
A Comprehensive Analysis of Chloroplast Genome Provides New Insights into the Evolution of the Genus Chrysosplenium


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
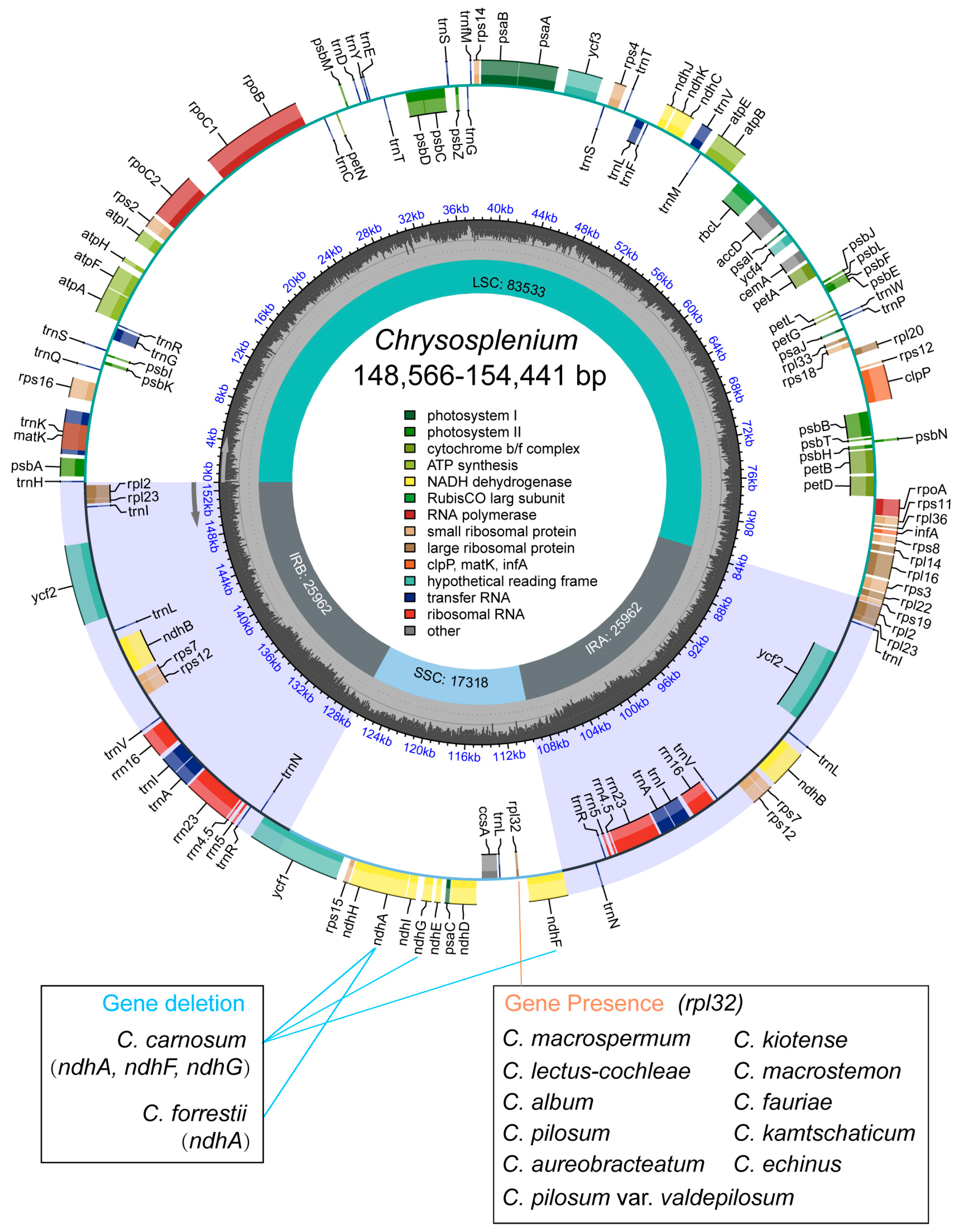
2.1. Structural Characterization of the Chloroplast Genome of Chrysosplenium
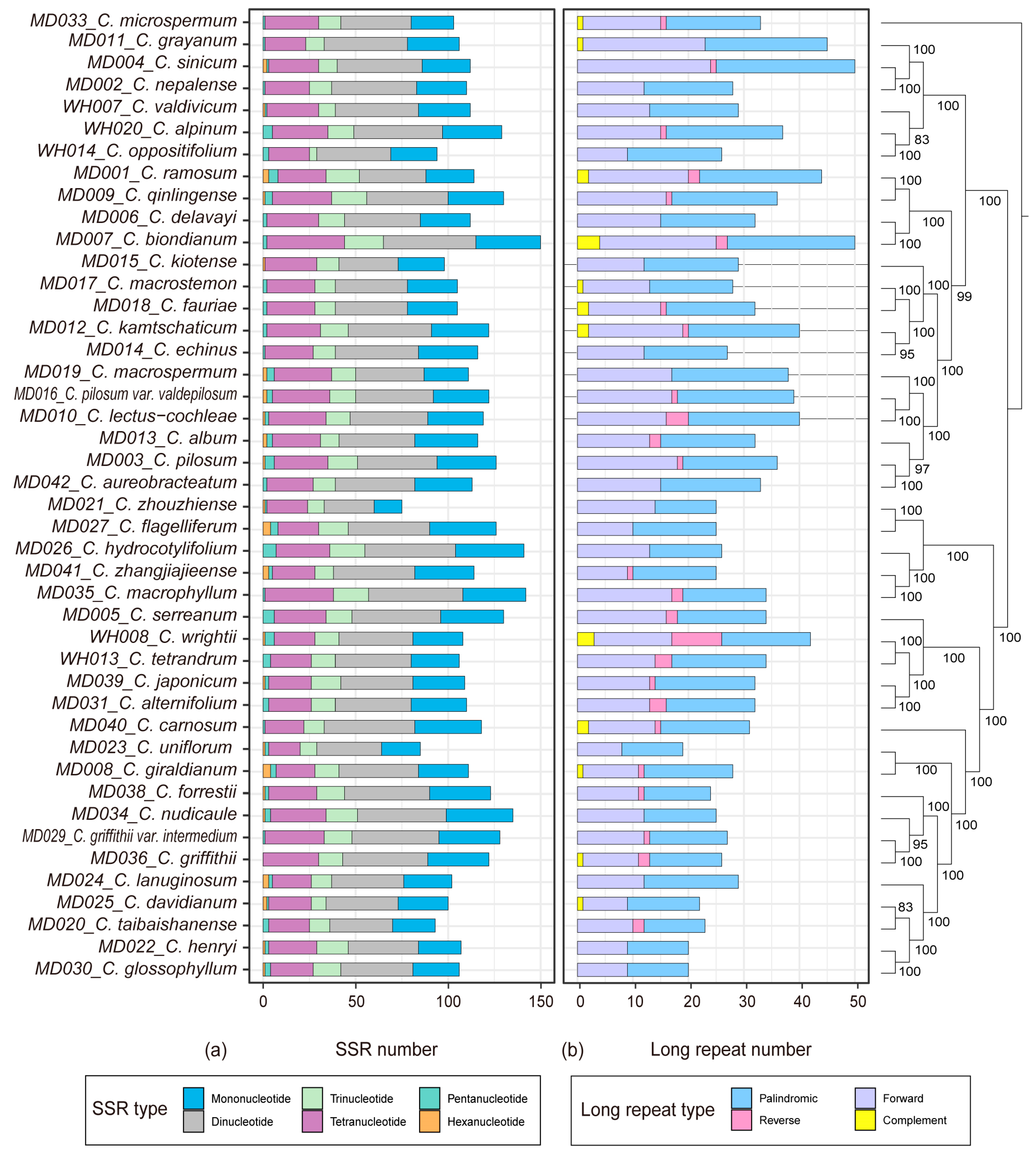
2.2. Repeat Identification
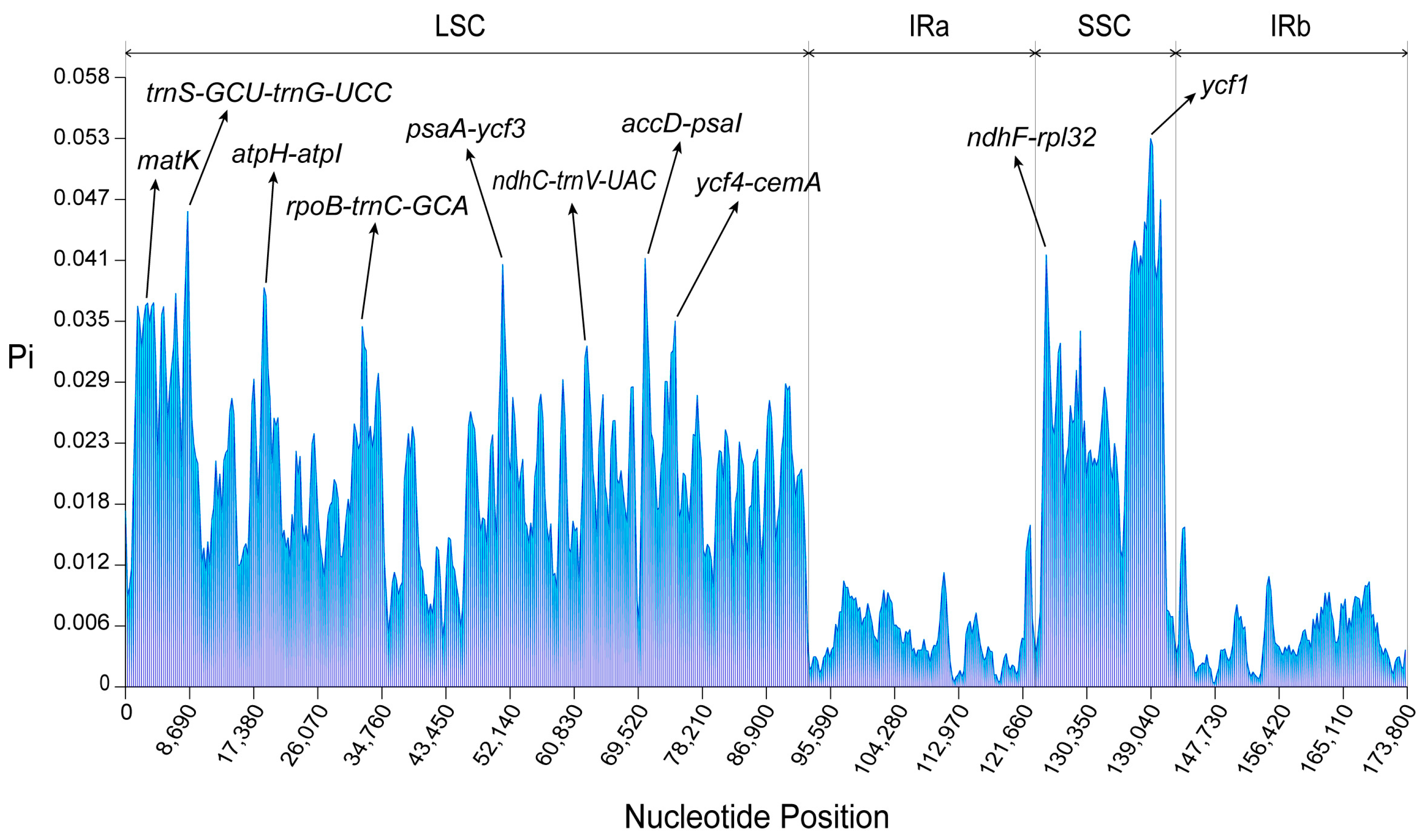
2.3. Divergence Hotspots and Rearrangement Analysis
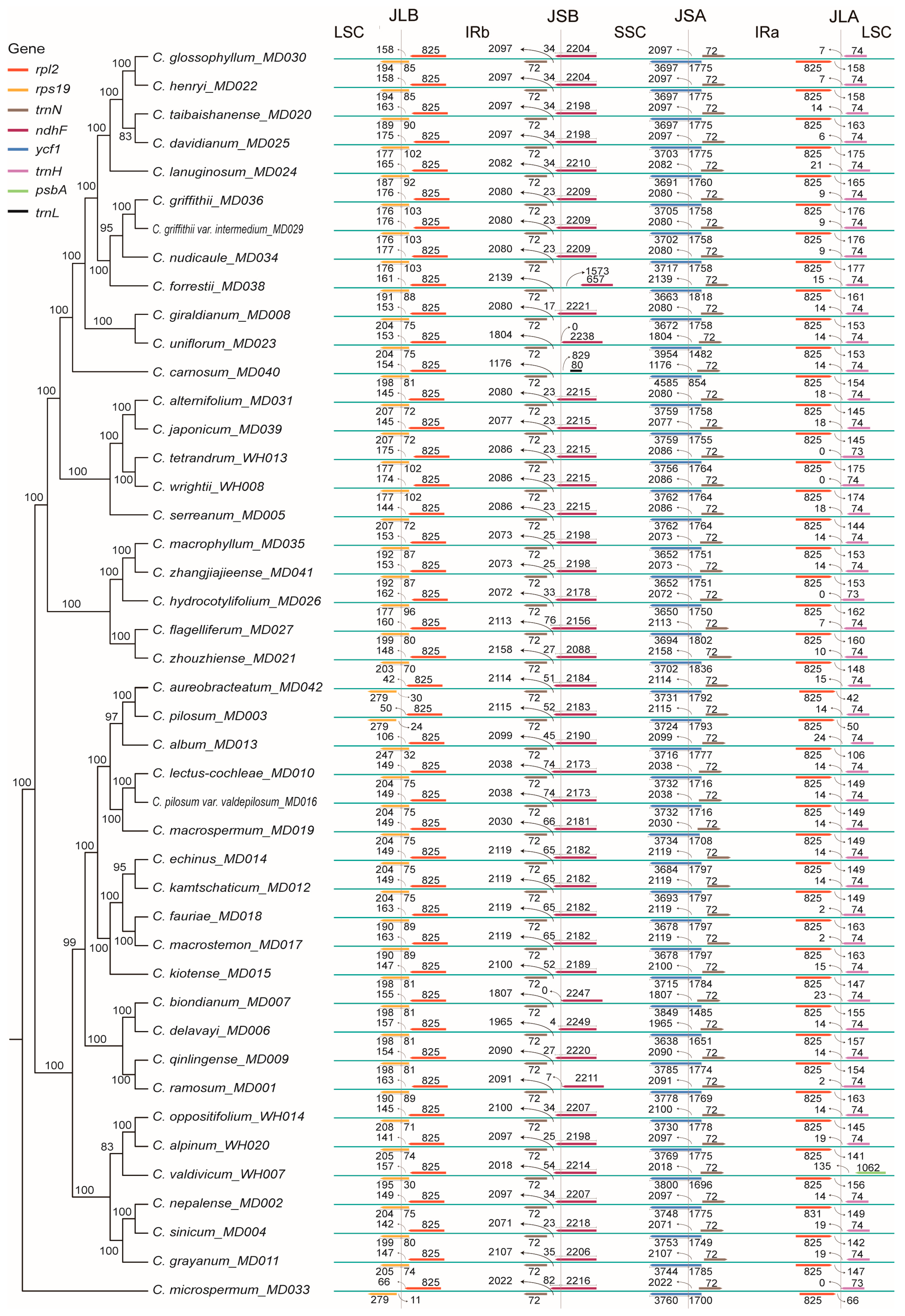
2.4. Dynamic Analysis of the IR Boundary
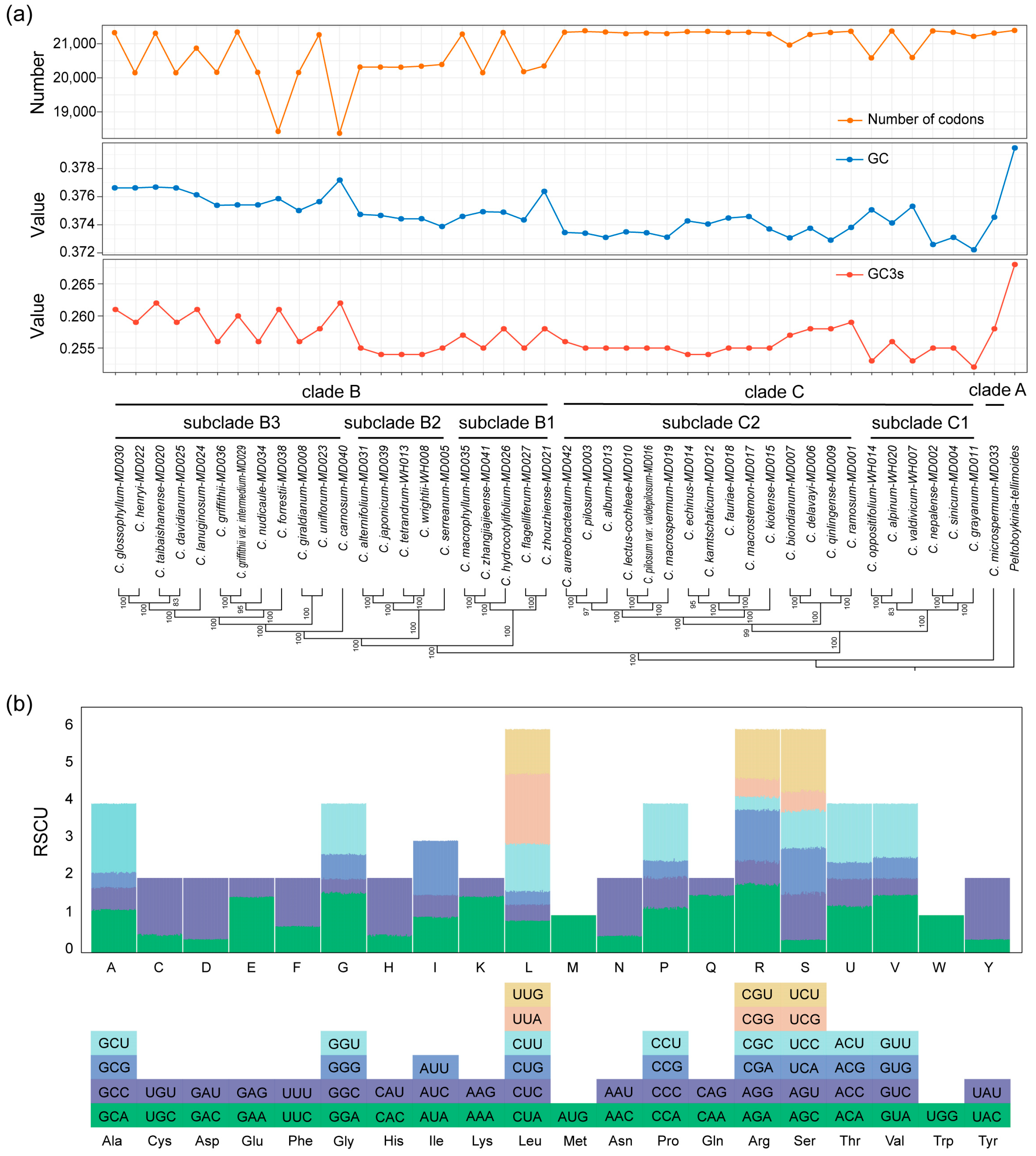
2.5. Codon Usage Analysis
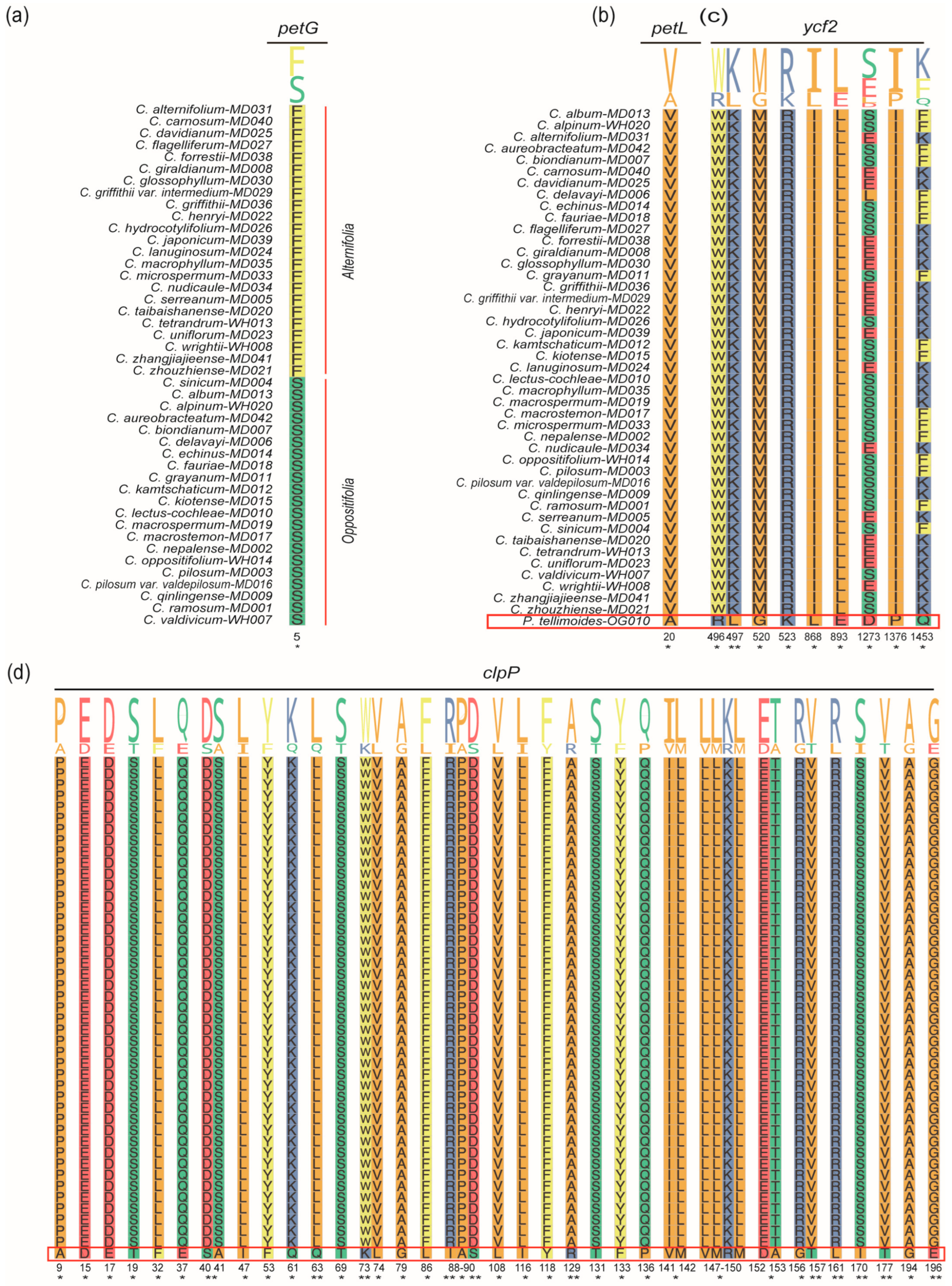
2.6. Selective Pressure Analyses
2.7. Phylogenetic Analysis
3. Discussion
3.1. Chloroplast Genome Evolution within Chrysosplenium Species
3.2. Selection Pressure Analysis of Chrysosplenium Species
3.3. Phylogenetic Relationships of Chrysosplenium Species
4. Materials and Methods
4.1. Sampling, DNA Extraction, and Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Repeat Structure Identification
4.4. Codon Usage Analysis
4.5. Sequence Variation Analysis
4.6. Selective Pressure Analysis
4.7. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gitzendanner, M.A.; Soltis, P.S.; Wong, G.K.S.; Ruhfel, B.R.; Soltis, D.E. Plastid phylogenomic analysis of green plants: A billion years of evolutionary history. Am. J. Bot. 2018, 105, 291–301. [Google Scholar] [CrossRef]
- Ruhfel, B.R.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J.G. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Ecol. Evol. 2014, 14, 23. [Google Scholar] [CrossRef]
- Hara, H. Synopsis of the genus Chrysosplenium L. J. Fac. Sci. Univ. Tokyo 1957, 7, 1–90. [Google Scholar]
