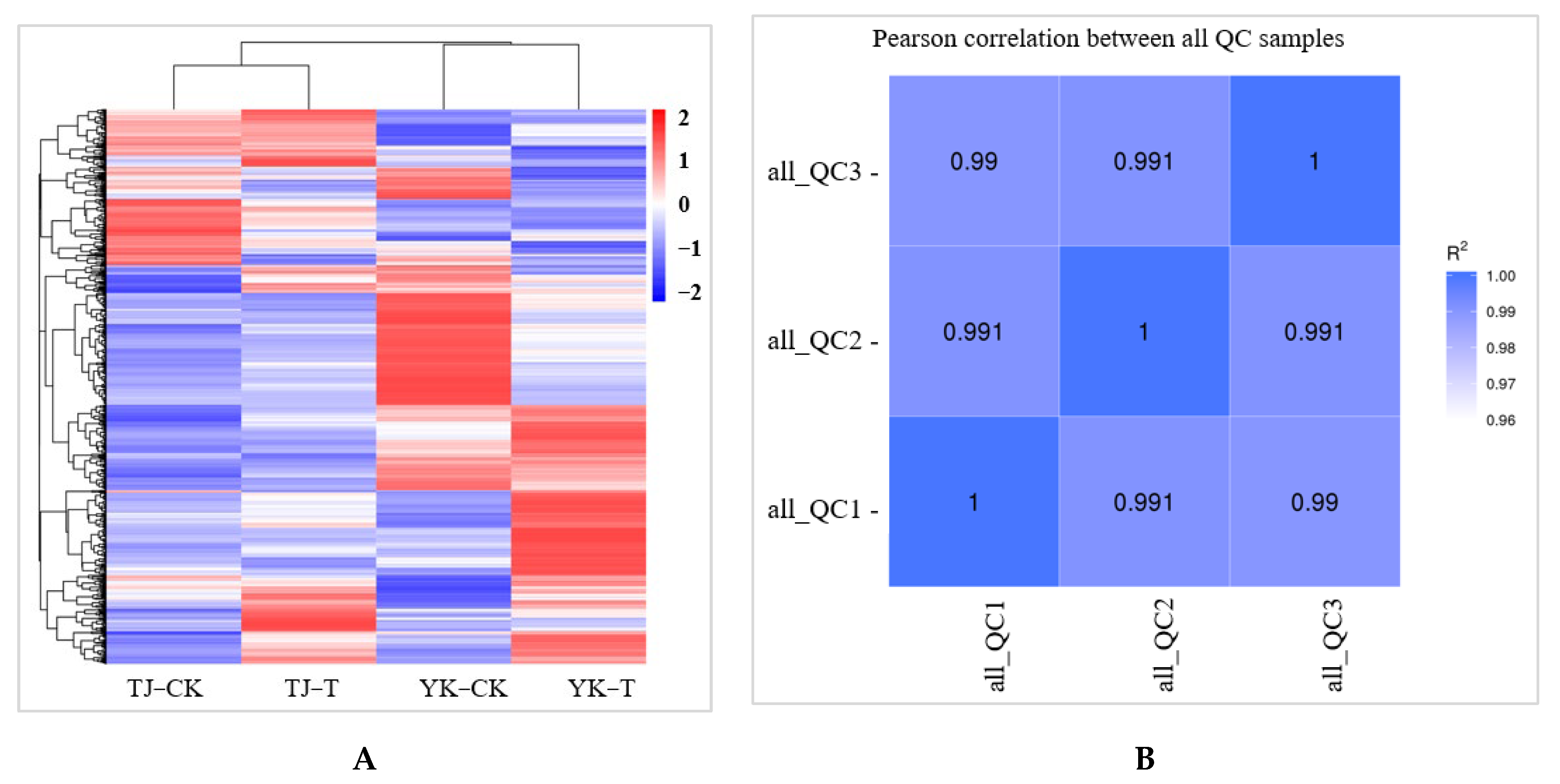
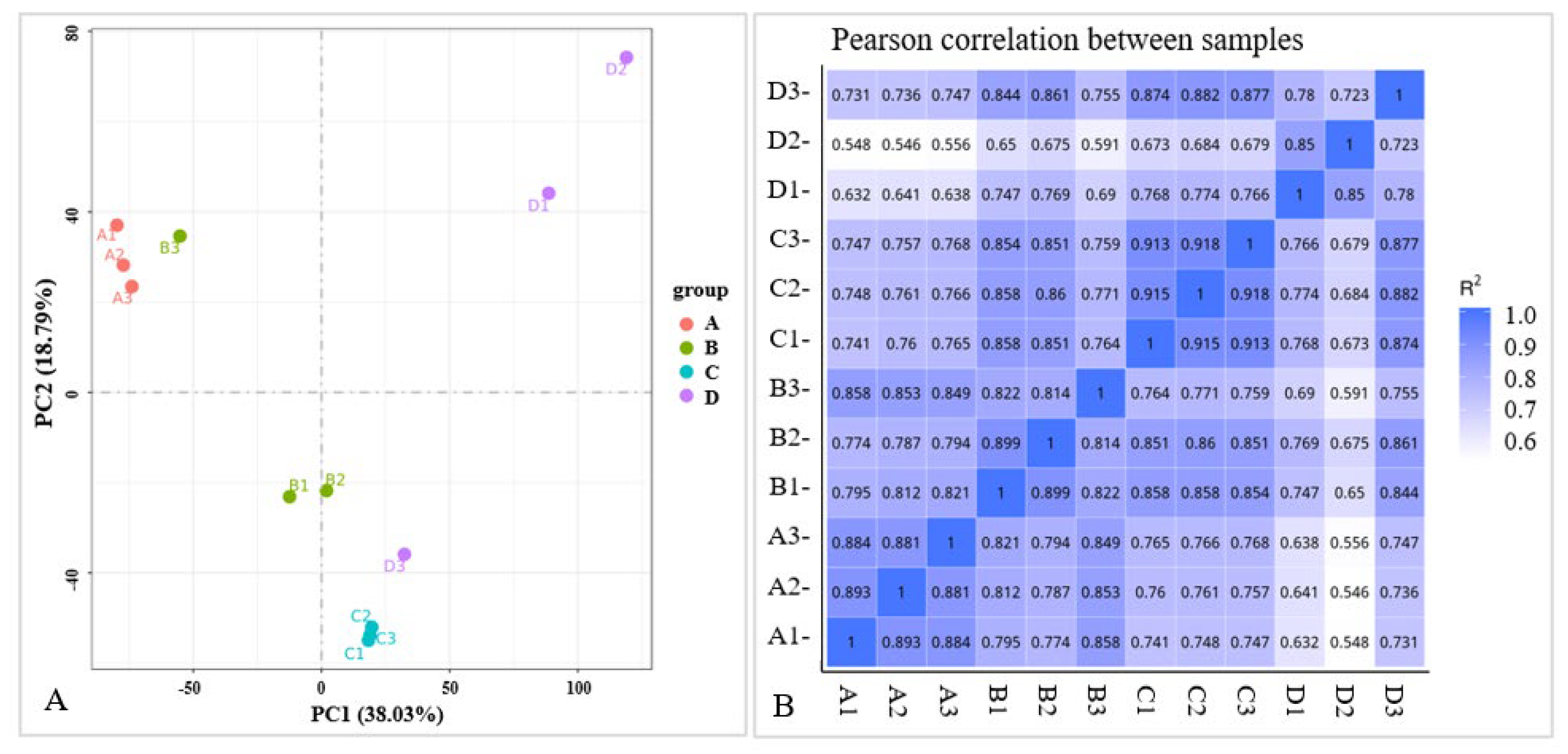
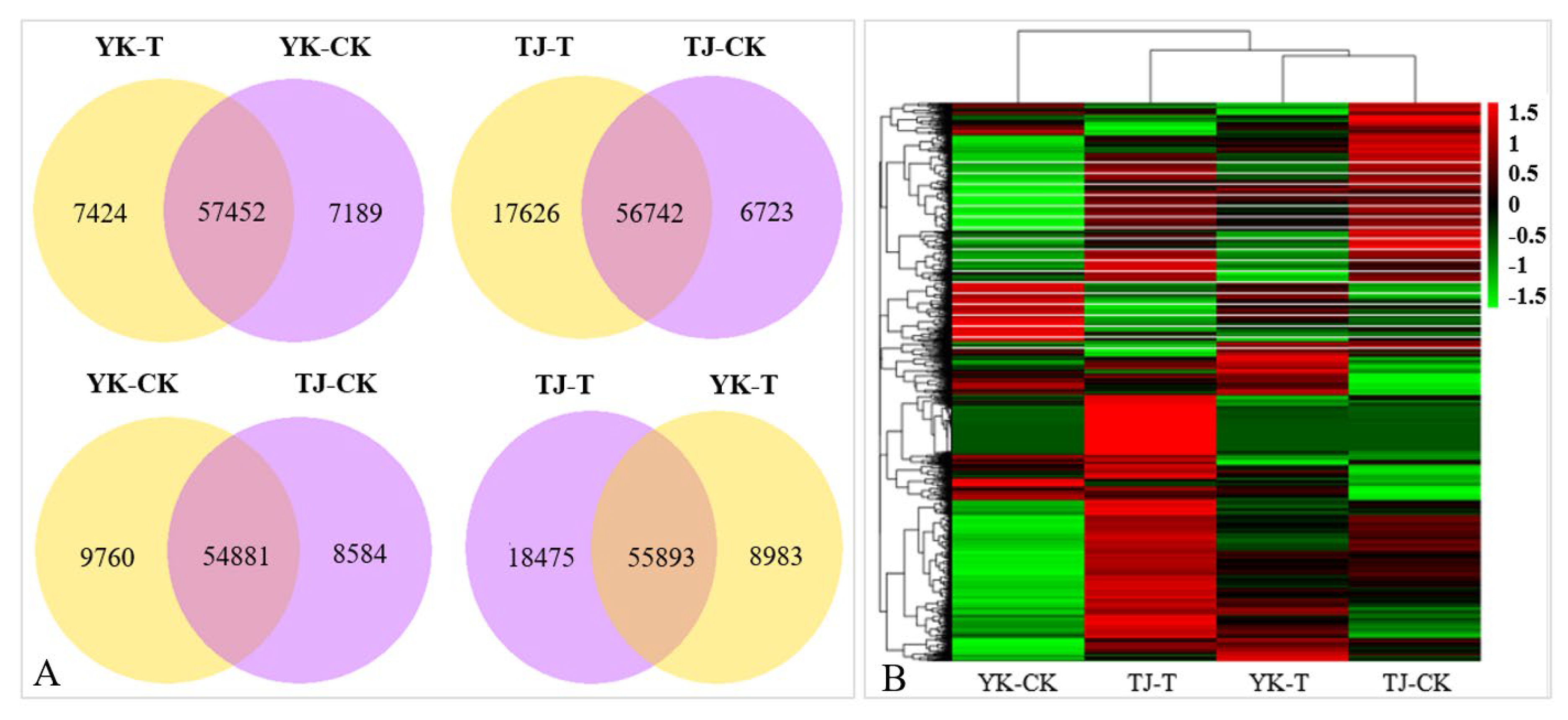
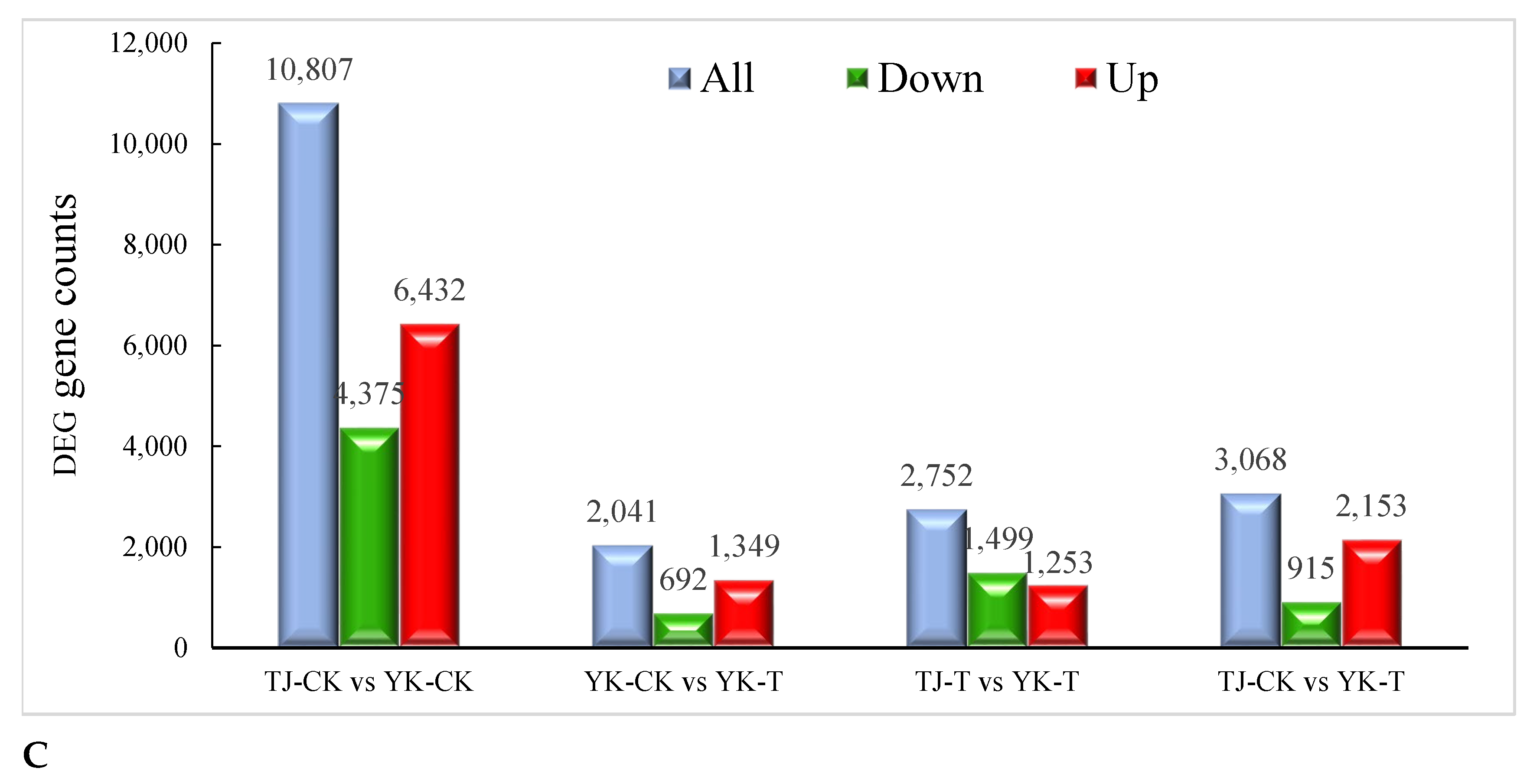
2.2.2. Transcriptome-Wide Identification of DEGs and Functional Analysis
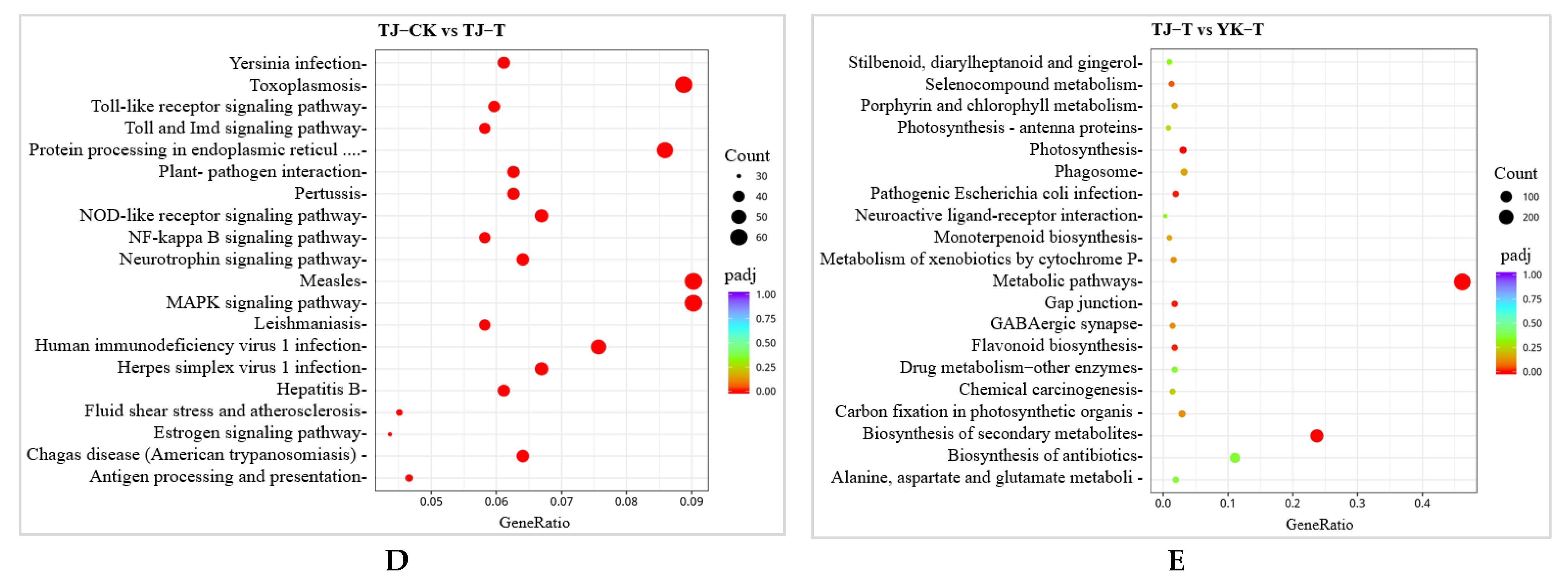
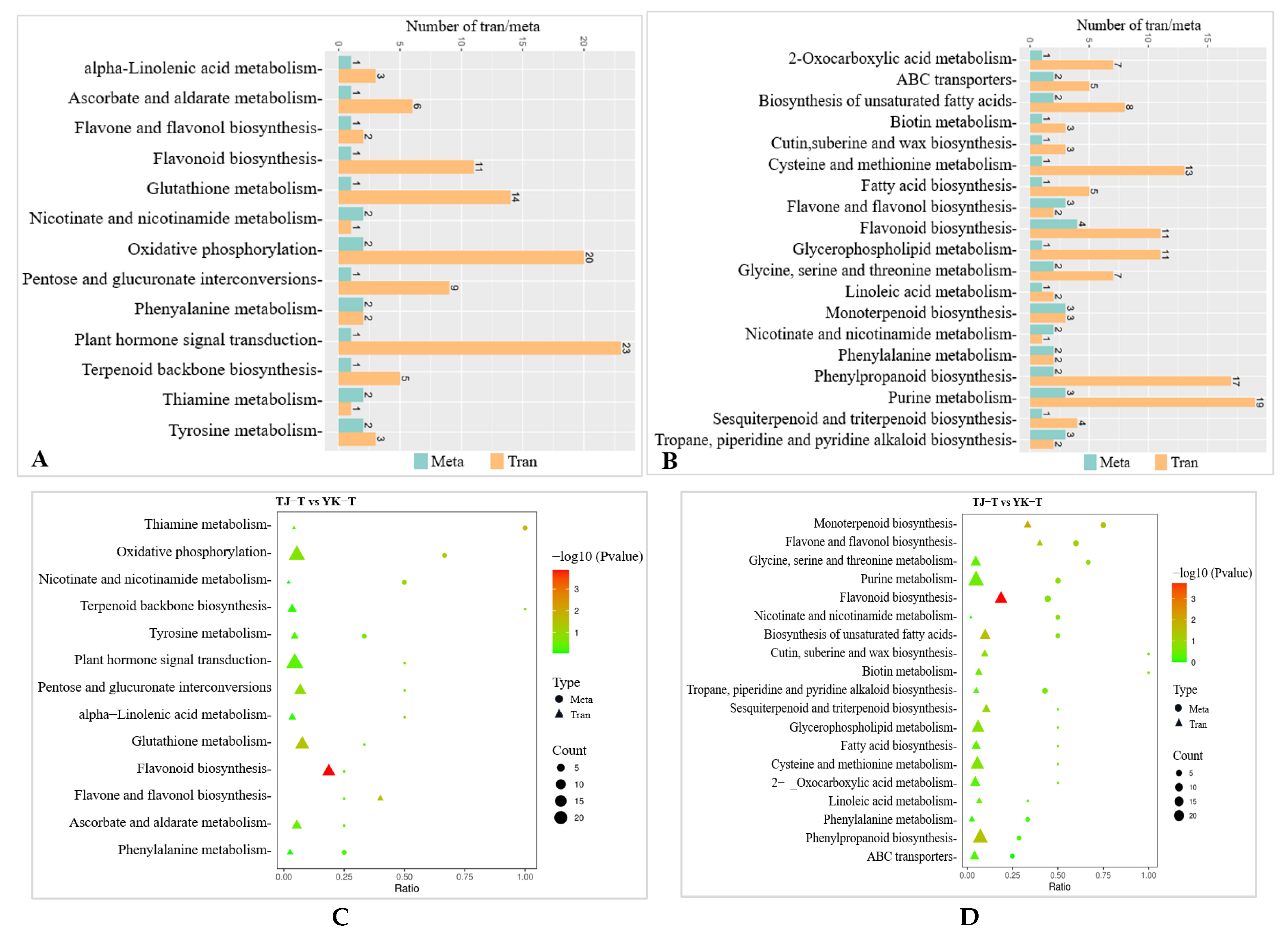
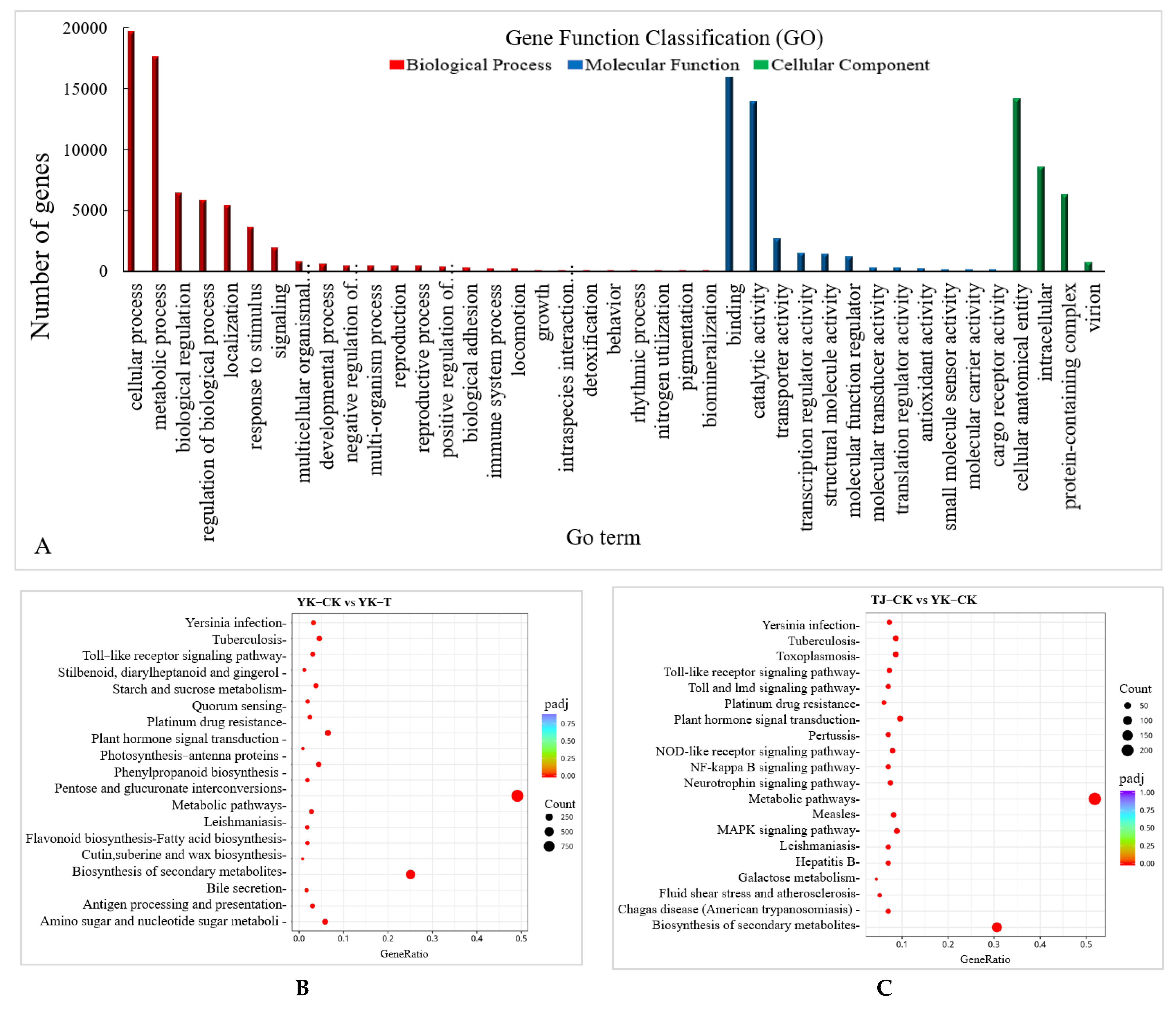
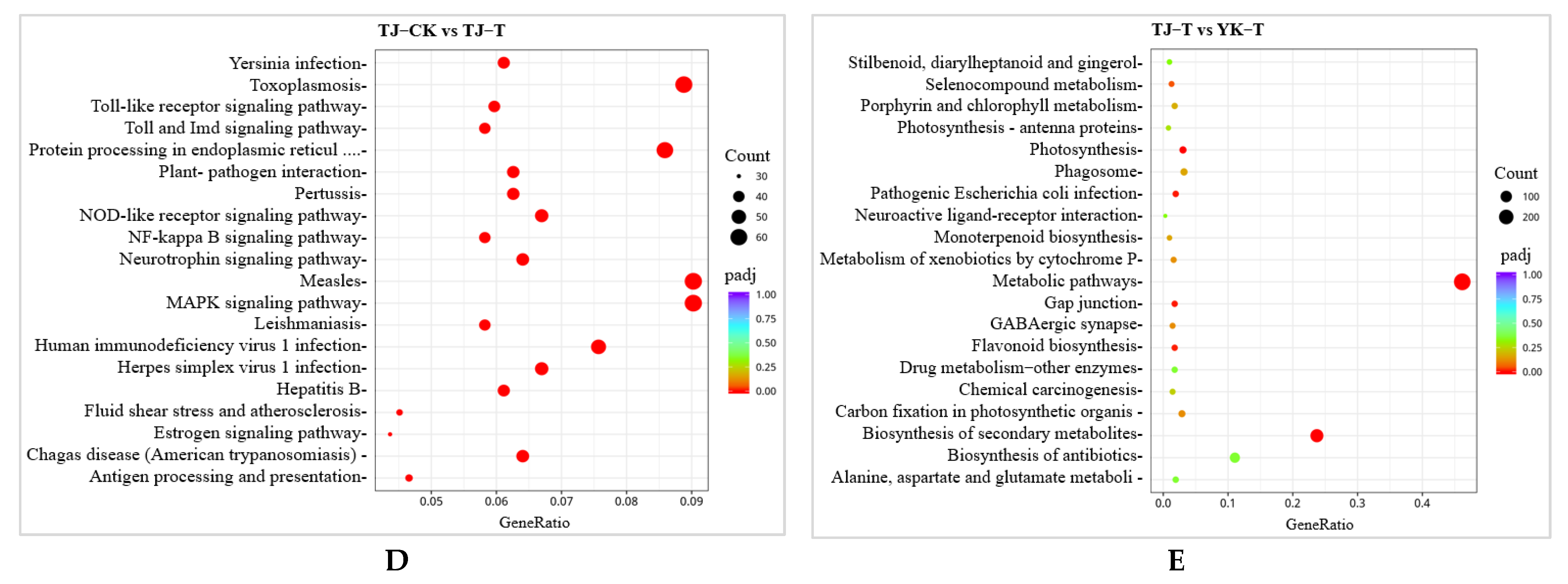
2.2.3. GO and KEGG Pathway Enrichment Analysis of DEGs in ‘Tengjiao’ and ‘Youkang’ Infected with C. zanthoxyli
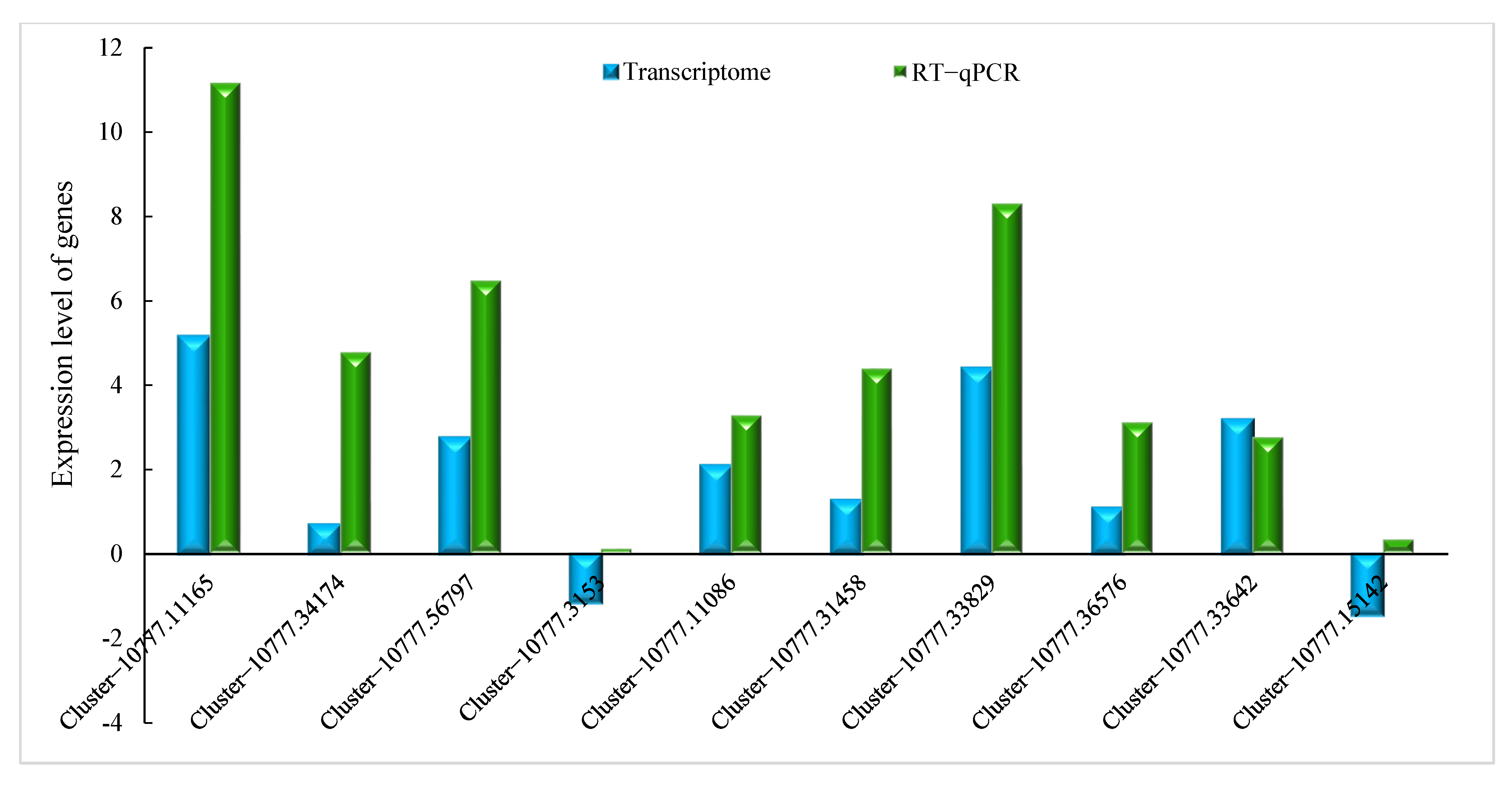
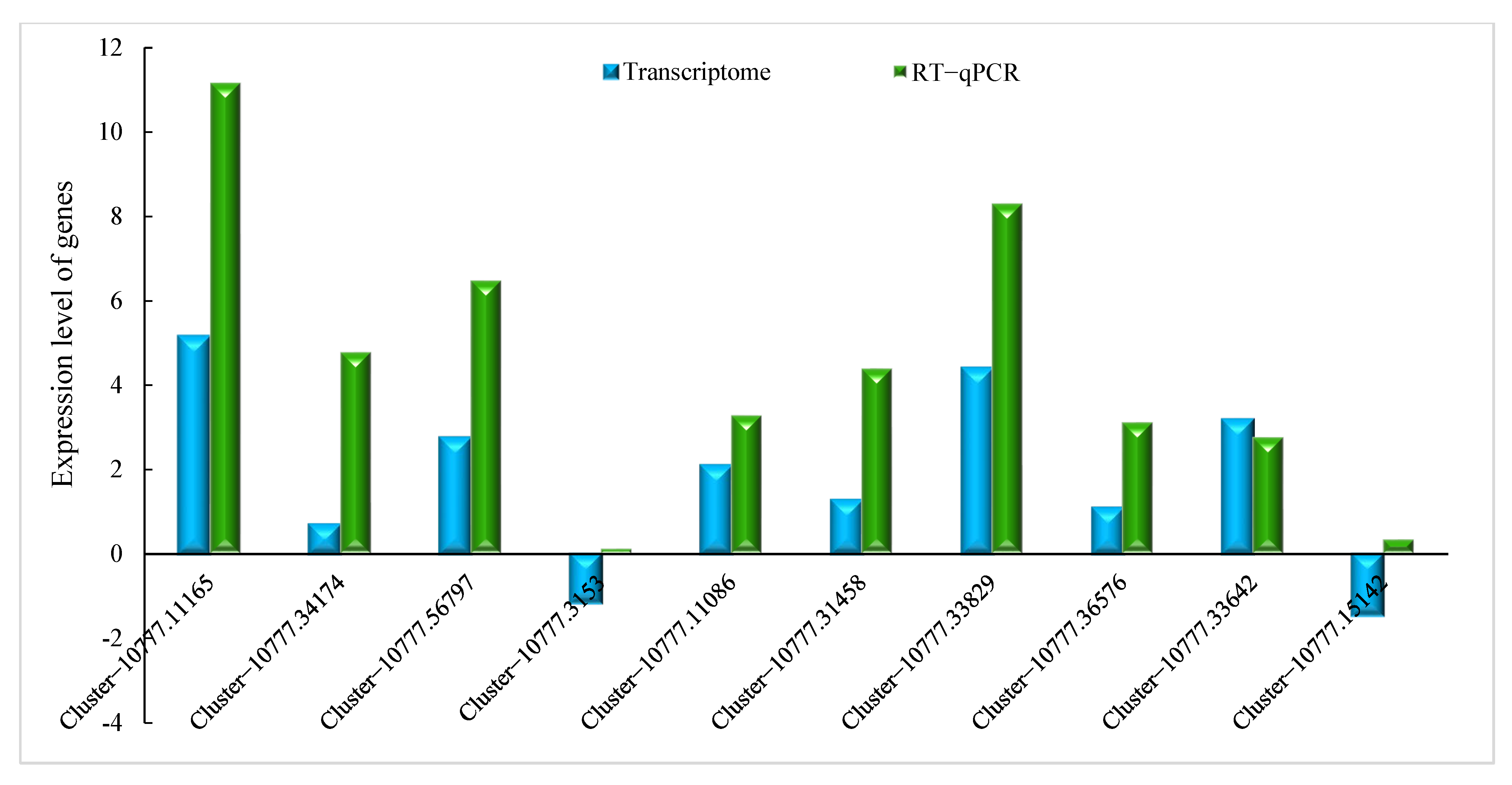
2.2.4. Verification by qRT-PCR
2.3. Metabolome Data Analysis
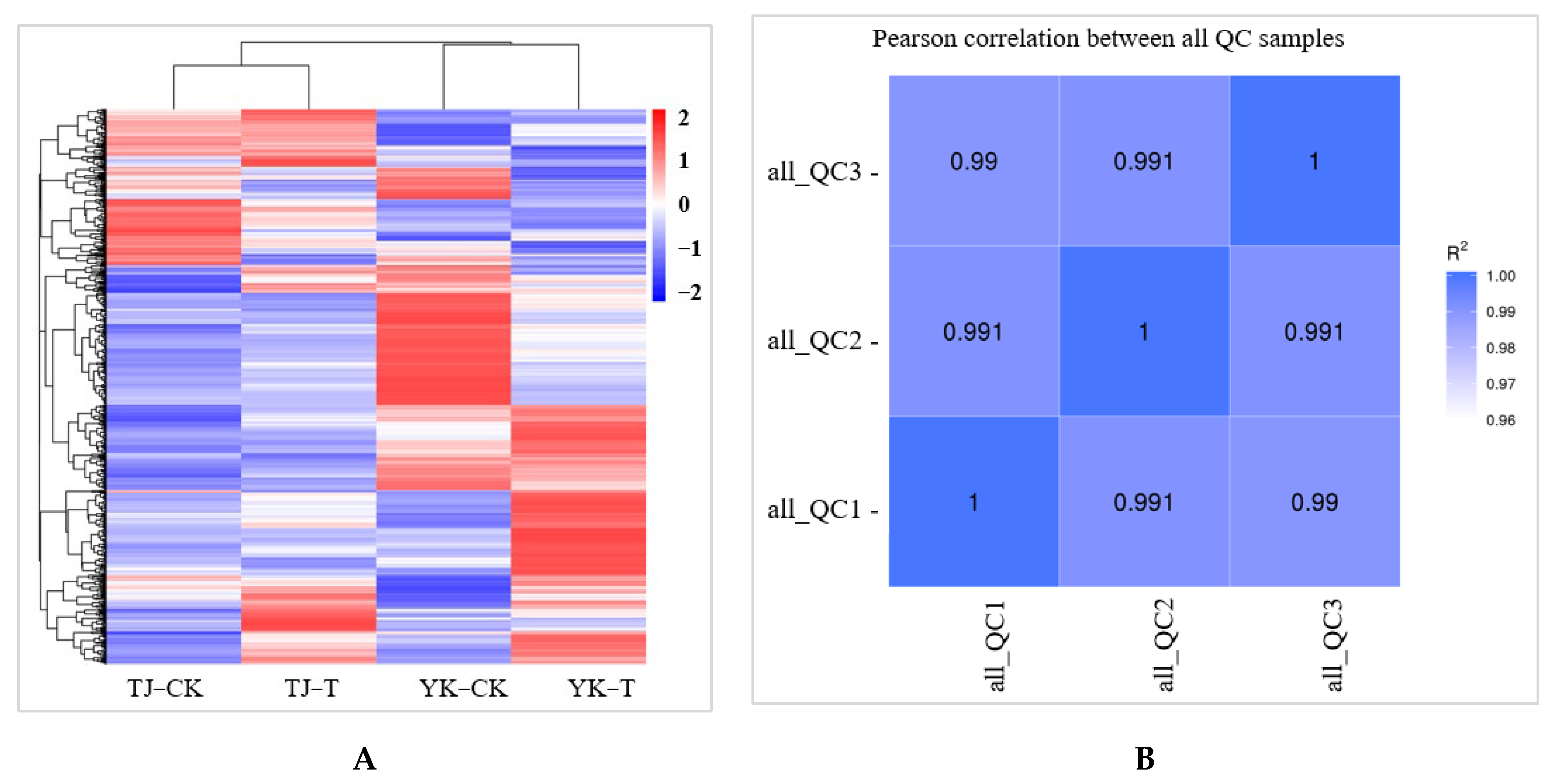
2.3.1. Data Quality Control
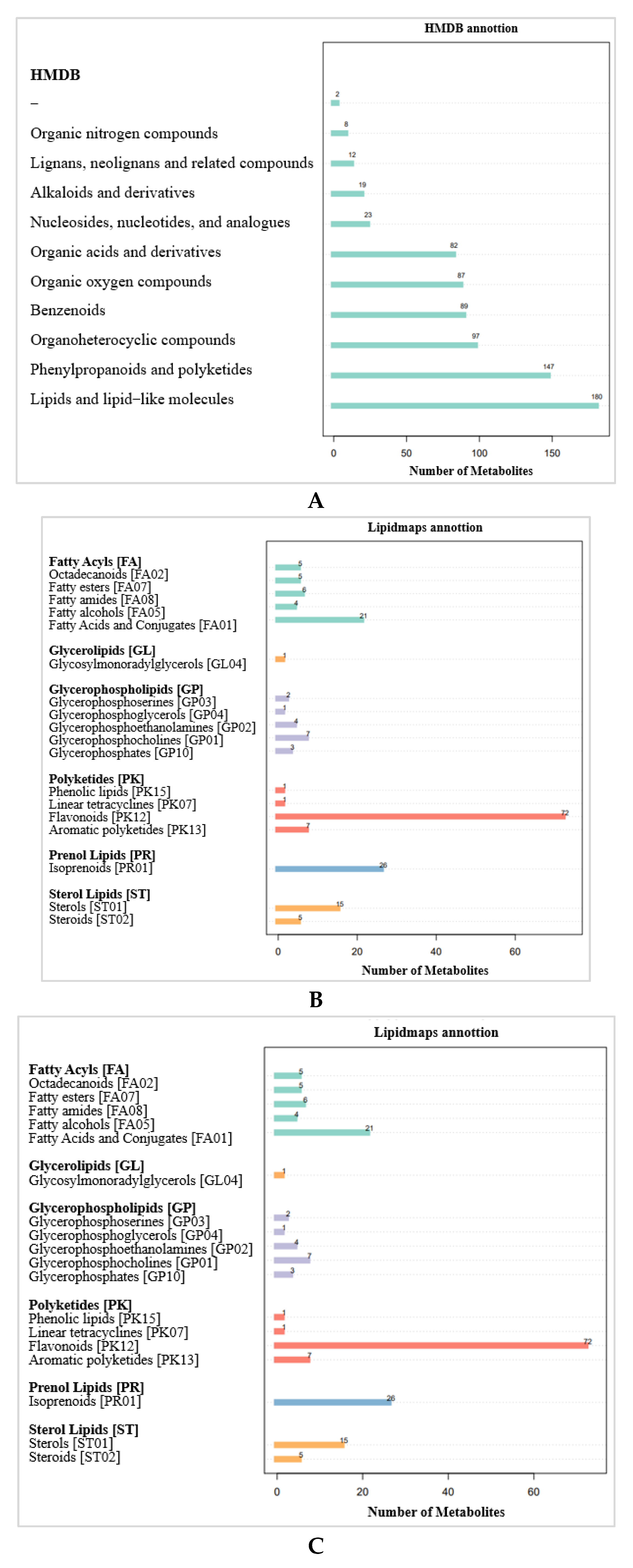
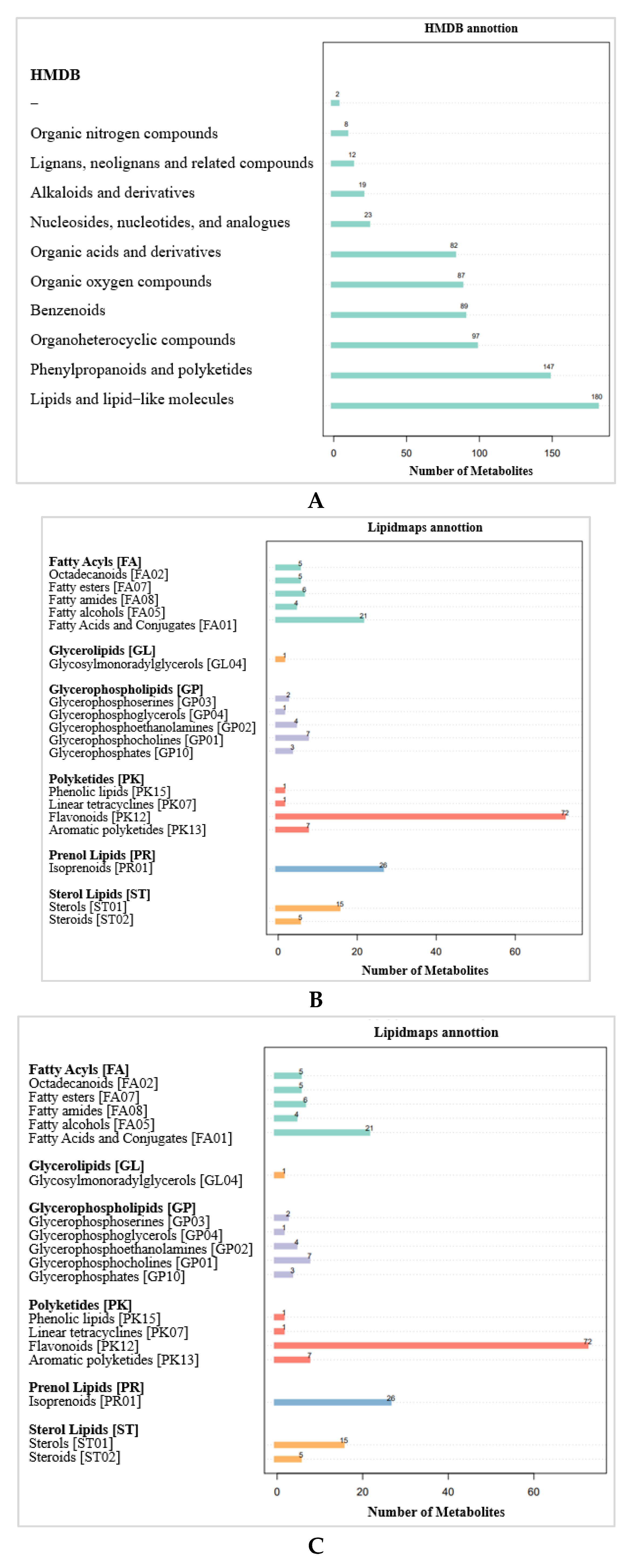
2.3.2. Metabolite Pathways and Classification Notes
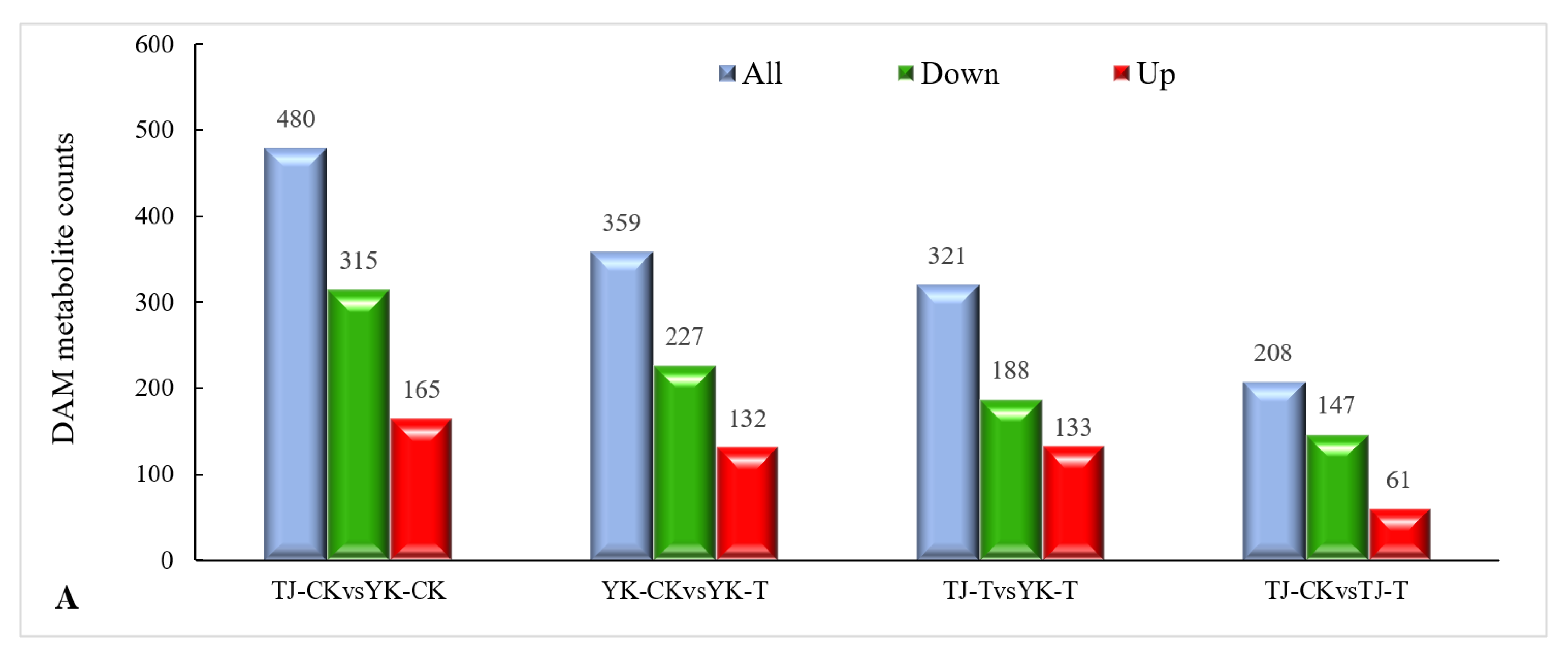
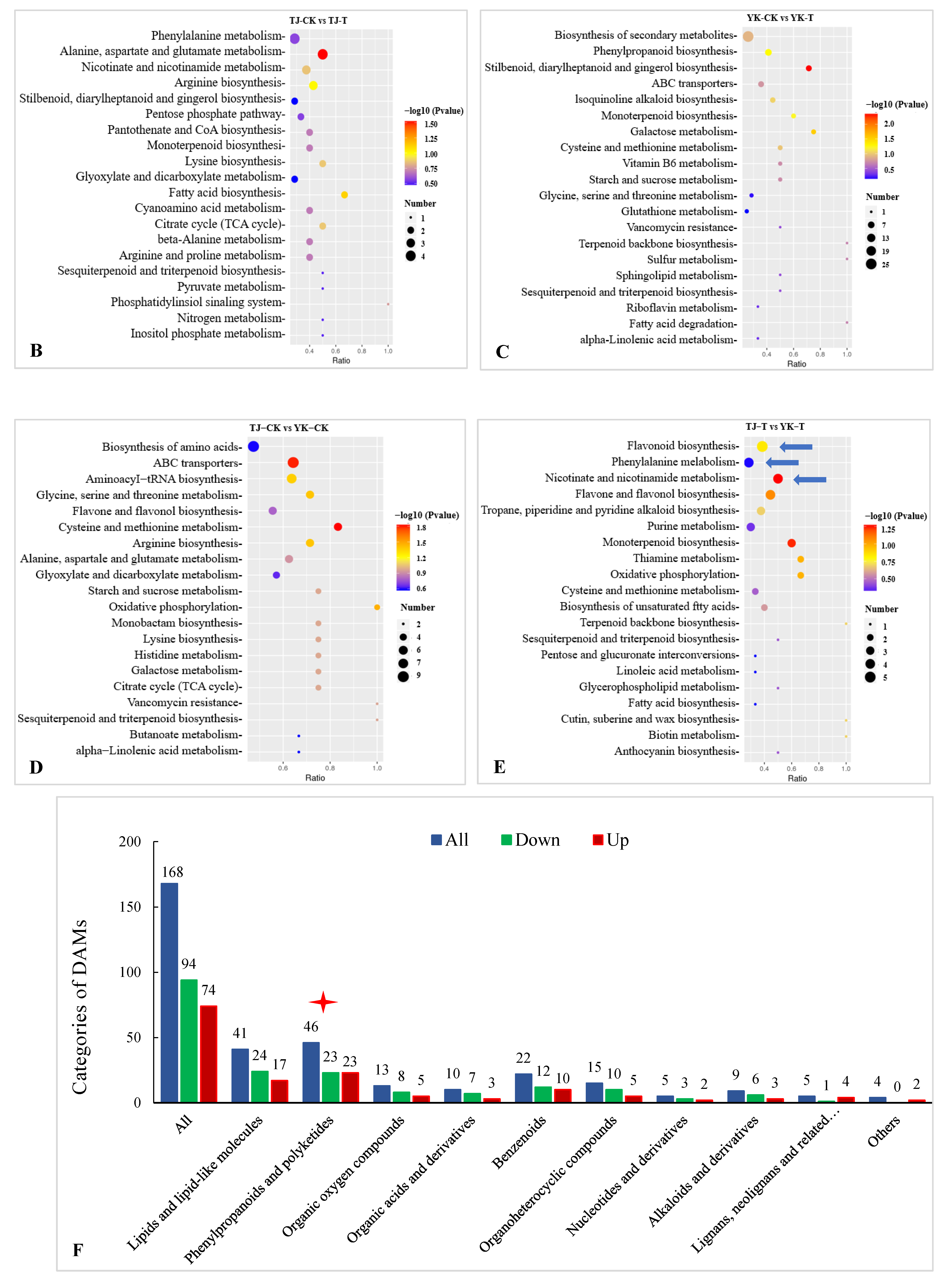
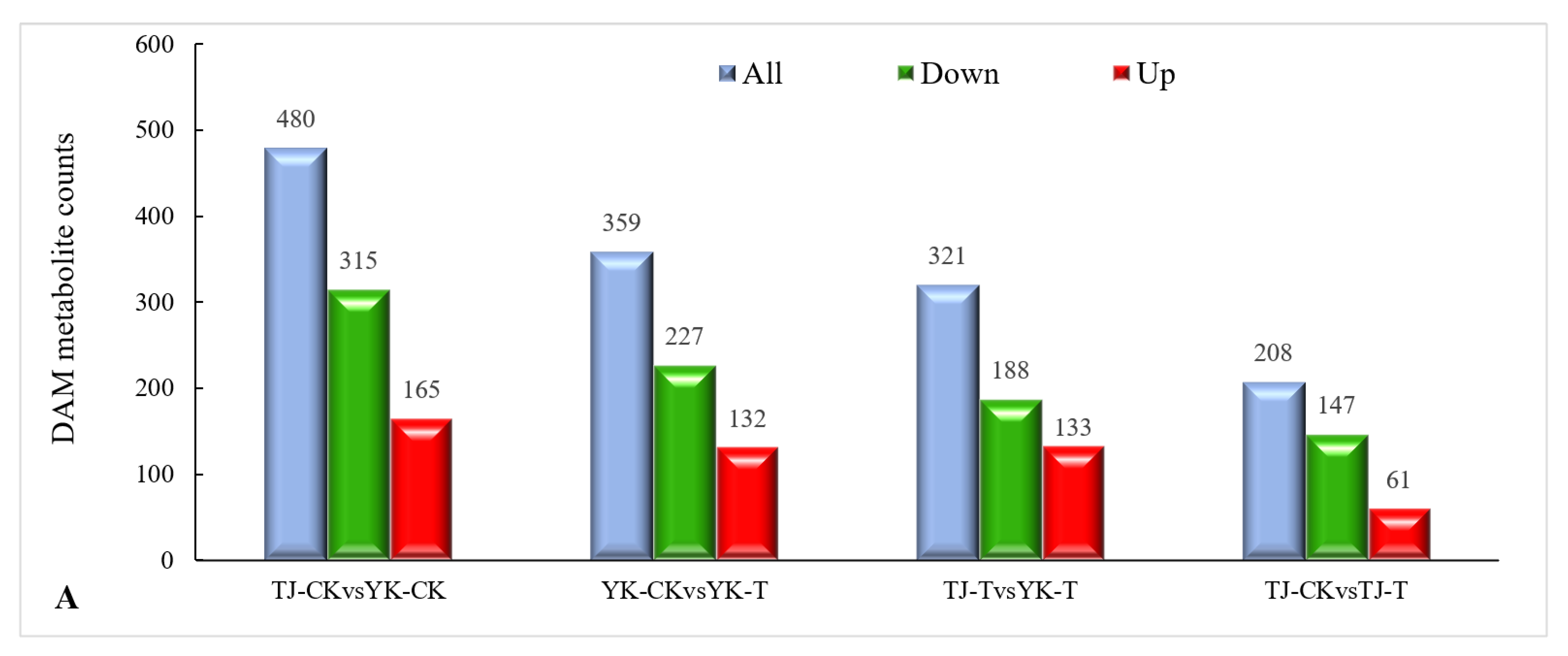
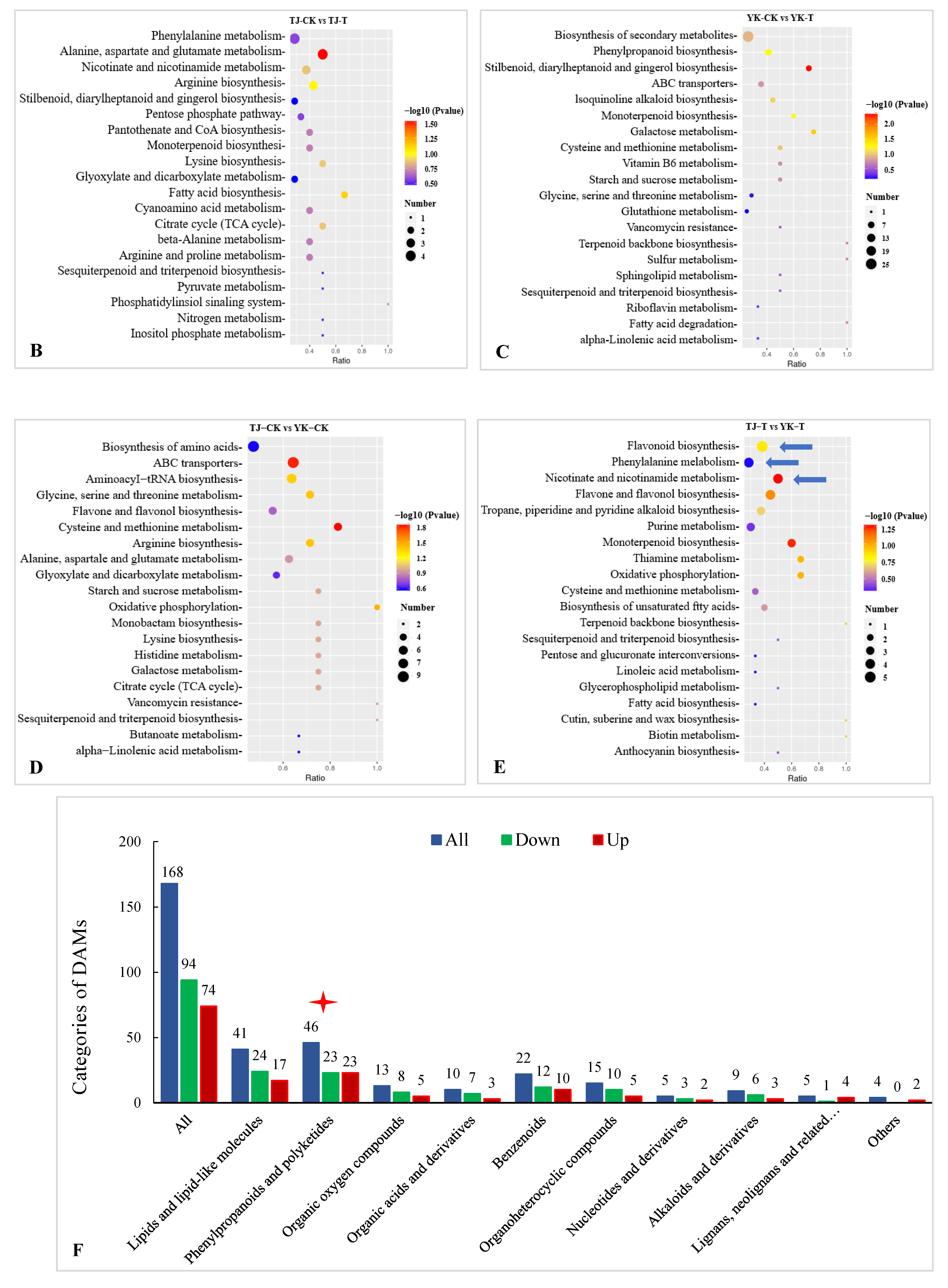
2.3.3. DAM Screening and KEGG Pathway Annotation
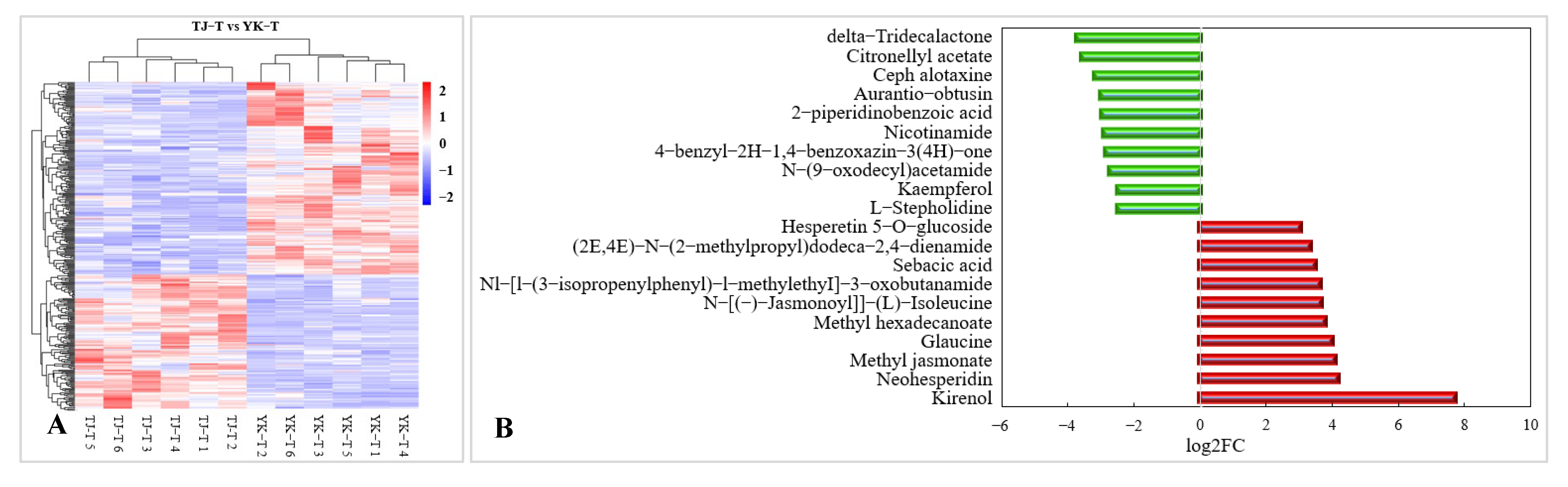
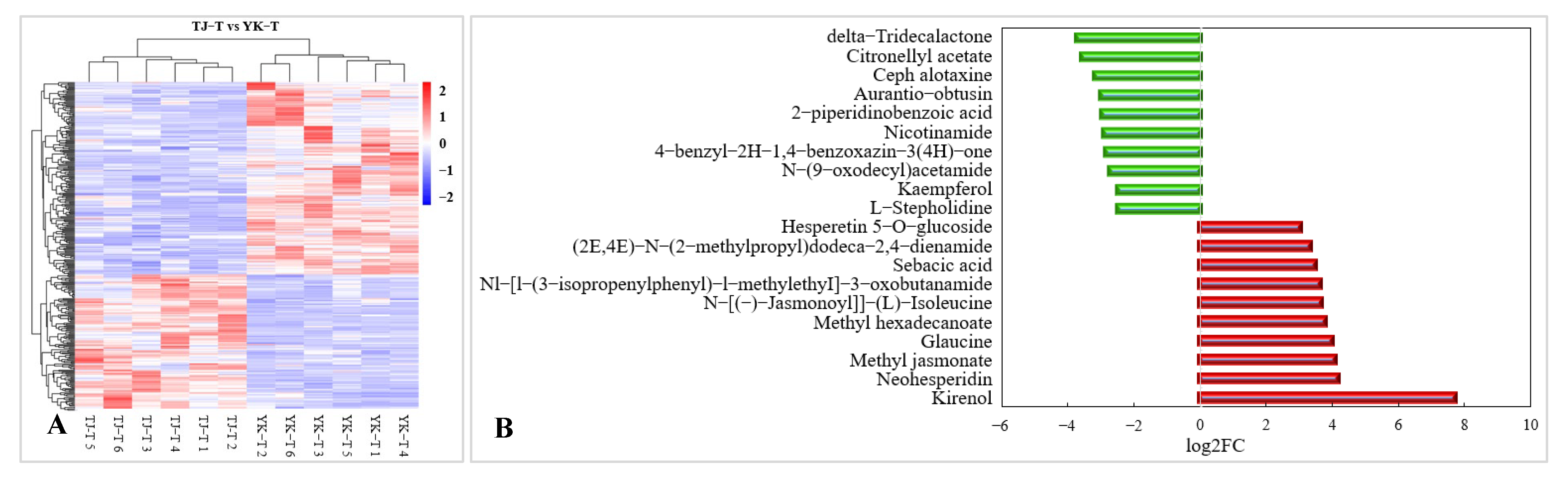
2.3.4. Metabolomic Analysis of TJ and YK Infected by C. zanthoxyli
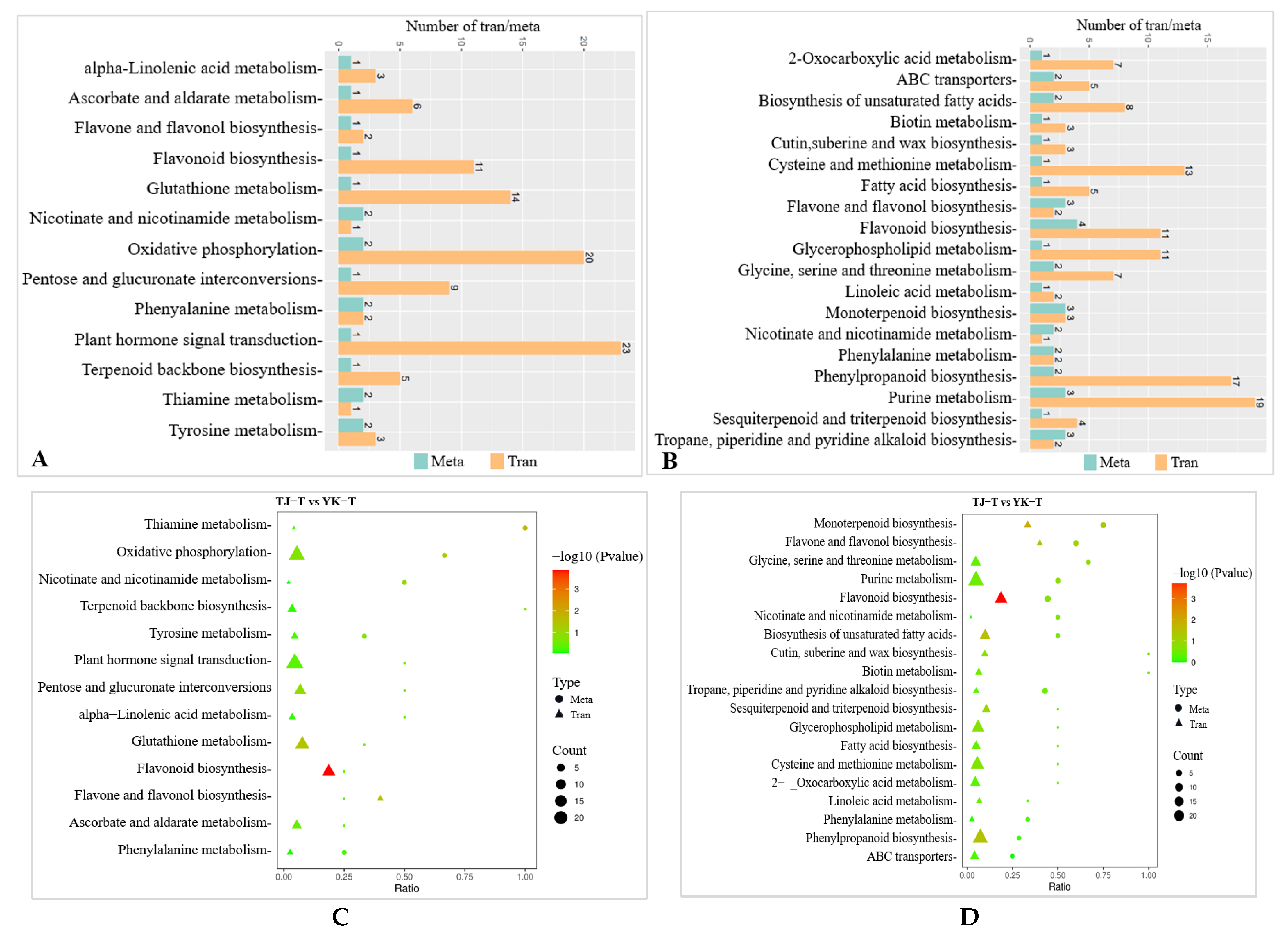
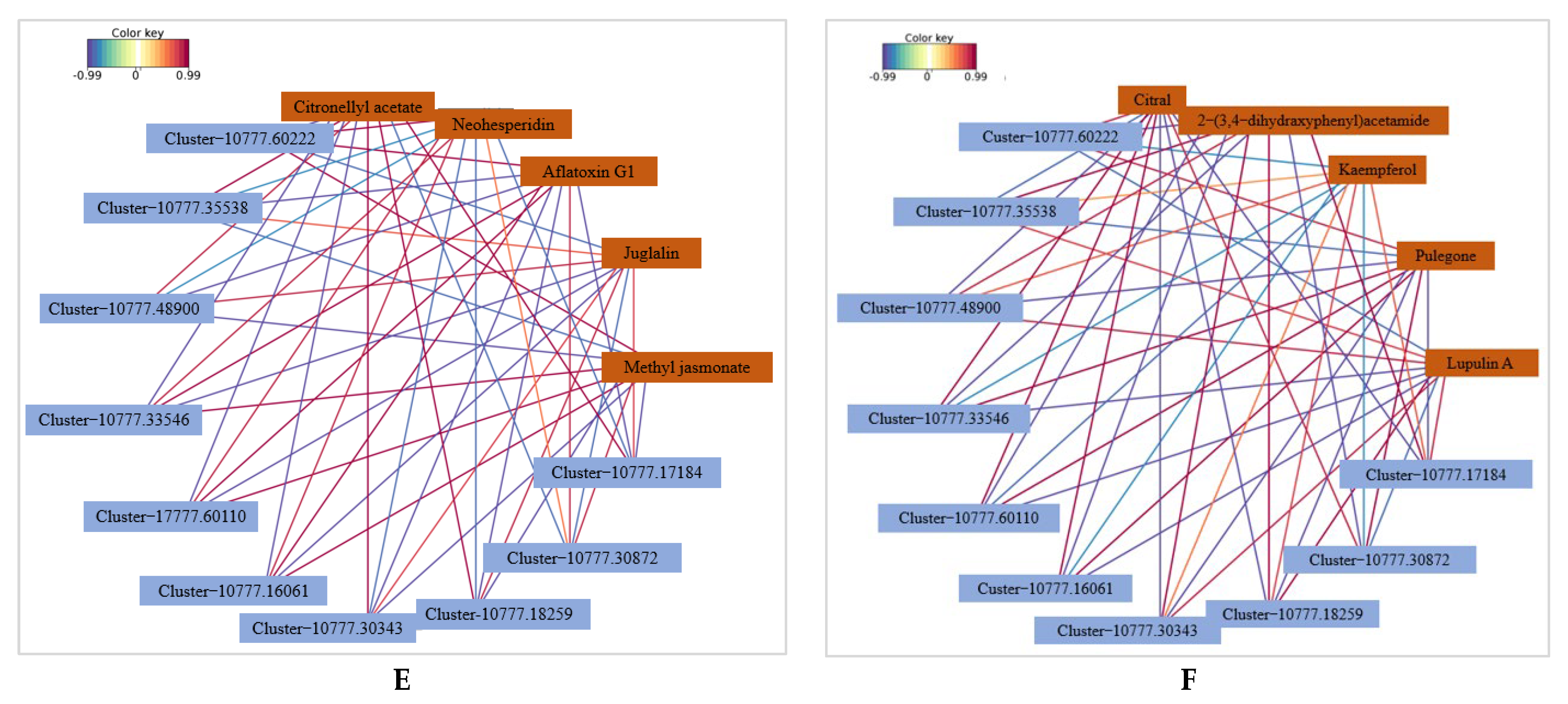
2.3.5. Transcriptome and Metabolome Association Analysis
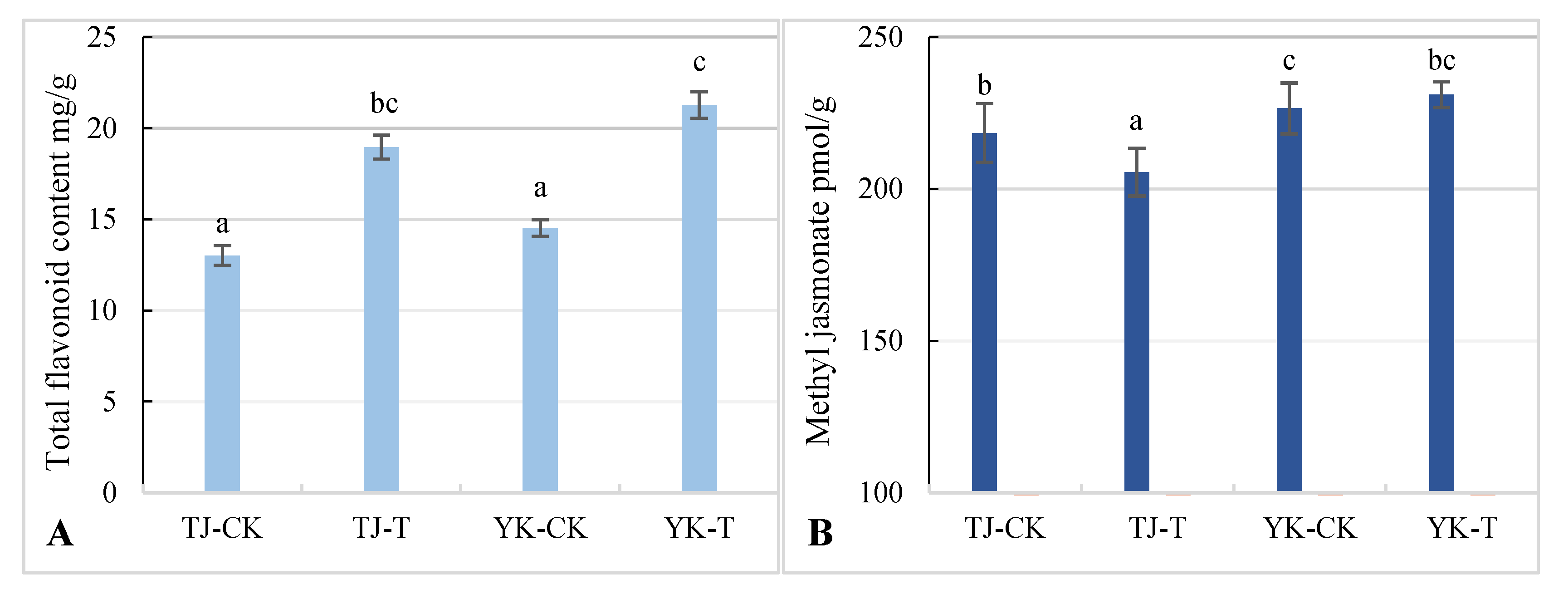
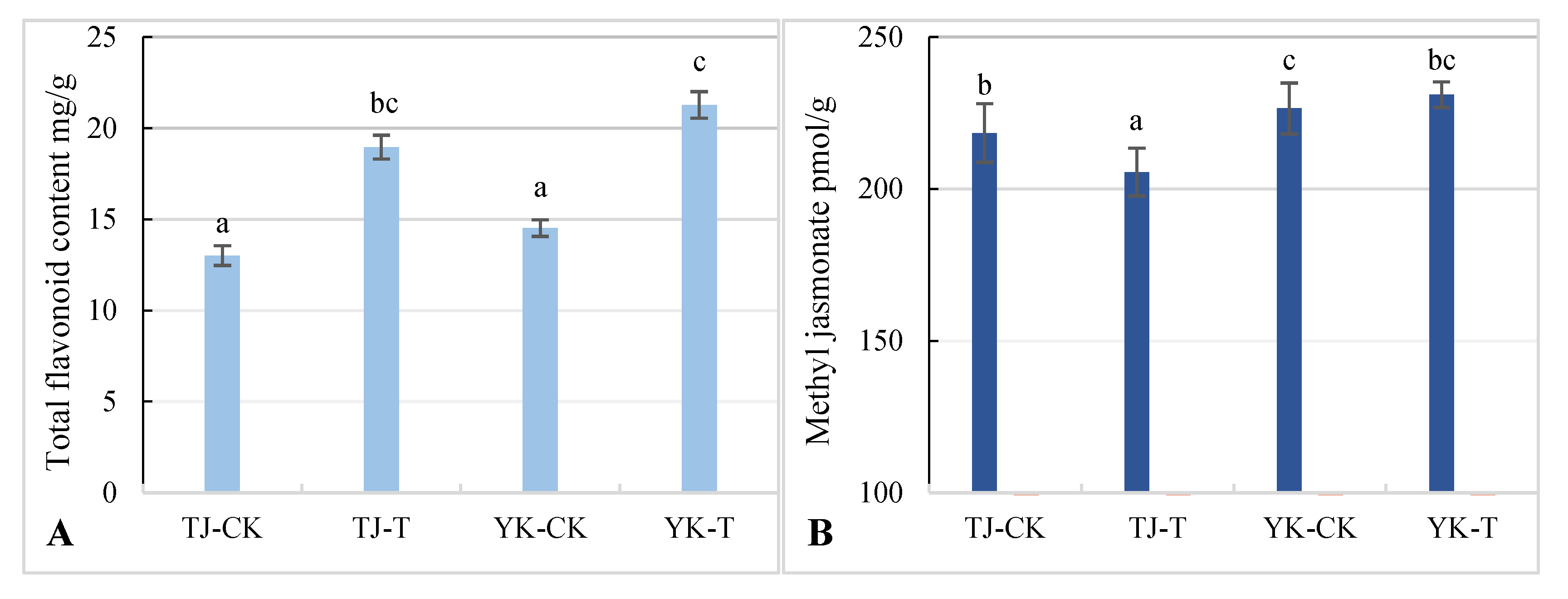
2.4. Differences in Flavonoid and Methyl Jasmonate Content in Z. armatum Leaves
3. Discussion
3.1. DEGs in Z. armatum Leaves Responsive to C. zanthoxyli Infection
3.2. Metabolome Analysis Reveals the Potential Metabolites
3.3. Integrated Analysis of the Transcriptome and Metabolome Reveals the Potential Disease Resistance Mechanism
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. Inoculation of Chinese Pepper Leaves with C. zanthoxyli
4.3. Transcriptome Sequencing and Data Analysis
4.4. Metabolite Extraction and Detection
4.5. Data Analysis/Widely Targeted Metabolome Analysis
4.6. Correlation Analysis of Transcriptomic and Metabolomic Data
4.7. qRT-PCR Analysis
4.8. Quantitation of Total Flavonoid Content and Methyl Jasmonate in Z. armatum Leaves
