
Platelets in Renal Disease


Abstract
1. Introduction
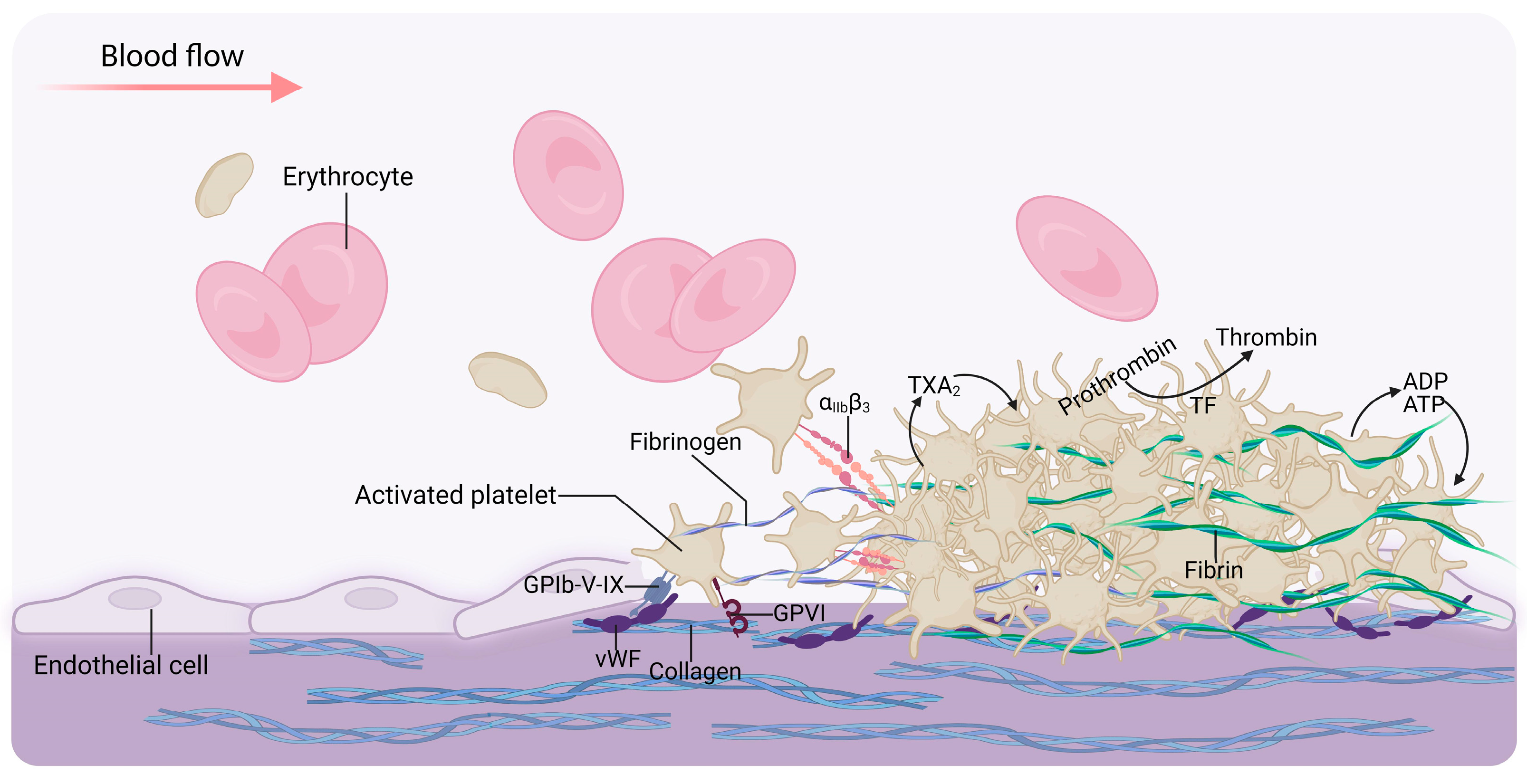
2. Platelet Biology
3. The Mechanisms of Platelet Abnormalities in Kidney Disease
3.1. Abnormalities in Antiplatelet Factors
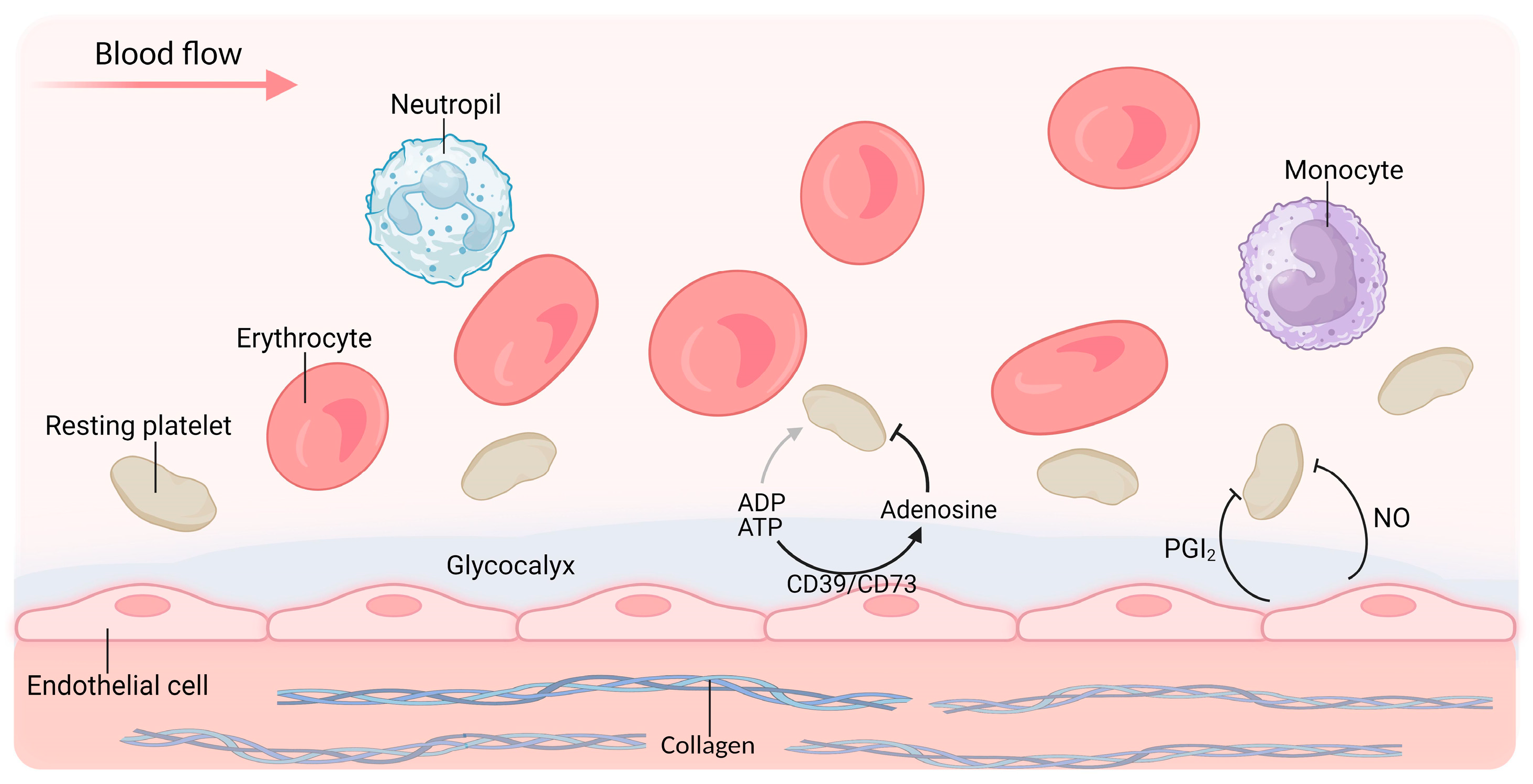
3.1.1. Glycocalyx
3.1.2. NO
3.1.3. PGI2
3.1.4. CD39/CD73
3.2. Abnormalities in Platelet-Activating Mediators
3.2.1. TF
3.2.2. DAMPs and PAMPs
3.2.3. Uremic Toxin Accumulation
3.3. Anemia and Hemodynamic Changes
3.4. Other Influencing Factors
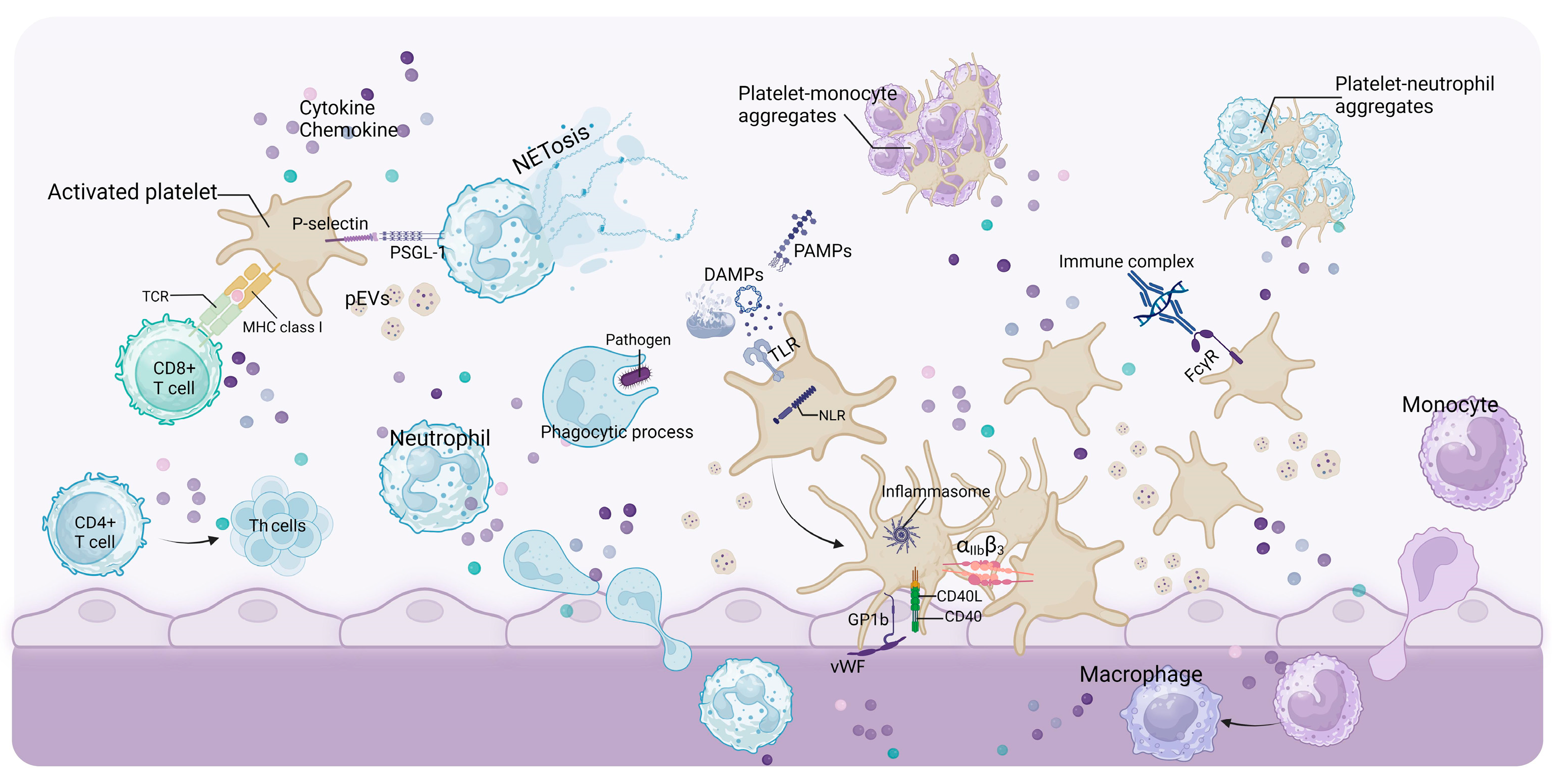
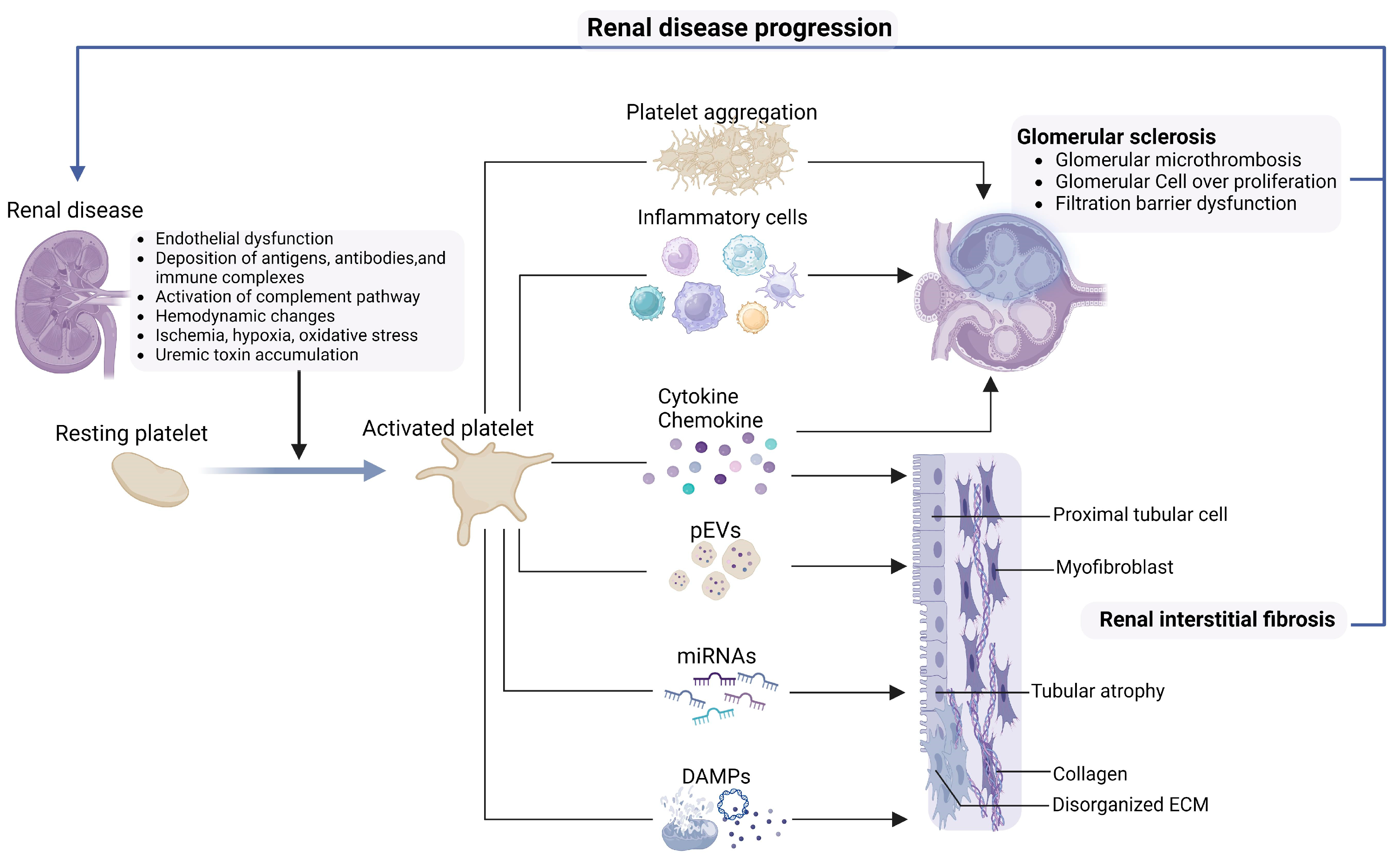
4. Platelet in Inducing Progression of Renal Disease
4.1. Glomerular Microthrombus
4.2. Platelets Promote Renal Inflammation and Fibrosis
4.2.1. CD40/CD40L(CD154)
4.2.2. Adhesion Molecules
4.2.3. Chemokine
4.2.4. Protease-Activated Receptors (PARs) and Platelet-Activating Factor (PAF)
4.3. Other Components Derived from Platelets
4.3.1. Platelet-Derived DAMPs
4.3.2. Growth Factor and Chemokine
4.3.3. MicroRNAs (miRNAs)
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interests
References
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute Kidney Injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A Single Number for Advocacy and Communication—Worldwide More than 850 Million Individuals Have Kidney Diseases. Kidney Int. 2019, 96, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern. Med. 2017, 167, ITC66–ITC80. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P. Epidemiology of Chronic Kidney Disease: An Update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of Maladaptive Repair after AKI Leading to Accelerated Kidney Ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Herzlinger, D.; Hurtado, R. Patterning the Renal Vascular Bed. Semin. Cell Dev. Biol. 2014, 36, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; Meta, E.; Borri, M.; Luo, Y.; Li, X.; Rabelink, T.J.; Carmeliet, P. Phenotypic Diversity and Metabolic Specialization of Renal Endothelial Cells. Nat. Rev. Nephrol. 2021, 17, 441–464. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Basso, P.J.; Andrade-Oliveira, V.; Câmara, N.O.S. Targeting Immune Cell Metabolism in Kidney Diseases. Nat. Rev. Nephrol. 2021, 17, 465–480. [Google Scholar] [CrossRef]
- Boccardo, P.; Remuzzi, G.; Galbusera, M. Platelet Dysfunction in Renal Failure. Semin. Thromb. Hemost. 2004, 30, 579–589. [Google Scholar] [CrossRef]
- Hvas, A.-M. Crucial Stepping Stones in Platelet History. Semin. Thromb. Hemost. 2023, 49, 272–278. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet Biology and Functions: New Concepts and Clinical Perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Plé, H.; Maltais, M.; Corduan, A.; Rousseau, G.; Madore, F.; Provost, P. Alteration of the Platelet Transcriptome in Chronic Kidney Disease. Thromb. Haemost. 2012, 108, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-H.; Sim, E.-H.; Goh, R.-Y.; Park, J.-I.; Han, J.-Y. Platelet Activation: The Mechanisms and Potential Biomarkers. Biomed. Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef] [PubMed]
- Rendu, F.; Brohard-Bohn, B. The Platelet Release Reaction: Granules’ Constituents, Secretion and Functions. Platelets 2001, 12, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Gremmel, T.; Frelinger, A.L.; Michelson, A.D. Platelet Physiology. Semin. Thromb. Hemost. 2016, 42, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Holinstat, M. Normal Platelet Function. Cancer Metastasis Rev. 2017, 36, 195–198. [Google Scholar] [CrossRef]
- Melki, I.; Allaeys, I.; Tessandier, N.; Lévesque, T.; Cloutier, N.; Laroche, A.; Vernoux, N.; Becker, Y.; Benk-Fortin, H.; Zufferey, A.; et al. Platelets Release Mitochondrial Antigens in Systemic Lupus Erythematosus. Sci. Transl. Med. 2021, 13, eaav5928. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S.; Kim, S. An Insight into Recent Advances on Platelet Function in Health and Disease. Int. J. Mol. Sci. 2022, 23, 6022. [Google Scholar] [CrossRef]
- Ribatti, D.; Crivellato, E. Giulio Bizzozero and the Discovery of Platelets. Leuk. Res. 2007, 31, 1339–1341. [Google Scholar] [CrossRef]
- Ghoshal, K.; Bhattacharyya, M. Overview of Platelet Physiology: Its Hemostatic and Nonhemostatic Role in Disease Pathogenesis. Sci. World J. 2014, 2014, 781857. [Google Scholar] [CrossRef] [PubMed]
- Brott, T.; Stump, D. Overview of Hemostasis and Thrombosis. Semin. Neurol. 1991, 11, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Broos, K.; Feys, H.B.; De Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Platelets at Work in Primary Hemostasis. Blood Rev. 2011, 25, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.R.; Storey, R.F. The Role of Platelets in Inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Prb, D.; Ac, Q.-T.; Lb, M.; Mbm, P.; Sv, R.; Fb, A.; Ed, H. Innate Immune Receptors in Platelets and Platelet-Leukocyte Interactions. J. Leukoc. Biol. 2020, 108, 1157–1182. [Google Scholar] [CrossRef]
- Kral, J.B.; Schrottmaier, W.C.; Salzmann, M.; Assinger, A. Platelet Interaction with Innate Immune Cells. Transfus. Med. Hemother. 2016, 43, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Yoder, M.C. Renal Endothelial Dysfunction in Acute Kidney Ischemia Reperfusion Injury. Cardiovasc. Hematol. Disord. Drug Targets 2014, 14, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Brunet, P.; Gondouin, B.; Duval-Sabatier, A.; Dou, L.; Cerini, C.; Dignat-George, F.; Jourde-Chiche, N.; Argiles, A.; Burtey, S. Does Uremia Cause Vascular Dysfunction? Kidney Blood Press. Res. 2011, 34, 284–290. [Google Scholar] [CrossRef]
- da Cunha, R.S.; Santos, A.F.; Barreto, F.C.; Stinghen, A.E.M. How Do Uremic Toxins Affect the Endothelium? Toxins 2020, 12, 412. [Google Scholar] [CrossRef]
- Harper, S.J.; Bates, D.O. Endothelial Permeability in Uremia. Kidney Int. Suppl. 2003, 63, S41–S44. [Google Scholar] [CrossRef]
- van der Poll, T.; Parker, R.I. Platelet Activation and Endothelial Cell Dysfunction. Crit. Care Clin. 2020, 36, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The Glycocalyx: A Novel Diagnostic and Therapeutic Target in Sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H. Derangement of the Endothelial Glycocalyx in Sepsis. J. Thromb. Haemost. 2019, 17, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Jedlicka, J.; Becker, B.F.; Chappell, D. Endothelial Glycocalyx. Crit. Care Clin. 2020, 36, 217–232. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Neves, F.M.; Araújo, C.B.; de Freitas, D.F.; Arruda, B.F.T.; de Macêdo Filho, L.J.M.; Salles, V.B.; Meneses, G.C.; Martins, A.M.C.; Libório, A.B. Fibroblast Growth Factor 23, Endothelium Biomarkers and Acute Kidney Injury in Critically-Ill Patients. J. Transl. Med. 2019, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Molema, G.; Zijlstra, J.G.; van Meurs, M.; Kamps, J.A.A.M. Renal Microvascular Endothelial Cell Responses in Sepsis-Induced Acute Kidney Injury. Nat. Rev. Nephrol. 2022, 18, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N. Endothelial Toxicity of High Glucose and Its By-Products in Diabetic Kidney Disease. Toxins 2019, 11, 578. [Google Scholar] [CrossRef]