- Wu, Z.; Liao, R.; Yang, T.; Dong, X.; Lan, D.; Qin, R.; Liu, H. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genom. 2020, 21, 621. [Google Scholar] [CrossRef]
- Pan, J.; Ohba, H. Chrysosplenium L; Science Press: Beijing, China, 2001; Volume 8, pp. 346–358. [Google Scholar]
- Fu, L.; Liao, R.; Lan, D.; Wen, F.; Liu, H. A new species of Chrysosplenium (Saxifragaceae) from Shaanxi, north-western China. PhytoKeys 2020, 159, 127–135. [Google Scholar] [CrossRef]
- Liu, H.; Luo, J.; Liu, Q.; Lan, D.; Qin, R.; Yu, X. A new species of Chrysosplenium (Saxifragaceae) from Zhangjiajie, Hunan, central China. Phytotaxa 2016, 277, 287–292. [Google Scholar] [CrossRef]
- Maximowicz, C.J. Diagnoses plantarum novarum asiaticarum scripsit. Bull. Acad. Imp. Sci. St.-Pétersbg. 1877, 23, 340–350. [Google Scholar]
- Franchet, A.R. Monographie du genere Chrysosplenium Tournfort; Nouvelles Archives du Muséum d’ histoire naturelle publiées par les professeurs-administrateurs de cet établissement: Pair, France, 1890; Volume 2, pp. 87–114. [Google Scholar]
- Jintang, P. A study on the genus Chrysosplenium L. from China. J. Univ. Chin. Acad. Sci. 1986, 24, 81–97. [Google Scholar]
- Jintang, P. A study on the genus Chrysosplenium L. from China (continued). J. Syst. Evol. 1986, 24, 203–214. [Google Scholar]
- Fu, L.; Yang, T.; Lan, D.; Wen, F.; Liu, H. Chrysosplenium sangzhiense (Saxifragaceae), a new species from Hunan, China. PhytoKeys 2021, 176, 21–32. [Google Scholar] [CrossRef]
- Koldaeva, M.N. Chrysosplenium fallax (Saxifragaceae), a new species from the Russian Far East. Phytotaxa 2021, 491, 35–46. [Google Scholar] [CrossRef]
- Nakazawa, M.; Wakabayashi, M.; Ono, M.; Murata, J. Molecular phylogenetic analysis of Chrysosplenium (Saxifragaceae) in Japan. J. Plant Res. 1997, 110, 265–274. [Google Scholar] [CrossRef]