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wang, M.; Tong, S.; Max, T.; Xi, Z.; Liu, J. The chromosome-level genome assembly of Sichuan pepper provides insights into apomixis, drought tolerance, and alkaloidbio synthesis. Mol. Ecol. Resour. 2021, 21, 2533–2545. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Liu, Z.; Cheng, J.; Li, Z.; Tian, L.; Liu, M.; Yang, T.; Liu, Y.; Liu, Y.; Dai, H.; et al. Zanthoxylum-specific whole genome duplication and recent activity of transposable elements in the highly repetitive paleotetraploid Z. bungeanum genome. Hortic. Res. 2021, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhou, M.; He, H.; Zhang, C.; Zou, X.; He, B.; Yuan, S. On the occurrence of economic plant rusts and its pathogenic factors in the four areas of northeast sichuan. J. China West Norm. Univ. 2017, 38, 365–372. [Google Scholar] [CrossRef]
- Qing, C.Y.; Liang, H.P.; Zhu, T.H. Investigation on Diseases of Zanthoxylum bungeanum in Sichuan Province. World J. For. 2020, 9, 26–34. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.; Pretorius, Z.; Hammond-Kosack, K.; Di Pietro, A.; Spanu, P.; Rudd, J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Lorrain, C.; Marchal, C.; Hacquard, S.; Delaruelle, C.; Pétrowski, J.; Petre, B.; Hecker, A.; Frey, P.; Duplessis, S. The rust fungus Melampsora larici-populina expresses a conserved genetic program and distinct sets of secreted protein genes during infection of its two host plants, larch and poplar. Mol. Plant Microbe Interact. 2018, 31, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Deb, D.; Engel, B.A.; Harbor, J.; Hahn, L.; Lim, K.J.; Zhai, T. Investigating potential water quality impacts of fungicides used to combat soybean rust in indiana. Water Air Soil Pollut. 2010, 207, 273–288. [Google Scholar] [CrossRef]
- Horsfield, P.A.; Wicks, B.T.; Wilson, D. Field evaluation of fungicides for the control of rust, brown rot, shot hole and scab in almonds. Australas. Plant Pathol. 2010, 39, 112–119. [Google Scholar] [CrossRef]
- Hawkins, N.J.; Bass, C.; Dixon, A.; Neve, P. The evolutionary origins of pesticide resistance. Biol. Rev. 2019, 94, 135–155. [Google Scholar] [CrossRef]
- Westwood, J.H.; Charudattan, R.; Duke, S.O.; Fennimore, S.A.; Marrone, P.; Slaughter, D.C. Weed management in 2050: Perspectives on the future of weed science. Weed Sci. 2018, 66, 275–285. [Google Scholar] [CrossRef]
- Mansfeld, B.; Colle, M.; Kang, Y.; Jones, A.; Grumet, R. Transcriptomic and metabolomic analyses of cucumber fruit peels reveal a developmental increase in terpenoid glycosides associated with age-related resistance to Phytophthora capsici. Hortic. Res. 2017, 4, 17022. [Google Scholar] [CrossRef] [PubMed]