- Mahtal, N.; Lenoir, O.; Tharaux, P.-L. Glomerular Endothelial Cell Crosstalk With Podocytes in Diabetic Kidney Disease. Front. Med. 2021, 8, 659013. [Google Scholar] [CrossRef]
- Regier, M.; Drost, C.C.; Rauen, M.; Pavenstädt, H.; Rovas, A.; Kümpers, P.; Vink, H.; Long, R.M.; Linke, W.A.; Nofer, J.-R.; et al. A Dietary Supplement Containing Fucoidan Preserves Endothelial Glycocalyx through ERK/MAPK Signaling and Protects against Damage Induced by CKD Serum. Int. J. Mol. Sci. 2022, 23, 15520. [Google Scholar] [CrossRef]
- Liew, H.; Roberts, M.A.; Pope, A.; McMahon, L.P. Endothelial Glycocalyx Damage in Kidney Disease Correlates with Uraemic Toxins and Endothelial Dysfunction. BMC Nephrol. 2021, 22, 21. [Google Scholar] [CrossRef]
- Ermert, K.; Buhl, E.M.; Klinkhammer, B.M.; Floege, J.; Boor, P. Reduction of Endothelial Glycocalyx on Peritubular Capillaries in Chronic Kidney Disease. Am. J. Pathol. 2023, 193, 138–147. [Google Scholar] [CrossRef] [PubMed]
- de Melo Bezerra Cavalcante, C.T.; Castelo Branco, K.M.; Pinto Júnior, V.C.; Meneses, G.C.; de Oliveira Neves, F.M.; de Souza, N.M.G.; Penaforte, K.L.; Martins, A.M.C.; Libório, A.B. Syndecan-1 Improves Severe Acute Kidney Injury Prediction after Pediatric Cardiac Surgery. J. Thorac. Cardiovasc. Surg. 2016, 152, 178–186.e2. [Google Scholar] [CrossRef] [PubMed]
- Chelazzi, C.; Villa, G.; Mancinelli, P.; De Gaudio, A.R.; Adembri, C. Glycocalyx and Sepsis-Induced Alterations in Vascular Permeability. Crit. Care 2015, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Adembri, C.; Sgambati, E.; Vitali, L.; Selmi, V.; Margheri, M.; Tani, A.; Bonaccini, L.; Nosi, D.; Caldini, A.L.; Formigli, L.; et al. Sepsis Induces Albuminuria and Alterations in the Glomerular Filtration Barrier: A Morphofunctional Study in the Rat. Crit. Care 2011, 15, R277. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Brettner, F.; Doerfler, N.; Jacob, M.; Rehm, M.; Bruegger, D.; Conzen, P.; Jacob, B.; Becker, B.F. Protection of Glycocalyx Decreases Platelet Adhesion after Ischaemia/Reperfusion: An Animal Study. Eur. J. Anaesthesiol. 2014, 31, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Jacob, M.; Hofmann-Kiefer, K.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B.F. Antithrombin Reduces Shedding of the Endothelial Glycocalyx Following Ischaemia/Reperfusion. Cardiovasc. Res. 2009, 83, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Heindl, B.; Jacob, M.; Annecke, T.; Chen, C.; Rehm, M.; Conzen, P.; Becker, B.F. Sevoflurane Reduces Leukocyte and Platelet Adhesion after Ischemia-Reperfusion by Protecting the Endothelial Glycocalyx. Anesthesiology 2011, 115, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Chandra, M.; Tuteja, R.; Misra, M.K. Nitric Oxide as a Unique Bioactive Signaling Messenger in Physiology and Pathophysiology. J. Biomed. Biotechnol. 2004, 2004, 227–237. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. An L-Arginine/Nitric Oxide Pathway Present in Human Platelets Regulates Aggregation. Proc. Natl. Acad. Sci. USA 1990, 87, 5193–5197. [Google Scholar] [CrossRef]
- Marietta, M.; Facchinetti, F.; Neri, I.; Piccinini, F.; Volpe, A.; Torelli, G. L-Arginine Infusion Decreases Platelet Aggregation through an Intraplatelet Nitric Oxide Release. Thromb. Res. 1997, 88, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Cortese-Krott, M.M.; Amabile, N.; Hundsdörfer, C.; Kröncke, K.-D.; Kelm, M.; Heiss, C. Circulating Microparticles Carry a Functional Endothelial Nitric Oxide Synthase That Is Decreased in Patients with Endothelial Dysfunction. J. Am. Heart Assoc. 2012, 2, e003764. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Forzano, I.; Avvisato, R.; Varzideh, F.; Jankauskas, S.S.; Cioppa, A.; Mone, P.; Salemme, L.; Kansakar, U.; Tesorio, T.; Trimarco, V.; et al. L-Arginine in Diabetes: Clinical and Preclinical Evidence. Cardiovasc. Diabetol. 2023, 22, 89. [Google Scholar] [CrossRef] [PubMed]
- Semenikhina, M.; Stefanenko, M.; Spires, D.R.; Ilatovskaya, D.V.; Palygin, O. Nitric-Oxide-Mediated Signaling in Podocyte Pathophysiology. Biomolecules 2022, 12, 745. [Google Scholar] [CrossRef] [PubMed]
- Lee, J. Nitric Oxide in the Kidney: Its Physiological Role and Pathophysiological Implications. Electrolyte Blood Press. 2008, 6, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Yamasowa, H.; Shimizu, S.; Inoue, T.; Takaoka, M.; Matsumura, Y. Endothelial Nitric Oxide Contributes to the Renal Protective Effects of Ischemic Preconditioning. J. Pharmacol. Exp. Ther. 2005, 312, 153–159. [Google Scholar] [CrossRef]
- Piepot, H.A.; Boer, C.; Groeneveld, A.B.; Van Lambalgen, A.A.; Sipkema, P. Lipopolysaccharide Impairs Endothelial Nitric Oxide Synthesis in Rat Renal Arteries. Kidney Int. 2000, 57, 2502–2510. [Google Scholar] [CrossRef][Green Version]
- Wang, W.; Mitra, A.; Poole, B.; Falk, S.; Lucia, M.S.; Tayal, S.; Schrier, R. Endothelial Nitric Oxide Synthase-Deficient Mice Exhibit Increased Susceptibility to Endotoxin-Induced Acute Renal Failure. Am. J. Physiol. Ren. Physiol. 2004, 287, F1044–F1048. [Google Scholar] [CrossRef]
- Kwon, O.; Hong, S.-M.; Ramesh, G. Diminished NO Generation by Injured Endothelium and Loss of Macula Densa nNOS May Contribute to Sustained Acute Kidney Injury after Ischemia-Reperfusion. Am. J. Physiol. Ren. Physiol. 2009, 296, F25–F33. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.B.; Florquin, S.; Roelofs, J.J.T.H. The Role of Platelets in Acute Kidney Injury. Nat. Rev. Nephrol. 2018, 14, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Pieper, G.M. Review of Alterations in Endothelial Nitric Oxide Production in Diabetes: Protective Role of Arginine on Endothelial Dysfunction. Hypertension 1998, 31, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Han, M.; Rezaei, A.; Li, D.; Wu, G.; Ma, X. L-Arginine Modulates Glucose and Lipid Metabolism in Obesity and Diabetes. Curr. Protein Pept. Sci. 2017, 18, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Nappo, F.; De Angelis, L.; Paolisso, G.; Tagliamonte, M.R.; Giugliano, D. Hemodynamic Effects of Acute Hyperglycemia in Type 2 Diabetic Patients. Diabetes Care 2000, 23, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Madore, F.; Prud’homme, L.; Austin, J.S.; Blaise, G.; Francoeur, M.; Léveillé, M.; Prud’homme, M.; Vinay, P. Impact of Nitric Oxide on Blood Pressure in Hemodialysis Patients. Am. J. Kidney Dis. 1997, 30, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, K.; Kohno, M.; Yoshikawa, J. Nitric Oxide Mediates the Cardiovascular Instability of Haemodialysis Patients. Curr. Opin. Nephrol. Hypertens. 1996, 5, 359–363. [Google Scholar] [CrossRef]