- Soltis, D.E.; Tago-Nakazawa, M.; Xiang, Q.Y.; Kawano, S.; Murata, J.; Wakabayashi, M.; Hibsch-Jetter, C. Phylogenetic relationships and evolution in Chrysosplenium (Saxifragaceae) based on matK sequence data. Am. J. Bot. 2001, 88, 883–893. [Google Scholar] [CrossRef]
- Xiang, C.; Gitzendanner, M.A.; Soltis, D.E.; Peng, H.; Lei, L. Phylogenetic placement of the enigmatic and critically endangered genus Saniculiphyllum (Saxifragaceae) inferred from combined analysis of plastid and nuclear DNA sequences. Mol. Phylogenet. Evol. 2012, 64, 357–367. [Google Scholar] [CrossRef]
- Tkach, N.; Röser, M.; Hoffmann, M.H. Molecular phylogenetics, character evolution and systematics of the genus Micranthes (Saxifragaceae). Bot. J. Linn. Soc. 2015, 178, 47–66. [Google Scholar] [CrossRef]
- Deng, J.; Drew, B.T.; Mavrodiev, E.V.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E. Phylogeny, divergence times, and historical biogeography of the angiosperm family Saxifragaceae. Mol. Phylogenet. Evol. 2015, 83, 86–98. [Google Scholar] [CrossRef]
- Folk, R.A.; Stubbs, R.L.; Mort, M.E.; Cellinese, N.; Allen, J.M.; Soltis, P.S.; Soltis, D.E.; Guralnick, R.P. Rates of niche and phenotype evolution lag behind diversification in a temperate radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 10874–10882. [Google Scholar] [CrossRef]
- Kim, Y.-i.; Lee, J.; Kim, Y. The complete chloroplast genome of a Korean endemic plant Chrysosplenium aureobracteatum Y.I. Kim & Y.D. Kim (Saxifragaceae). Mitochondrial DNA Part B 2018, 3, 380–381. [Google Scholar]
- Yan, W.; Liu, H.; Yang, T.; Liao, R.; Qin, R. Complete chloroplast genome sequence of Chrysosplenium ramosum and Chrysosplenium alternifolium (Saxifragaceae). Mitochondrial DNA Part B 2020, 5, 2843–2844. [Google Scholar] [CrossRef]