- Sade, D.; Shriki, O.; Cuadros-Inostroza, A.; Tohge, T.; Semel, Y.; Haviv, Y.; Willmitzer, L.; Fernie, A.R.; Czosnek, H.; Brotman, Y. Comparative metabolomics and transcriptomics of plant response to Tomato yellow leaf curl virus infection in resistant and susceptible tomato cultivars. Metabolomics 2015, 11, 81–97. [Google Scholar] [CrossRef]
- Ullah, R.; Akhtar, K.P.; Moffett, P.; Shah, T.A.; Hussain, M.; Khan, M.K.R.; Mansoor, S.; Saeed, M. Analysis of the resistance of Gossypium herbaceum to cotton leaf curl kokhran virus strain burewala and cotton leaf curl multan betasatellite. J. Plant Pathol. 2018, 100, 313–316. [Google Scholar] [CrossRef]
- Feussner, I.; Polle, A. What the transcriptome does not tell—Proteomics and metabolomics are closer to the plants’ patho-phenotype. Curr. Opin. Plant Biol. 2015, 26, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.E.; Kim, J.G.; Fischer, C.R.; Mehta, N.; Cosima, D.S.; Wemmer, K.; Mudgett, M.B.; Sattely, E. A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell 2020, 180, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Jedrzej, S.; Samuel, B.; Sayantan, P.; Prashant, S.; Pablo, D.C.; Justin, L.; Avinash, K.; Nir, S.; Sagit, M.; Arnaud, B.; et al. Analysis of wild tomato introgression lines elucidates the genetic basis of transcriptome and metabolome variation underlying fruit traits and pathogen response. Nat. Genet. 2020, 52, 1111–1121. [Google Scholar] [CrossRef]
- Li, P.Q.; Ruan, Z.; Fei, Z.X.; Yan, J.J.; Tang, G.H. Integrated Transcriptome and Metabolome Analysis Revealed That Flavonoid Biosynthesis May Dominate the Resistance of Zanthoxylum bungeanum against Stem Canker. J. Agric. Food Chem. 2021, 69, 6360–6378. [Google Scholar] [CrossRef]
- Kim, D.R.; Jeon, C.W.; Cho, G.; Thomashow, L.S.; Weller, D.M.; Paik, M.J.; Lee, Y.B.; Kwak, Y.S. Glutamic acid reshapes the plant microbiota to protect plants against pathogens. Microbiome 2021, 9, 244. [Google Scholar] [CrossRef]
- Li, S.N.; Lin, D.X.; Zhang, Y.W.; Deng, M.; Chen, Y.X.; Lv, B.; Li, B.S.; Lei, Y.; Wang, Y.P.; Zhao, L.; et al. Genome-edited powdery mildew resistance in wheat without growth penalties. Nature 2022, 602, 455–460. [Google Scholar] [CrossRef]
- Dang, J.L.; Horvath, D.M.; Staskawicz, B.J. Pivoting the plant immune system from dissection to deployment. Science 2013, 341, 746–751. [Google Scholar] [CrossRef]