- Mårtensson, L.; Hegbrant, J.; Thysell, H. Generation of Nitrate during Dialysis as a Measure of Nitric Oxide Synthesis. Artif. Organs 1997, 21, 163–167. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Uremic Bleeding: Closing the Circle after 30 Years of Controversies? Blood 1999, 94, 2569–2574. [Google Scholar] [CrossRef]
- Noris, M.; Benigni, A.; Boccardo, P.; Aiello, S.; Gaspari, F.; Todeschini, M.; Figliuzzi, M.; Remuzzi, G. Enhanced Nitric Oxide Synthesis in Uremia: Implications for Platelet Dysfunction and Dialysis Hypotension. Kidney Int. 1993, 44, 445–450. [Google Scholar] [CrossRef][Green Version]
- Brunini, T.M.C.; da Silva, C.D.; Siqueira, M.A.S.; Moss, M.B.; Santos, S.F.F.; Mendes-Ribeiro, A.C. Uremia, Atherothrombosis and Malnutrition: The Role of L-Arginine-Nitric Oxide Pathway. Cardiovasc. Hematol. Disord. Drug Targets 2006, 6, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Hirata, Y.; Kakoki, M.; Nagata, D.; Momomura, S.-i.; Sugimoto, T.; Tagawa, H.; Omata, M. Increased Excretion of Nitric Oxide in Exhaled Air of Patients with Chronic Renal Failure. Clin. Sci. 1999, 96, 67–74. [Google Scholar] [CrossRef]
- Hörl, W.H. Hemodialysis Membranes: Interleukins, Biocompatibility, and Middle Molecules. J. Am. Soc. Nephrol. 2002, 13 (Suppl. S1), S62–S71. [Google Scholar] [PubMed]
- Pereira, B.J.; Dinarello, C.A. Production of Cytokines and Cytokine Inhibitory Proteins in Patients on Dialysis. Nephrol. Dial. Transplant. 1994, 9 (Suppl. S2), 60–71. [Google Scholar]
- Brunini, T.M.C.; Mendes-Ribeiro, A.C.; Ellory, J.C.; Mann, G.E. Platelet Nitric Oxide Synthesis in Uremia and Malnutrition: A Role for L-Arginine Supplementation in Vascular Protection? Cardiovasc. Res. 2007, 73, 359–367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mendes Ribeiro, A.C.; Brunini, T.M.; Ellory, J.C.; Mann, G.E. Abnormalities in L-Arginine Transport and Nitric Oxide Biosynthesis in Chronic Renal and Heart Failure. Cardiovasc. Res. 2001, 49, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.R.; Kaitwatcharachai, C.; Levin, N.W. Nitric Oxide and Hemodialysis. Semin. Dial. 2004, 17, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R. Prostacyclin. J. R. Soc. Med. 1983, 76, 245–249. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Nemenoff, R. Renal Tubular Arachidonic Acid Metabolism. Kidney Int. 1991, 39, 438–449. [Google Scholar] [CrossRef]
- Walter, U.; Geiger, J.; Haffner, C.; Markert, T.; Nehls, C.; Silber, R.E.; Schanzenbächer, P. Platelet-Vessel Wall Interactions, Focal Adhesions, and the Mechanism of Action of Endothelial Factors. Agents Actions Suppl. 1995, 45, 255–268. [Google Scholar] [CrossRef]
- Dusting, G.J.; MacDonald, P.S. Prostacyclin and Vascular Function: Implications for Hypertension and Atherosclerosis. Pharmacol. Ther. 1990, 48, 323–344. [Google Scholar] [CrossRef] [PubMed]
- Stitham, J.; Hwa, J. Prostacyclin, Atherothrombosis and Diabetes Mellitus: Physiologic and Clinical Considerations. Curr. Mol. Med. 2016, 16, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, C.; Yabuki, T.; Shimonishi, M.; Wada, M.; Hatae, T.; Ohkawara, S.; Takeda, J.; Kinoshita, T.; Okabe, M.; Tanabe, T. Prostacyclin-Deficient Mice Develop Ischemic Renal Disorders, Including Nephrosclerosis and Renal Infarction. Circulation 2002, 106, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zolty, E.; Falk, S.; Summer, S.; Stearman, R.; Geraci, M.; Schrier, R. Prostacyclin in Endotoxemia-Induced Acute Kidney Injury: Cyclooxygenase Inhibition and Renal Prostacyclin Synthase Transgenic Mice. Am. J. Physiology. Ren. Physiol. 2007, 293, F1131–F1136. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, G.; Cavenaghi, A.E.; Mecca, G.; Donati, M.B.; de Gaetano, G. Prostacyclin-like Activity and Bleeding in Renal Failure. Lancet 1977, 2, 1195–1197. [Google Scholar] [CrossRef] [PubMed]
- Kyrle, P.A.; Stockenhuber, F.; Brenner, B.; Gössinger, H.; Korninger, C.; Pabinger, I.; Sunder-Plassmann, G.; Balcke, P.; Lechner, K. Evidence for an Increased Generation of Prostacyclin in the Microvasculature and an Impairment of the Platelet Alpha-Granule Release in Chronic Renal Failure. Thromb. Haemost. 1988, 60, 205–208. [Google Scholar] [PubMed]
- Zoja, C.; Viganò, G.; Bergamelli, A.; Benigni, A.; de Gaetano, G.; Remuzzi, G. Prolonged Bleeding Time and Increased Vascular Prostacyclin in Rats with Chronic Renal Failure: Effects of Conjugated Estrogens. J. Lab. Clin. Med. 1988, 112, 380–386. [Google Scholar]
- Defreyn, G.; Dauden, M.V.; Machin, S.J.; Vermylen, J. A Plasma Factor in Uraemia Which Stimulates Prostacyclin Release from Cultured Endothelial Cells. Thromb. Res. 1980, 19, 695–699. [Google Scholar] [CrossRef]
- Atkinson, B.; Dwyer, K.; Enjyoji, K.; Robson, S.C. Ecto-Nucleotidases of the CD39/NTPDase Family Modulate Platelet Activation and Thrombus Formation: Potential as Therapeutic Targets. Blood Cells Mol. Dis. 2006, 36, 217–222. [Google Scholar] [CrossRef]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating Platelet and Coagulation Activation in Fibrin Clot Formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef]