- Yan, W.; Yang, T.; Liao, R.; Wu, Z.; Qin, R.; Liu, H. Complete chloroplast genome sequence of Chrysosplenium macrophyllum and Chrysosplenium flagelliferum (Saxifragaceae). Mitochondrial DNA Part B 2020, 5, 2040–2041. [Google Scholar] [CrossRef]
- Lan, Z.; Shi, Y.; Yin, Q.; Gao, R.; Liu, C.; Wang, W.; Tian, X.; Liu, J.; Nong, Y.; Xiang, L.; et al. Comparative and phylogenetic analysis of complete chloroplast genomes from five Artemisia species. Front. Plant Sci. 2022, 13, 1049209. [Google Scholar] [CrossRef]
- Peng, J.Y.; Zhang, X.S.; Zhang, D.G.; Wang, Y.; Deng, T.; Huang, X.H.; Kuang, T.H.; Zhou, Q. Newly reported chloroplast genome of Sinosenecio albonervius Y. Liu & Q. E. Yang and comparative analyses with other Sinosenecio species. BMC Genom. 2022, 23, 639. [Google Scholar]
- Yan, K.; Ran, J.; Bao, S.; Li, Y.; Islam, R.; Zhang, N.; Zhao, W.; Ma, Y.; Sun, C. The Complete Chloroplast Genome Sequence of Eupatorium fortunei: Genome Organization and Comparison with Related Species. Genes 2022, 14, 64. [Google Scholar] [CrossRef]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D.; et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef]
- Gantt, J.S.; Baldauf, S.L.; Calie, P.J.; Weeden, N.F.; Palmer, J.D. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 1991, 10, 3073–3078. [Google Scholar] [CrossRef]
- Yagi, Y.; Shiina, T. Recent advances in the study of chloroplast gene expression and its evolution. Front. Plant Sci. 2014, 5, 61. [Google Scholar] [CrossRef]