- Stotz, H.U.; Mitrousia, G.K.; de Wit, P.J.; Fitt, B.D. Effector-triggered defence against apoplastic fungal pathogens. Trends Plant Sci. 2014, 19, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tang, C.L.; Fan, X.; He, M.Y.; Gan, P.F.; Zhang, S.; Hu, Z.Y.; Wang, X.D.; Yan, T.; Shu, W.X.; et al. lnactivation of a wheat protein kinase gene confersbroad spectrum resistance to rust fungi. Cell 2022, 185, 2961–2974.e19. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhu, L.; Zhou, J.M.; Ronald, P.C.; Li, S.; Li, J.; Chen, X. A single transcription factor promotes both yield and immunity in rice. Science 2018, 7, 361. [Google Scholar] [CrossRef]
- Karki, H.S.; Halterman, D.A. Phytophthora infestans (late blight) infection assay in a detached leaf of potato. Bio-protocol 2021, 11, e3926. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Y.; Zhang, X.X.; Chen, Z.D.; Xia, Y.Q.; Wang, L.; Sun, Y.J.; Zhang, M.M.; Xiao, Y.; Han, Z.F.; et al. Plant receptor-like protein activation by a microbial glycoside hydrolase. Nature 2022, 610, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Garcia, B.J.; Furches, A.; Tuskan, G.A.; Jacobson, D. Interactions of Plants with Bacteria and Fungi: Molecular and Epigenetic Plasticity of the Host. Front. Plant Sci. 2019, 10, 862. [Google Scholar] [CrossRef]
- Yang, Y.X.; Ahammed, G.J.; Wu, C.; Fan, S.Y.; Zhou, Y.H. Crosstalk among jasmonate, salicylate and ethylene signaling pathways in plant disease and immune responses. Curr. Prot. Pept. Sci. 2015, 16, 450–461. [Google Scholar] [CrossRef]
- Boller, T.; He, S.Y. Innate immunity in plants: An arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science 2009, 324, 742–744. [Google Scholar] [CrossRef]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Mu, Q.; Wang, X.; Zhang, J.; Yu, H.; Huang, T.; He, Y.; Dai, S.; Meng, X. Multilayered synergistic regulation of phytoalexin biosynthesis by ethylene, jasmonate, and MAPK signaling pathways in Arabidopsis. Plant Cell 2022, 34, 3066–3087. [Google Scholar] [CrossRef]