- Kishore, B.K.; Robson, S.C.; Dwyer, K.M. CD39-Adenosinergic Axis in Renal Pathophysiology and Therapeutics. Purinergic Signal. 2018, 14, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Roberts, V.; Lu, B.; Rajakumar, S.; Cowan, P.J.; Dwyer, K.M. The CD39-Adenosinergic Axis in the Pathogenesis of Renal Ischemia-Reperfusion Injury. Purinergic Signal. 2013, 9, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Kaczmarek, E.; Siegel, J.B.; Candinas, D.; Koziak, K.; Millan, M.; Hancock, W.W.; Bach, F.H. Loss of ATP Diphosphohydrolase Activity with Endothelial Cell Activation. J. Exp. Med. 1997, 185, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Köhler, D.; Eckle, T.; Faigle, M.; Grenz, A.; Mittelbronn, M.; Laucher, S.; Hart, M.L.; Robson, S.C.; Müller, C.E.; Eltzschig, H.K. CD39/Ectonucleoside Triphosphate Diphosphohydrolase 1 Provides Myocardial Protection during Cardiac Ischemia/Reperfusion Injury. Circulation 2007, 116, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Imai, M.; Nowak-Machen, M.; Guckelberger, O.; Enjyoji, K.; Wu, Y.; Khalpey, Z.; Berberat, P.; Munasinghe, J.; Robson, S.C. Liver Damage and Systemic Inflammatory Responses Are Exacerbated by the Genetic Deletion of CD39 in Total Hepatic Ischemia. Purinergic Signal. 2011, 7, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Guckelberger, O.; Sun, X.F.; Sévigny, J.; Imai, M.; Kaczmarek, E.; Enjyoji, K.; Kruskal, J.B.; Robson, S.C. Beneficial Effects of CD39/Ecto-Nucleoside Triphosphate Diphosphohydrolase-1 in Murine Intestinal Ischemia-Reperfusion Injury. Thromb. Haemost. 2004, 91, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Grenz, A.; Zhang, H.; Hermes, M.; Eckle, T.; Klingel, K.; Huang, D.Y.; Müller, C.E.; Robson, S.C.; Osswald, H.; Eltzschig, H.K. Contribution of E-NTPDase1 (CD39) to Renal Protection from Ischemia-Reperfusion Injury. FASEB J. 2007, 21, 2863–2873. [Google Scholar] [CrossRef]
- Crikis, S.; Lu, B.; Murray-Segal, L.M.; Selan, C.; Robson, S.C.; D’Apice, A.J.F.; Nandurkar, H.H.; Cowan, P.J.; Dwyer, K.M. Transgenic Overexpression of CD39 Protects against Renal Ischemia-Reperfusion and Transplant Vascular Injury. Am. J. Transpl. 2010, 10, 2586–2595. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; Wang, W.; Dai, Y.; Ning, C.; Luo, R.; Sun, K.; Glover, L.; Grenz, A.; Sun, H.; et al. Elevated Ecto-5’-Nucleotidase-Mediated Increased Renal Adenosine Signaling via A2B Adenosine Receptor Contributes to Chronic Hypertension. Circ. Res. 2013, 112, 1466–1478. [Google Scholar] [CrossRef]
- Lunkes, G.I.; Lunkes, D.; Stefanello, F.; Morsch, A.; Morsch, V.M.; Mazzanti, C.M.; Schetinger, M.R.C. Enzymes That Hydrolyze Adenine Nucleotides in Diabetes and Associated Pathologies. Thromb. Res. 2003, 109, 189–194. [Google Scholar] [CrossRef]
- Lunkes, G.I.; Lunkes, D.S.; Leal, D.; Araújo, M.d.C.; Corrêa, M.; Becker, L.; da Rosa, C.S.; Morsch, V.M.; Schetinger, M.R.C. Effect of High Glucose Levels in Human Platelet NTPDase and 5’-Nucleotidase Activities. Diabetes Res. Clin. Pract. 2008, 81, 351–357. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arter. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Erlich, J.; Fearns, C.; Mathison, J.; Ulevitch, R.J.; Mackman, N. Lipopolysaccharide Induction of Tissue Factor Expression in Rabbits. Infect. Immun. 1999, 67, 2540–2546. [Google Scholar] [CrossRef] [PubMed]
- Ushigome, H.; Sano, H.; Okamoto, M.; Kadotani, Y.; Nakamura, K.; Akioka, K.; Yoshimura, R.; Ohmori, Y.; Yoshimura, N. The Role of Tissue Factor in Renal Ischemic Reperfusion Injury of the Rat. J. Surg. Res. 2002, 102, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Welty-Wolf, K.E.; Carraway, M.S.; Miller, D.L.; Ortel, T.L.; Ezban, M.; Ghio, A.J.; Idell, S.; Piantadosi, C.A. Coagulation Blockade Prevents Sepsis-Induced Respiratory and Renal Failure in Baboons. Am. J. Respir. Crit. Care Med. 2001, 164, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic Uremic Solutes Increase Tissue Factor Production in Endothelial Cells by the Aryl Hydrocarbon Receptor Pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of Tissue Factor Induction by the Uremic Toxin Indole-3 Acetic Acid through Aryl Hydrocarbon Receptor/Nuclear Factor-Kappa B Signaling Pathway in Human Endothelial Cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl Sulfate Induces Platelet Hyperactivity and Contributes to Chronic Kidney Disease-Associated Thrombosis in Mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A Unified Theory of Sepsis-Induced Acute Kidney Injury: Inflammation, Microcirculatory Dysfunction, Bioenergetics, and the Tubular Cell Adaptation to Injury. Shock 2014, 41, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Härter, L.; Mica, L.; Stocker, R.; Trentz, O.; Keel, M. Increased Expression of Toll-like Receptor-2 and -4 on Leukocytes from Patients with Sepsis. Shock 2004, 22, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Viemann, D.; Dubbel, G.; Schleifenbaum, S.; Harms, E.; Sorg, C.; Roth, J. Expression of Toll-like Receptors in Neonatal Sepsis. Pediatr. Res. 2005, 58, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, G. TLR4 Inhibitor Alleviates Sepsis-Induced Organ Failure by Inhibiting Platelet mtROS Production, Autophagy, and GPIIb/IIIa Expression. J. Bioenerg. Biomembr. 2022, 54, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, G.M.; Castoldi, A.; Braga, T.T.; Câmara, N.O.S. New Roles for Innate Immune Response in Acute and Chronic Kidney Injuries. Scand. J. Immunol. 2011, 73, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.B.; Pulskens, W.P.; Butter, L.M.; Florquin, S.; Juffermans, N.P.; Roelofs, J.J.T.H.; Leemans, J.C. Mitochondrial DNA Is Released in Urine of SIRS Patients With Acute Kidney Injury and Correlates With Severity of Renal Dysfunction. Shock 2018, 49, 301. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.B.; Pulskens, W.P.C.; Uil, M.; Claessen, N.; Nieuwenhuizen, G.; Standaar, D.; Hau, C.M.; Nieuwland, R.; Florquin, S.; Bemelman, F.J.; et al. Urinary Mitochondrial DNA Associates with Delayed Graft Function Following Renal Transplantation. Nephrol. Dial. Transpl. 2020, 35, 1320–1327. [Google Scholar] [CrossRef]
- Zhao, Z.; Hu, Z.; Zeng, R.; Yao, Y. HMGB1 in Kidney Diseases. Life Sci. 2020, 259, 118203. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.-S.; Borst, O.; et al. Platelet-Derived HMGB1 Is a Critical Mediator of Thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.-C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets Release Mitochondria Serving as Substrate for Bactericidal Group IIA-Secreted Phospholipase A2 to Promote Inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, T.; Qiao, R. Pyroptosis in Platelets: Thrombocytopenia and Inflammation. J. Clin. Lab. Anal. 2023, 37, e24852. [Google Scholar] [CrossRef] [PubMed]
- Baaten, C.C.F.M.J.; Schröer, J.R.; Floege, J.; Marx, N.; Jankowski, J.; Berger, M.; Noels, H. Platelet Abnormalities in CKD and Their Implications for Antiplatelet Therapy. Clin. J. Am. Soc. Nephrol. 2022, 17, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Karbowska, M.; Kaminski, T.W.; Marcinczyk, N.; Misztal, T.; Rusak, T.; Smyk, L.; Pawlak, D. The Uremic Toxin Indoxyl Sulfate Accelerates Thrombotic Response after Vascular Injury in Animal Models. Toxins 2017, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Corken, A.L.; Kumar, A.; Davis, C.L.; Ware, J.; Arthur, J.M. Role of Platelets in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2021, 32, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Ferro, C.J.; Mark, P.B.; Kanbay, M.; Sarafidis, P.; Heine, G.H.; Rossignol, P.; Massy, Z.A.; Mallamaci, F.; Valdivielso, J.M.; Malyszko, J.; et al. Lipid Management in Patients with Chronic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 727–749. [Google Scholar] [CrossRef] [PubMed]