- Lin, C.S.; Chen, J.J.W.; Chiu, C.C.; Hsiao, H.C.W.; Yang, C.J.; Jin, X.H.; Leebens-Mack, J.; de Pamphilis, C.W.; Huang, Y.T.; Yang, L.H.; et al. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. Cell Mol. Biol. 2017, 90, 994–1006. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Dong, J.; Zhang, T.; Xiao, H. Comparative chloroplast genomes and phylogenetic analysis of Aquilegia. Appl. Plant Sci. 2021, 9, e11412. [Google Scholar] [CrossRef]
- Yan, C.; Du, J.; Gao, L.; Li, Y.; Hou, X. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 2019, 699, 24–36. [Google Scholar] [CrossRef]
- Zhong, Q.; Yang, S.; Sun, X.; Wang, L.; Li, Y. The complete chloroplast genome of the Jerusalem artichoke (Helianthus tuberosus L.) and an adaptive evolutionary analysis of the ycf2 gene. PeerJ 2019, 7, e7596. [Google Scholar] [CrossRef]
- Drescher, A.; Ruf, S.; Calsa, T., Jr.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. Cell Mol. Biol. 2000, 22, 97–104. [Google Scholar] [CrossRef]
- Kikuchi, S.; Asakura, Y.; Imai, M.; Nakahira, Y.; Kotani, Y.; Hashiguchi, Y.; Nakai, Y.; Takafuji, K.; Bédard, J.; Hirabayashi-Ishioka, Y.; et al. A Ycf2-FtsHi Heteromeric AAA-ATPase Complex Is Required for Chloroplast Protein Import. Plant Cell 2018, 30, 2677–2703. [Google Scholar] [CrossRef]
- Wang, Q.M.; Cui, J.; Dai, H.; Zhou, Y.; Li, N.; Zhang, Z. Comparative transcriptome profiling of genes and pathways involved in leaf-patterning of Clivia miniata var. variegata. Gene 2018, 677, 280–288. [Google Scholar] [CrossRef]
- Zhang, Y.; An, D.; Li, C.; Zhao, Z.; Wang, W. The complete chloroplast genome of greater duckweed (Spirodela polyrhiza 7498) using PacBio long reads: Insights into the chloroplast evolution and transcription regulation. BMC Genom. 2020, 21, 76. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, J.; Bai, M.; Zhang, G.; Liu, Z. The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 2021, 21, 248. [Google Scholar] [CrossRef]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef]