- Seshime, Y.; Juvvadi, P.R.; Fujii, I.; Kitamoto, K. Genomic evidences for the existence of a phenylpropanoid metabolic pathway in Aspergillus oryzae. Biochem. Biophys. Res. Commun. 2005, 337, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Stobiecki, M.; Kachlicki, P. Isolation and identification of flavonoids. In The Science of Flavonoids; Grotewold, E., Ed.; Springer: New York, NY, USA, 2006; pp. 47–70. [Google Scholar] [CrossRef]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Watanabe, M.; Hoefgen, R.; Fernie, A.R. The evolution of phenylpropanoid metabolism in the green lineage. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 123–152. [Google Scholar] [CrossRef]
- Brenda, W.S. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Kachroo, A.; Robin, G.P. Systemic signaling during plant defense. Curr. Opin. Plant Biol. 2013, 16, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Takahashi, S.; Waki, T. Formation of flavonoid metabolons: Functional significance of protein-protein interactions and impact on flavonoid chemodiversity. Front. Plant Sci. 2019, 10, 821. [Google Scholar] [CrossRef]
- Xu, D.; Deng, Y.; Xi, P.; Yu, G.; Wang, Q.; Zeng, Q.; Jiang, Z.; Gao, L. Fulvic acid-induced disease resistance to Botrytis cinerea in table grapes may be mediated by regulating phenylpropanoid metabolism. Food Chem. 2019, 286, 226–233. [Google Scholar] [CrossRef]
- Shadle, G.L.; Wesley, S.V.; Korth, K.L.; Chen, F.; Lamb, C.; Dixon, R.A. Phenylpropanoid compounds and disease resistance in transgenic tobacco with altered expression of l -phenylalanine ammonia-lyase. Phytochemistry 2003, 64, 153–161. [Google Scholar] [CrossRef]
- Pan, L.Y.; Zhou, J.; Sun, Y.; Qiao, B.X.; Wan, T.; Guo, R.Q.; Zhang, J.; Shan, D.Q.; Cai, Y.L. Comparative transcriptome and metabolome analyses of cherry leaves spot disease caused by Alternaria alternata. Front. Plant Sci. 2023, 14, 1129515. [Google Scholar] [CrossRef]
- Gre, M.E.; Altn, N.; Erdoan, O.; Zkan, I.; Yüksel, B. Genetics of verticillium wilt resistance in cotton. Rom. Agric. Res. 2017, 34, 31–36. [Google Scholar]
- Zhang, P.; Zhu, Y.; Zhou, S. Comparative analysis of powdery mildew resistant and susceptible cultivated cucumber (Cucumis sativus L.) varieties to reveal the metabolic responses to Sphaerotheca fuliginea infection. BMC Plant Biol. 2001, 21, 91. [Google Scholar] [CrossRef]
- Gill, U.S.; Uppalapati, S.R.; Gallego, L.; Ishiga, Y.; Dixon, R.A.; Mysore, K. Metabolic flux towards the (iso) flavonoid pathway in lignin modified alfalfa lines induces resistance against Fusarium oxysporum f. sp. medicaginis. Plant Cell Environ. 2018, 41, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Wu, P.F.; Cao, Y.Q.; Yang, B.B.; Liu, L.X.; Chen, J.J.; Zhuo, R.Y.; Yao, X.H. Overexpression of dihydroflavonol 4-reductase boosts flavonoid production involved in the anthracnose resistance. Front. Plant Sci. 2022, 13, 1038467. [Google Scholar] [CrossRef]