- Livio, M.; Gotti, E.; Marchesi, D.; Mecca, G.; Remuzzi, G.; de Gaetano, G. Uraemic Bleeding: Role of Anaemia and Beneficial Effect of Red Cell Transfusions. Lancet 1982, 2, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Turitto, V.T.; Baumgartner, H.R. Platelet Interaction with Subendothelium in a Perfusion System: Physical Role of Red Blood Cells. Microvasc. Res. 1975, 9, 335–344. [Google Scholar] [CrossRef]
- Gaarder, A.; Jonsen, J.; Laland, S.; Hellem, A.; Owren, P.A. Adenosine Diphosphate in Red Cells as a Factor in the Adhesiveness of Human Blood Platelets. Nature 1961, 192, 531–532. [Google Scholar] [CrossRef]
- Santos, M.T.; Valles, J.; Marcus, A.J.; Safier, L.B.; Broekman, M.J.; Islam, N.; Ullman, H.L.; Eiroa, A.M.; Aznar, J. Enhancement of Platelet Reactivity and Modulation of Eicosanoid Production by Intact Erythrocytes. A new approach to platelet activation and recruitment. J. Clin. Investig. 1991, 87, 571–580. [Google Scholar] [CrossRef]
- Willems, C.; Stel, H.V.; van Aken, W.G.; van Mourik, J.A. Binding and Inactivation of Prostacyclin (PGI2) by Human Erythrocytes. Br. J. Haematol. 1983, 54, 43–52. [Google Scholar] [CrossRef]
- Azarov, I.; Huang, K.T.; Basu, S.; Gladwin, M.T.; Hogg, N.; Kim-Shapiro, D.B. Nitric Oxide Scavenging by Red Blood Cells as a Function of Hematocrit and Oxygenation. J. Biol. Chem. 2005, 280, 39024–39032. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. CHOIR Investigators Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease. N. Engl. J. Med. 2006, 355, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Zahr, F.; Moguilanski, D.; Grate, S.; Semaan, R.W.; Lemieux, N.; Lee, J.S.; Cortese-Hassett, A.; Mulukutla, S.; Rao, S.V.; et al. Impact of Anemia on Platelet Response to Clopidogrel in Patients Undergoing Percutaneous Coronary Stenting. Am. J. Cardiol. 2012, 109, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Giustino, G.; Kirtane, A.J.; Baber, U.; Généreux, P.; Witzenbichler, B.; Neumann, F.-J.; Weisz, G.; Maehara, A.; Rinaldi, M.J.; Metzger, C.; et al. Impact of Anemia on Platelet Reactivity and Ischemic and Bleeding Risk: From the Assessment of Dual Antiplatelet Therapy With Drug-Eluting Stents Study. Am. J. Cardiol. 2016, 117, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Tanaka, T.; Nangaku, M. Future Perspectives of Anemia Management in Chronic Kidney Disease Using Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitors. Pharmacol. Ther. 2022, 239, 108272. [Google Scholar] [CrossRef] [PubMed]
- Grzeszczak, W.; Szczyra, D.; Śnit, M. Whether Prolyl Hydroxylase Blocker—Roxadustat—In the Treatment of Anemia in Patients with Chronic Kidney Disease Is the Future? Int. J. Environ. Res. Public Health 2021, 18, 1612. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Haase, V.H.; Hao, C.-M. Updates on Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors in the Treatment of Renal Anemia. Kidney Dis. 2023, 9, 1–11. [Google Scholar] [CrossRef]
- Ogawa, C.; Tsuchiya, K.; Tomosugi, N.; Maeda, K. Threshold of Serum Ferritin to Discriminate against Those at Greater Risk of Platelet Increase during Treatment with Hypoxia-Inducible Factor Prolyl Hydroxylase Domain Inhibitor. Acta Haematol. 2022, 145, 412–418. [Google Scholar] [CrossRef]
- Yoshida, T.; Okumura, T.; Matsuo, Y.; Okuyama, T.; Michiura, T.; Kaibori, M.; Umezaki, N.; Bono, H.; Hirota, K.; Sekimoto, M. Activation of Transcription Factor HIF Inhibits IL-1β-Induced NO Production in Primary Cultured Rat Hepatocytes. Nitric Oxide 2022, 124, 1–14. [Google Scholar] [CrossRef]
- Hu, X.; Xie, J.; Chen, N. Hypoxia-Inducible Factor-Proline Hydroxylase Inhibitor in the Treatment of Renal Anemia. Kidney Dis. 2021, 7, 1–9. [Google Scholar] [CrossRef]
- Yamamoto, T.; Tada, T.; Brodsky, S.V.; Tanaka, H.; Noiri, E.; Kajiya, F.; Goligorsky, M.S. Intravital Videomicroscopy of Peritubular Capillaries in Renal Ischemia. Am. J. Physiol. Ren. Physiol. 2002, 282, F1150–F1155. [Google Scholar] [CrossRef] [PubMed]
- Scholz, H.; Boivin, F.J.; Schmidt-Ott, K.M.; Bachmann, S.; Eckardt, K.-U.; Scholl, U.I.; Persson, P.B. Kidney Physiology and Susceptibility to Acute Kidney Injury: Implications for Renoprotection. Nat. Rev. Nephrol. 2021, 17, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Kurtcuoglu, V. Renal Blood Flow and Oxygenation. Pflug. Arch. 2022, 474, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Rustiasari, U.J.; Roelofs, J.J. The Role of Platelets in Diabetic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 8270. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Urbanus, R.T.; Ten Cate, H.; de Groot, P.G.; de Laat, B.; Heemskerk, J.W.M.; Roest, M. Platelet Activation Mechanisms and Consequences of Immune Thrombocytopenia. Cells 2021, 10, 3386. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Nishi, H. The Role of the Complement System in Kidney Glomerular Capillary Thrombosis. Front. Immunol. 2022, 13, 981375. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E. Hemodialysis-Associated Acute Systemic Reactions and Heparin-Induced Thrombocytopenia. Thromb. Res. 2012, 129, 405–406. [Google Scholar] [CrossRef]
- Honda, T.; Hirakawa, Y.; Nangaku, M. The Role of Oxidative Stress and Hypoxia in Renal Disease. Kidney Res. Clin. Pract. 2019, 38, 414–426. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Kobayashi, S.; Satoh, M.; Namikoshi, T.; Haruna, Y.; Fujimoto, S.; Arakawa, S.; Komai, N.; Tomita, N.; Sasaki, T.; Kashihara, N. Blockade of Serotonin 2A Receptor Improves Glomerular Endothelial Function in Rats with Streptozotocin-Induced Diabetic Nephropathy. Clin. Exp. Nephrol. 2008, 12, 119–125. [Google Scholar] [CrossRef]
- Macconi, D.; Noris, M.; Benfenati, E.; Quaglia, R.; Pagliarino, G.; Remuzzi, G. Increased Urinary Excretion of Platelet Activating Factor in Mice with Lupus Nephritis. Life Sci. 1991, 48, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L. Platelets in Glomerular Disease. Nephron 1997, 77, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, G. Eicosanoids and Platelet Activating Factor as Possible Mediators of Injury in Experimental Nephropathies. Adv. Exp. Med. Biol. 1989, 259, 221–247. [Google Scholar] [CrossRef] [PubMed]