- Huang, R.; Xie, X.; Chen, A.; Li, F.; Tian, E.; Chao, Z. The chloroplast genomes of four Bupleurum (Apiaceae) species endemic to Southwestern China, a diversity center of the genus, as well as their evolutionary implications and phylogenetic inferences. BMC Genom. 2021, 22, 714. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Li, W.Y.; Peng, Y.; Gao, J. Comparative chloroplast genome analysis of Ficus (Moraceae): Insight into adaptive evolution and mutational hotspot regions. Front. Plant Sci. 2022, 13, 965335. [Google Scholar] [CrossRef]
- Moghaddam, M.; Ohta, A.; Shimizu, M.; Terauchi, R.; Kazempour-Osaloo, S. The complete chloroplast genome of Onobrychis gaubae (Fabaceae-Papilionoideae): Comparative analysis with related IR-lacking clade species. BMC Plant Biol. 2022, 22, 75. [Google Scholar] [CrossRef]
- Liu, K.; Sun, L.; Meng, W.; Zhu, H.; Zhang, D.; Wang, J. Comparative genomics and phylogenetic perspectives of six fertile Lycoris species endemic to East Asia based on plastome characterization. Nord. J. Bot. 2022, 4, e03412. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Erixon, P.; Oxelman, B. Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS ONE 2008, 3, e1386. [Google Scholar] [CrossRef]
- Rosendahl, C.; Gortner, R.; Burr, G. A Monograph on the Genus Heuchera; University of Minnesota Press: Minneapolis, MN, USA, 1936; pp. 1–192. [Google Scholar]
- Spongberg, S.A. The Genera of Saxifragaceae in the Southeastern United States. J. Arnold Arbor. 1972, 53, 409–498. [Google Scholar] [CrossRef]