- Lorenc-Kukuła, K.; Wróbel-Kwiatkowska, M.; Starzycki, M.; Szopa, J. Engineering flax with increased flavonoid content and thus Fusarium resistance. Physiol. Mol. Plant Pathol. 2007, 70, 38–48. [Google Scholar] [CrossRef]
- Treutter, D. Significance of flavonoids in plant resistance: A review. Environ. Chem. Lett. 2006, 4, 147–157. [Google Scholar] [CrossRef]
- Shah, J. Lipids, lipases, and lipid-modifying enzymes in plant disease resistance. Annu. Rev. Phytopathol. 2005, s43, 229–260. [Google Scholar] [CrossRef]
- Reina-Pinto, J.J.; Yephremov, A. Surface lipids and plant defenses. Plant Physiol. Biochem. 2009, 47, 540–549. [Google Scholar] [CrossRef]
- Kachroo, A.; Kachroo, P. Fatty acid-derived signals in plant defense. Annu. Rev. Phytopathol. 2009, 47, 153–176. [Google Scholar] [CrossRef]
- Farmer, E.E.; Almeras, E.; Krishnamurthy, V. Jasmonates and related oxylipins in plant responses to pathogenesis and herbivory. Curr. Opin. Plant. Biol. 2003, 6, 372–378. [Google Scholar] [CrossRef]
- Palmer, I.A.; Chen, H.; Chen, J.; Chang, M.; Li, M.; Liu, F.Q.; Zheng, Q.F. Novel Salicylic Acid Analogs Induce a Potent Defense Response in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 3356. [Google Scholar] [CrossRef]
- Lalithya, K.A.; Manjunatha, G.; Raju, B.; Kulkarni, M.S.; Lokesh, V. Plant growth regulators and signal molecules enhance resistance against bacterial blight disease of pomegranate. J. Phytopathol. 2017, 165, 727–736. [Google Scholar] [CrossRef]
- Sirhindi, G.; Mushtaq, R.; Gill, S.S.; Sharma, P.; Abd_Allah, E.F.; Ahmad, P. Jasmonic acid and methyl jasmonate modulate growth, photosynthetic activity and expression of photosystem II subunit genes in Brassica oleracea L. Sci. Rep. 2020, 10, 9322. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.S.; Lei, L.; Liu, Z.X.; Zhou, S.; Luo, J. Selection of a subspecies-specific diterpene gene cluster implicated in rice disease resistance. Nat. Plants 2020, 6, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Bong, S.J.; Park, J.S.; Park, Y.K.; Arasu, M.V.; Al-Dhabi, N.A.; Park, S.U. De novo transcriptome analysis and glucosinolate profiling in watercress (Nasturtium officinale R. Br.). BMC Genom. 2017, 18, 401. [Google Scholar] [CrossRef] [PubMed]