- Amenta, P.S.; Katz, S.M. Platelets in Renal Scleroderma. Arch. Pathol. Lab. Med. 1983, 107, 439–440. [Google Scholar] [PubMed]
- Suthanthiran, M.; Strom, T.B. Renal Transplantation. N. Engl. J. Med. 1994, 331, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kant, K.S.; Pollak, V.E.; Weiss, M.A.; Glueck, H.I.; Miller, A.N.; Hess, E.V. Glomerular Thrombosis in Systemic Lupus Erythematosus: Prevalence and Significance. Medicine 1981, 60, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Buelli, S.; Zoja, C.; Remuzzi, G.; Morigi, M. Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Microorganisms 2019, 7, 15. [Google Scholar] [CrossRef]
- Duffy, J.L.; Cinque, T.; Grishman, E.; Churg, J. Intraglomerular Fibrin, Platelet Aggregation, and Subendothelial Deposits in Lipoid Nephrosis. J. Clin. Investig. 1970, 49, 251–258. [Google Scholar] [CrossRef]
- Nakajima, M.; Hewitson, T.D.; Mathews, D.C.; Kincaid-Smith, P. Platelet-Derived Growth Factor Mesangial Deposits in Mesangial IgA Glomerulonephritis. Nephrol. Dial. Transpl. 1991, 6, 11–16. [Google Scholar] [CrossRef]
- Duffus, P.; Parbtani, A.; Frampton, G.; Cameron, J.S. Intraglomerular Localization of Platelet Related Antigens, Platelet Factor 4 and Beta-Thromboglobulin in Glomerulonephritis. Clin. Nephrol. 1982, 17, 288–297. [Google Scholar]
- Li, S.; Wang, F.; Sun, D. The Renal Microcirculation in Chronic Kidney Disease: Novel Diagnostic Methods and Therapeutic Perspectives. Cell Biosci. 2021, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Kuligowski, M.P.; Kwan, R.Y.Q.; Westein, E.; Jackson, S.P.; Kitching, A.R.; Hickey, M.J. Platelet Recruitment to the Inflamed Glomerulus Occurs via an alphaIIbbeta3/GPVI-Dependent Pathway. Am. J. Pathol. 2010, 177, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Imanishi, M.; Okamura, M.; Hosoi, M.; Okada, N.; Konishi, Y.; Morikawa, T.; Miura, K.; Nakatani, T.; Fujii, S. Role for Thromboxane A2 from Glomerular Thrombi in Nephropathy with Type 2 Diabetic Rats. Life Sci. 2003, 72, 2695–2705. [Google Scholar] [CrossRef] [PubMed]
- Tönshoff, B.; Busch, C.; Schweer, H.; Schärer, K.; Seyberth, H.W. In Vivo Prostanoid Formation during Acute Renal Allograft Rejection. Nephrol. Dial. Transpl. 1993, 8, 631–636. [Google Scholar]
- Kelley, V.E.; Sneve, S.; Musinski, S. Increased Renal Thromboxane Production in Murine Lupus Nephritis. J. Clin. Investig. 1986, 77, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.C.; de Fijter, J.H. Cyclosporine-Induced Renal Dysfunction. Transplant. Proc. 2004, 36, 224S–228S. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.W.; Coffman, T.M. A Genetic Approach for Studying the Role of Thromboxane A2 in the Kidney. Kidney Int. Suppl. 1998, 67, S84–S87. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Valdivielso, J.M. New Therapeutic Targets in Chronic Kidney Disease Progression and Renal Fibrosis. Expert Opin. Ther. Targets 2020, 24, 655–670. [Google Scholar] [CrossRef]
- Inwald, D.P.; McDowall, A.; Peters, M.J.; Callard, R.E.; Klein, N.J. CD40 Is Constitutively Expressed on Platelets and Provides a Novel Mechanism for Platelet Activation. Circ. Res. 2003, 92, 1041–1048. [Google Scholar] [CrossRef]
- Ferroni, P.; Santilli, F.; Guadagni, F.; Basili, S.; Davì, G. Contribution of Platelet-Derived CD40 Ligand to Inflammation, Thrombosis and Neoangiogenesis. Curr. Med. Chem. 2007, 14, 2170–2180. [Google Scholar] [CrossRef]
- Rigothier, C.; Daculsi, R.; Lepreux, S.; Auguste, P.; Villeneuve, J.; Dewitte, A.; Doudnikoff, E.; Saleem, M.; Bourget, C.; Combe, C.; et al. CD154 Induces Matrix Metalloproteinase-9 Secretion in Human Podocytes. J. Cell. Biochem. 2016, 117, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.X.; Alderson, H.; Ritchie, J.; Kalra, P.A.; Xie, Y.; Ren, K.; Nguyen, H.; Chen, T.; Brewster, P.; Gupta, R.; et al. Circulating CD40 and sCD40L Predict Changes in Renal Function in Subjects with Chronic Kidney Disease. Sci. Rep. 2017, 7, 7942. [Google Scholar] [CrossRef] [PubMed]
- Mörtberg, J.; Lundwall, K.; Mobarrez, F.; Wallén, H.; Jacobson, S.H.; Spaak, J. Increased Concentrations of Platelet- and Endothelial-Derived Microparticles in Patients with Myocardial Infarction and Reduced Renal Function—A Descriptive Study. BMC Nephrol. 2019, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Lajer, M.; Tarnow, I.; Michelson, A.D.; Jorsal, A.; Frelinger, A.L.; Parving, H.-H.; Rossing, P.; Tarnow, L. Soluble CD40 Ligand Is Elevated in Type 1 Diabetic Nephropathy but Not Predictive of Mortality, Cardiovascular Events or Kidney Function. Platelets 2010, 21, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, F.; Giannini, C.; Verrotti, A.; Mezzetti, A.; Mohn, A. Increased Concentrations of Soluble CD40 Ligand May Help to Identify Type 1 Diabetic Adolescents and Young Adults at Risk for Developing Persistent Microalbuminuria. Diabetes Metab. Res. Rev. 2008, 24, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wu, H.; Fang, X.; He, J.; Zhu, F. Platelet, a Key Regulator of Innate and Adaptive Immunity. Front. Med. 2023, 10, 1074878. [Google Scholar] [CrossRef] [PubMed]
- Elangbam, C.S.; Qualls, C.W.; Dahlgren, R.R. Cell Adhesion Molecules--Update. Vet Pathol 1997, 34, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.D.; Burrow, C.R. Cystic Diseases of the Kidney: Role of Adhesion Molecules in Normal and Abnormal Tubulogenesis. Exp. Nephrol. 1999, 7, 114–124. [Google Scholar] [CrossRef]
- Rivero, A.; Mora, C.; Muros, M.; García, J.; Herrera, H.; Navarro-González, J.F. Pathogenic Perspectives for the Role of Inflammation in Diabetic Nephropathy. Clin. Sci. 2009, 116, 479–492. [Google Scholar] [CrossRef]
- Omoto, S.; Nomura, S.; Shouzu, A.; Hayakawa, T.; Shimizu, H.; Miyake, Y.; Yonemoto, T.; Nishikawa, M.; Fukuhara, S.; Inada, M. Significance of Platelet-Derived Microparticles and Activated Platelets in Diabetic Nephropathy. Nephron 1999, 81, 271–277. [Google Scholar] [CrossRef]