- Soltis, D.E.; Soltis, P.S. Intergeneric Hybridization between Conimitella williamsii and Mitella stauropetala (Saxifragaceae). Syst. Bot. 1986, 11, 293–297. [Google Scholar] [CrossRef]
- Soltis, D.E.; Bohm, B.A. Chromosomal and flavonoid chemical confirmation of intergeneric hybridization between Tolmiea and Tellima (Saxifragaceae). Can. J. Bot. 1985, 63, 1309–1312. [Google Scholar] [CrossRef]
- Liu, L.; Du, Y.; Folk, R.A.; Wang, S.; Soltis, D.E.; Shang, F.; Li, P. Plastome Evolution in Saxifragaceae and Multiple Plastid Capture Events Involving Heuchera and Tiarella. Front. Plant Sci. 2020, 11, 361. [Google Scholar] [CrossRef]
- Metzlaff, M.; Börner, T.; Hagemann, R. Variations of chloroplast DNAs in the genus Pelargonium and their biparental inheritance. Theor. Appl. Genet. 1981, 60, 37–41. [Google Scholar] [CrossRef]
- Matsushima, R.; Hu, Y.; Toyoda, K.; Sodmergen; Sakamoto, W. The Model Plant Medicago truncatula Exhibits Biparental Plastid Inheritance. Plant Cell Physiol. 2008, 49, 81–91. [Google Scholar] [CrossRef]
- Ruf, S.; Karcher, D.; Bock, R. Determining the transgene containment level provided by chloroplast transformation. Proc. Natl. Acad. Sci. USA 2007, 104, 6998–7002. [Google Scholar] [CrossRef]
- Azhagiri, A.K.; Maliga, P. Exceptional paternal inheritance of plastids in Arabidopsis suggests that low-frequency leakage of plastids via pollen may be universal in plants. Plant J. 2007, 52, 817–823. [Google Scholar] [CrossRef]
- Chung, K.P.; Gonzalez-Duran, E.; Ruf, S.; Endries, P.; Bock, R. Control of plastid inheritance by environmental and genetic factors. Nat. Plants 2023, 9, 68–80. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissues. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Simon, A. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2020. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 February 2023).