- Delteil, A.; Zhang, J.; Lessard, P.; Morel, J.B. Potential candidate genes for improving rice disease resistance. Rice 2010, 3, 56–71. [Google Scholar] [CrossRef]
- Pieterse, C.M.J.; Van Der Does, D.; Zamioudis, C.; Leon-Reyes, A.; Van Wees, S.C.M. Hormonal modulation of plant immunity. Annu. Rev. Cell Dev. Biol. 2012, 28, 489–521. [Google Scholar] [CrossRef]
- Nick, G.; Tony, R.; Mike, S.; Taylor, J.T. Physiological trade-offs associated with methyl jasmonate-induced resistance in pinus radiata. Can. J. For. Res. 2008, 38, 677–684. [Google Scholar]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Patra, B.; Pattanaik, S.; Schluttenhofer, C.; Yuan, L. A network of jasmonate-responsive bHLH factors modulate monoterpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 2018, 217, 1566–1581. [Google Scholar] [CrossRef]
- Doppler, M.; Kluger, B.; Bueschl, C.; Schneider, C.; Krska, R.; Delcambre, S.; Hiller, K.; Lemmens, M.; Schuhmacher, R. Stable isotopeassisted evaluation of different extraction solvents for untargeted metabolomics of plants. Int. J. Mol. Sci. 2016, 17, 1017. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data usingreal-8time quantitative PCR and the 2 −∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhu, T.; Han, S.; Li, S.; Liu, Y.; Lin, T.; Qiao, T. Changes in the Histology of Walnut (Juglans regia L.) Infected with Phomopsis capsici and Transcriptome and Metabolome Analysis. Int. J. Mol. Sci. 2023, 24, 4879. [Google Scholar] [CrossRef]
- Kaur, T.; Hussain, K.; Koul, S.; Vishwakarma, R.; Vyas, D. Evaluation of Nutritional and Antioxidant Status of Lepidium latifolium Linn.: A Novel Phytofood from Ladakh. PLoS ONE 2013, 8, e69112. [Google Scholar] [CrossRef] [PubMed]
- Zaveri, A.; Edwards, J.; Rochfort, S. Production of Primary Metabolites by Rhizopus stolonifer, Causal Agent of Almond Hull Rot Disease. Molecules 2022, 27, 7199. [Google Scholar] [CrossRef]
- Creelman, R.A.; Mullet, T.J.E. Jasmonic acid/methyl jasmonate accumulate in wounded soybean hypocotyls and modulate wound gene expression. Proc. Natl. Acad. Sci. USA 1992, 89, 4938–4941. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Error Rate | Q20 (%) | Q30 (%) | GC Content (%) | Total Map |
|---|---|---|---|---|---|---|---|
| YK-CK1 | 21,440,315 | 20,913,184 | 0.03 | 97.27 | 92.57 | 44.83 | 31,488,252 (75.28%) |