- Chen, J.; Tan, W. Platelet Activation and Immune Response in Diabetic Microangiopathy. Clin. Chim. Acta 2020, 507, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Shikata, K.; Matsuda, M.; Akiyama, K.; Sugimoto, H.; Kushiro, M.; Makino, H. Increased Expression of Selectins in Kidneys of Patients with Diabetic Nephropathy. Diabetologia 1998, 41, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Segawa, C.; Wada, T.; Takaeda, M.; Furuichi, K.; Matsuda, I.; Hisada, Y.; Ohta, S.; Takasawa, K.; Takeda, S.; Kobayashi, K.; et al. In Situ Expression and Soluble Form of P-Selectin in Human Glomerulonephritis. Kidney Int. 1997, 52, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Anders, H.-J. Thrombospondin Immune Regulation and the Kidney. Nephrol. Dial. Transpl. 2017, 32, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, M.A.; Rauova, L.; Poncz, M. Role of the Platelet Chemokine Platelet Factor 4 (PF4) in Hemostasis and Thrombosis. Thromb. Res. 2010, 125, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Xiang, S.C.; Rondina, M.T. Platelet Secretion in Inflammatory and Infectious Diseases. Platelets 2017, 28, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Carrier, M.; Rodger, M.A.; Fergusson, D.; Doucette, S.; Kovacs, M.J.; Moore, J.; Kelton, J.G.; Knoll, G.A. Increased Mortality in Hemodialysis Patients Having Specific Antibodies to the Platelet Factor 4-Heparin Complex. Kidney Int. 2008, 73, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Cavallo-Perin, P.; Romagnoli, R.; Ruiu, G.; Pagano, G. Plasma Beta-Thromboglobulin and Platelet Factor 4 Are Not Increased in Insulin-Dependent Diabetic Patients with Microalbuminuria. Acta Diabetol. 1994, 31, 130–132. [Google Scholar] [CrossRef]
- Soliman, S.A.; Haque, A.; Vanarsa, K.; Zhang, T.; Ismail, F.; Lee, K.H.; Pedroza, C.; Greenbaum, L.A.; Mason, S.; Hicks, M.J.; et al. Urine ALCAM, PF4 and VCAM-1 Surpass Conventional Metrics in Identifying Nephritis Disease Activity in Childhood-Onset Systemic Lupus Erythematosus. Front. Immunol. 2022, 13, 885307. [Google Scholar] [CrossRef]
- Whittall-Garcia, L.; Goliad, K.; Kim, M.; Bonilla, D.; Gladman, D.; Urowitz, M.; Fortin, P.R.; Atenafu, E.G.; Touma, Z.; Wither, J. Identification and Validation of a Urinary Biomarker Panel to Accurately Diagnose and Predict Response to Therapy in Lupus Nephritis. Front. Immunol. 2022, 13, 889931. [Google Scholar] [CrossRef]
- Mok, C.C.; Soliman, S.; Ho, L.Y.; Mohamed, F.A.; Mohamed, F.I.; Mohan, C. Urinary Angiostatin, CXCL4 and VCAM-1 as Biomarkers of Lupus Nephritis. Arthritis Res. Ther. 2018, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, Y.; Zhang, D.; Zhang, J.; Tian, Y. Platelet Factor 4 Protects Kidney Allograft in a Rat Kidney Transplantation Model. Inflammation 2015, 38, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial Dysfunction and Platelet Hyperactivity in Type 2 Diabetes Mellitus: Molecular Insights and Therapeutic Strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Kubisz, P.; Stančiaková, L.; Staško, J.; Galajda, P.; Mokáň, M. Endothelial and Platelet Markers in Diabetes Mellitus Type 2. World J. Diabetes 2015, 6, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Gurney, D.; Lip, G.Y.H.; Blann, A.D. A Reliable Plasma Marker of Platelet Activation: Does It Exist? Am. J. Hematol. 2002, 70, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Mamiya, S.; Satoh, M.; Takahashi, K.; Harada, T. Plasma Beta-Thromboglobulin and Platelet Factor 4 in Patients with Chronic Renal Failure and Effect of Hemodialysis. Tohoku J. Exp. Med. 1981, 135, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Andrassy, K.; Deppermann, D.; Ritz, E.; Koderisch, J.; Seelig, H. Different Effects of Renal Failure on Beta-Thromboglobulin and High Affinity Platelet Factor 4 (HA-PF4)-Concentrations. Thromb. Res. 1980, 18, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Hopper, A.H.; Tindall, H.; Davies, J.A. Urinary Beta-Thromboglobulin Correlates with Impairment of Renal Function in Patients with Diabetic Nephropathy. Thromb. Haemost. 1986, 56, 229–231. [Google Scholar] [CrossRef]
- Kahn, M.L.; Nakanishi-Matsui, M.; Shapiro, M.J.; Ishihara, H.; Coughlin, S.R. Protease-Activated Receptors 1 and 4 Mediate Activation of Human Platelets by Thrombin. J. Clin. Investig. 1999, 103, 879–887. [Google Scholar] [CrossRef]
- Rwibasira Rudinga, G.; Khan, G.J.; Kong, Y. Protease-Activated Receptor 4 (PAR4): A Promising Target for Antiplatelet Therapy. Int. J. Mol. Sci. 2018, 19, 573. [Google Scholar] [CrossRef]
- Bagang, N.; Gupta, K.; Singh, G.; Kanuri, S.H.; Mehan, S. Protease-Activated Receptors in Kidney Diseases: A Comprehensive Review of Pathological Roles, Therapeutic Outcomes and Challenges. Chem. Biol. Interact. 2023, 377, 110470. [Google Scholar] [CrossRef] [PubMed]
- Lok, S.W.Y.; Yiu, W.H.; Li, H.; Xue, R.; Zou, Y.; Li, B.; Chan, K.W.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; et al. The PAR-1 Antagonist Vorapaxar Ameliorates Kidney Injury and Tubulointerstitial Fibrosis. Clin. Sci. 2020, 134, 2873–2891. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.A.; Rondeau, E.; Chen, X.; Coughlin, S.R.; Holdsworth, S.R.; Tipping, P.G. Protease-Activated Receptor 1 Mediates Thrombin-Dependent, Cell-Mediated Renal Inflammation in Crescentic Glomerulonephritis. J. Exp. Med. 2000, 191, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G. Potential Role of Platelet-Activating Factor in Renal Pathophysiology. Kidney Int. 1986, 29, 469–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Correa-Costa, M.; Andrade-Oliveira, V.; Braga, T.T.; Castoldi, A.; Aguiar, C.F.; Origassa, C.S.T.; Rodas, A.C.D.; Hiyane, M.I.; Malheiros, D.M.A.C.; Rios, F.J.O.; et al. Activation of Platelet-Activating Factor Receptor Exacerbates Renal Inflammation and Promotes Fibrosis. Lab. Investig. 2014, 94, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Perico, N.; Remuzzi, A.; Dadan, J.; Battaglia, C.; Remuzzi, G. Platelet-Activating Factor Alters Glomerular Barrier Size Selectivity for Macromolecules in Rats. Am. J. Physiol. 1991, 261, F85–F90. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Macconi, D.; Riccardi, E.; Boccardo, P.; Zilio, P.; Bertani, T.; Remuzzi, G. Platelet-Activating Factor Receptor Blocking Reduces Proteinuria and Improves Survival in Lupus Autoimmune Mice. J. Pharmacol. Exp. Ther. 1991, 258, 601–606. [Google Scholar] [PubMed]
- Jansen, M.P.B.; Emal, D.; Teske, G.J.D.; Dessing, M.C.; Florquin, S.; Roelofs, J.J.T.H. Release of Extracellular DNA Influences Renal Ischemia Reperfusion Injury by Platelet Activation and Formation of Neutrophil Extracellular Traps. Kidney Int. 2017, 91, 352–364. [Google Scholar] [CrossRef]
- Salazar-Gonzalez, H.; Zepeda-Hernandez, A.; Melo, Z.; Saavedra-Mayorga, D.E.; Echavarria, R. Neutrophil Extracellular Traps in the Establishment and Progression of Renal Diseases. Medicina 2019, 55, 431. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef]