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; dePamphilis, C.W.; Yi, T.; Li, D. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.C.; Chen, H.M.; Jiang, M.; Wang, L.Q.; Wu, X.; Huang, L.F.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Qu, X.; Moore, M.J.; Li, D.; Yi, T. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Ankenbrand, M.J.; Hohlfeld, S.; Hackl, T.; Förster, F. AliTV—Interactive visualization of whole genome comparisons. PeerJ Comput. Sci. 2017, 3, e116. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.; Wu, Z.; Tie, J.; Qin, R.; Wang, J.; Liu, H. A Comprehensive Analysis of Chloroplast Genome Provides New Insights into the Evolution of the Genus Chrysosplenium. Int. J. Mol. Sci. 2023, 24, 14735. https://doi.org/10.3390/ijms241914735
Yang T, Wu Z, Tie J, Qin R, Wang J, Liu H. A Comprehensive Analysis of Chloroplast Genome Provides New Insights into the Evolution of the Genus Chrysosplenium. International Journal of Molecular Sciences. 2023; 24(19):14735. https://doi.org/10.3390/ijms241914735
Chicago/Turabian StyleYang, Tiange, Zhihua Wu, Jun Tie, Rui Qin, Jiangqing Wang, and Hong Liu. 2023. "A Comprehensive Analysis of Chloroplast Genome Provides New Insights into the Evolution of the Genus Chrysosplenium" International Journal of Molecular Sciences 24, no. 19: 14735. https://doi.org/10.3390/ijms241914735
APA StyleYang, T., Wu, Z., Tie, J., Qin, R., Wang, J., & Liu, H. (2023). A Comprehensive Analysis of Chloroplast Genome Provides New Insights into the Evolution of the Genus Chrysosplenium. International Journal of Molecular Sciences, 24(19), 14735. https://doi.org/10.3390/ijms241914735