| YK-CK2 | 23,545,476 | 22,713,650 | 0.03 | 97.54 | 93.12 | 44.24 | 33,838,906 (74.49%) |
| YK-CK3 | 22,641,971 | 22,074,057 | 0.03 | 97.32 | 92.62 | 44.5 | 32,675,810 (74.01%) |
| YK-T1 | 21,707,455 | 21,218,135 | 0.03 | 97.42 | 92.88 | 43.8 | 31,019,402 (73.10%) |
| YK-T2 | 21,024,152 | 20,494,901 | 0.03 | 97.26 | 92.52 | 44.2 | 30,387,514 (74.13%) |
| YK-T3 | 20,843,912 | 20,316,601 | 0.03 | 96.82 | 91.61 | 45.42 | 30,567,864 (75.23%) |
| TJ-CK1 | 20,666,347 | 20,205,862 | 0.03 | 96.88 | 91.72 | 43.78 | 29,411,490 (72.78%) |
| TJ-CK2 | 21,206,648 | 20,714,680 | 0.03 | 97.23 | 92.43 | 44.7 | 30,755,186 (74.24%) |
| TJ-CK3 | 22,574,829 | 22,034,849 | 0.03 | 97.1 | 92.2 | 45.06 | 32,950,058 (74.77%) |
| TJ-T1 | 22,062,869 | 21,543,733 | 0.03 | 97.09 | 92.19 | 43.99 | 31,528,218 (73.17%) |
| TJ-T2 | 22,507,430 | 21,693,488 | 0.03 | 97.21 | 92.43 | 43.74 | 32,443,130 (74.78%) |
| TJ-T3 | 22,944,166 | 22,082,650 | 0.03 | 97.47 | 92.96 | 43.84 | 33,163,596 (75.09%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Xu, X.; Yuan, H.; Li, S.; Lin, T.; Liu, Y.; Li, S.; Zhu, T. Integrated Transcriptome and Metabolome Analysis Reveals the Molecular Mechanism of Rust Resistance in Resistant (Youkang) and Susceptive (Tengjiao) Zanthoxylum armatum Cultivars. Int. J. Mol. Sci. 2023, 24, 14761. https://doi.org/10.3390/ijms241914761
Han S, Xu X, Yuan H, Li S, Lin T, Liu Y, Li S, Zhu T. Integrated Transcriptome and Metabolome Analysis Reveals the Molecular Mechanism of Rust Resistance in Resistant (Youkang) and Susceptive (Tengjiao) Zanthoxylum armatum Cultivars. International Journal of Molecular Sciences. 2023; 24(19):14761. https://doi.org/10.3390/ijms241914761
Chicago/Turabian StyleHan, Shan, Xiu Xu, Huan Yuan, Shujiang Li, Tiantian Lin, Yinggao Liu, Shuying Li, and Tianhui Zhu. 2023. "Integrated Transcriptome and Metabolome Analysis Reveals the Molecular Mechanism of Rust Resistance in Resistant (Youkang) and Susceptive (Tengjiao) Zanthoxylum armatum Cultivars" International Journal of Molecular Sciences 24, no. 19: 14761. https://doi.org/10.3390/ijms241914761
APA StyleHan, S., Xu, X., Yuan, H., Li, S., Lin, T., Liu, Y., Li, S., & Zhu, T. (2023). Integrated Transcriptome and Metabolome Analysis Reveals the Molecular Mechanism of Rust Resistance in Resistant (Youkang) and Susceptive (Tengjiao) Zanthoxylum armatum Cultivars. International Journal of Molecular Sciences, 24(19), 14761. https://doi.org/10.3390/ijms241914761