- Okubo, K.; Kurosawa, M.; Kamiya, M.; Urano, Y.; Suzuki, A.; Yamamoto, K.; Hase, K.; Homma, K.; Sasaki, J.; Miyauchi, H.; et al. Macrophage Extracellular Trap Formation Promoted by Platelet Activation Is a Key Mediator of Rhabdomyolysis-Induced Acute Kidney Injury. Nat. Med. 2018, 24, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Bellio, M. Platelet Extracellular Vesicles and the Secretory Interactome Join Forces in Health and Disease. Immunol. Rev. 2022, 312, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet Extracellular Vesicles: Beyond the Blood. Arter. Thromb. Vasc. Biol. 2021, 41, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Vajen, T.; Benedikter, B.J.; Heinzmann, A.C.A.; Vasina, E.M.; Henskens, Y.; Parsons, M.; Maguire, P.B.; Stassen, F.R.; Heemskerk, J.W.M.; Schurgers, L.J.; et al. Platelet Extracellular Vesicles Induce a Pro-Inflammatory Smooth Muscle Cell Phenotype. J. Extracell. Vesicles 2017, 6, 1322454. [Google Scholar] [CrossRef] [PubMed]
- Melki, I.; Allaeys, I.; Tessandier, N.; Mailhot, B.; Cloutier, N.; Campbell, R.A.; Rowley, J.W.; Salem, D.; Zufferey, A.; Laroche, A.; et al. FcγRIIA Expression Accelerates Nephritis and Increases Platelet Activation in Systemic Lupus Erythematosus. Blood 2020, 136, 2933–2945. [Google Scholar] [CrossRef] [PubMed]
- Scherlinger, M.; Richez, C.; Tsokos, G.C.; Boilard, E.; Blanco, P. The Role of Platelets in Immune-Mediated Inflammatory Diseases. Nat. Rev. Immunol. 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Yang, H.; Harris, H. Extracellular HMGB1 as a Therapeutic Target in Inflammatory Diseases. Expert Opin. Ther. Targets 2018, 22, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.R.; Chen, Q.; Haldeman, S.; Yazdani, H.; Hoffman, R.; Loughran, P.; Tsung, A.; Zuckerbraun, B.S.; Simmons, R.L.; Neal, M.D. Deep Vein Thrombosis in Mice Is Regulated by Platelet HMGB1 through Release of Neutrophil-Extracellular Traps and DNA. Sci. Rep. 2018, 8, 2068. [Google Scholar] [CrossRef]
- Pfeiler, S.; Stark, K.; Massberg, S.; Engelmann, B. Propagation of Thrombosis by Neutrophils and Extracellular Nucleosome Networks. Haematologica 2017, 102, 206–213. [Google Scholar] [CrossRef]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Brühl, M.-L.; et al. Disulfide HMGB1 Derived from Platelets Coordinates Venous Thrombosis in Mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Liu, P.; Li, F.; Xu, X.; Li, S.; Dong, X.; Chen, L.; Bai, B.; Wang, Y.; Qiu, M.; Dong, Y. 1,25(OH)2D3 Provides Protection against Diabetic Kidney Disease by Downregulating the TLR4-MyD88-NF-κB Pathway. Exp. Mol. Pathol. 2020, 114, 104434. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Sourris, K.; Ziemann, M.; Tieqiao, W.; Mohan, M.; McClelland, A.D.; Brennan, E.; Forbes, J.; Coughlan, M.; Harcourt, B.; et al. RAGE Deletion Confers Renoprotection by Reducing Responsiveness to Transforming Growth Factor-β and Increasing Resistance to Apoptosis. Diabetes 2018, 67, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Zhang, Y.; Wang, C.; Gu, Y.; Zhang, Y.; Li, X. Platelets Derived Transthyretin Participate in The Development of Sepsis Associated Acute Kidney Injury by Inducing Oxidative Stress and Apoptosis of Renal Tubular Epithelial Cells. Shock 2022, 57, 722. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Inoue, H.; Sasahara, M. Platelet-Derived Growth Factor and Renal Disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Ostendorf, T.; Eitner, F.; Floege, J. The PDGF Family in Renal Fibrosis. Pediatr. Nephrol. 2012, 27, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Kok, H.M.; Falke, L.L.; Goldschmeding, R.; Nguyen, T.Q. Targeting CTGF, EGF and PDGF Pathways to Prevent Progression of Kidney Disease. Nat. Rev. Nephrol. 2014, 10, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFβ. Int. J. Mol. Sci. 2021, 22, 4492. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Rowland-Jones, S.L. RANTES: A Versatile and Controversial Chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Mezzano, S.; Aros, C.; Droguett, A.; Burgos, M.E.; Ardiles, L.; Flores, C.; Schneider, H.; Ruiz-Ortega, M.; Egido, J. NF-kappaB Activation and Overexpression of Regulated Genes in Human Diabetic Nephropathy. Nephrol. Dial. Transpl. 2004, 19, 2505–2512. [Google Scholar] [CrossRef]
- Feng, S.-T.; Yang, Y.; Yang, J.-F.; Gao, Y.-M.; Cao, J.-Y.; Li, Z.-L.; Tang, T.-T.; Lv, L.-L.; Wang, B.; Wen, Y.; et al. Urinary Sediment CCL5 Messenger RNA as a Potential Prognostic Biomarker of Diabetic Nephropathy. Clin. Kidney J. 2022, 15, 534–544. [Google Scholar] [CrossRef]
- Gong, S.; Wang, C.; Xiong, J.; Zhao, J.; Yang, K. Activated Platelets, the Booster of Chronic Kidney Disease and Cardiovascular Complications. Kidney Dis. 2022, 8, 297–307. [Google Scholar] [CrossRef]
- Nastase, M.V.; Zeng-Brouwers, J.; Wygrecka, M.; Schaefer, L. Targeting Renal Fibrosis: Mechanisms and Drug Delivery Systems. Adv. Drug Deliv. Rev. 2018, 129, 295–307. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Granule | Type | Contents |
|---|---|---|
| α-granules | Adhesive proteins | P-selectin, Von Willebrand factor, Fibronectin, Vitronectin, Fibrinogen |
| Integral membrane proteins | Integrin αIIbβ3, GPIba-IX-V, GPVI, TLT-1 | |
| Chemokines | CXCL1, CXCL2, CXCL5, CXCL6, CXCL7, CXCL8 (IL 8), CXCL-12, CCL2, CCL3, CCL5 (RANTES), CCL7, IL1β, CD40L Proteases | |
| Growth factors | Transforming growth factor β (TGF-β), Platelet-derived growth factor (PDGF), Vascular endothelium growth factor (VEGF), Fibroblast growth factor (FGF), Insulin-like growth factor 1 (IGF-1), Epidermal growth factor (EGF) | |
| Coagulation factors | Factor V, Protein S, Factor XI, Factor XIII | |
| Immune mediators | Complement C3 precursor, Complement C4 precursor, Factor D, Factor H, C1 inhibitor, Immunoglobulins | |
| δ-granules | Nucleotides | ATP, ADP, GTP, GDP |
| Bivalent cations | Ca2+, Mg2+ | |
| Amines | Serotonin, Histamine | |
| Lysosome | Glycohydrolases | Heparinase, β-N-acetyl-glucosaminidase |
| Acid proteases | Aryl sulphatase, Cathepsins, Collagenase, Acid phosphatase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomchok, D.; Ge, R.-L.; Wuren, T. Platelets in Renal Disease. Int. J. Mol. Sci. 2023, 24, 14724. https://doi.org/10.3390/ijms241914724
Gomchok D, Ge R-L, Wuren T. Platelets in Renal Disease. International Journal of Molecular Sciences. 2023; 24(19):14724. https://doi.org/10.3390/ijms241914724
Chicago/Turabian StyleGomchok, Drolma, Ri-Li Ge, and Tana Wuren. 2023. "Platelets in Renal Disease" International Journal of Molecular Sciences 24, no. 19: 14724. https://doi.org/10.3390/ijms241914724
APA StyleGomchok, D., Ge, R.-L., & Wuren, T. (2023). Platelets in Renal Disease. International Journal of Molecular Sciences, 24(19), 14724. https://doi.org/10.3390/ijms241914724