
The Spectrum of Cognitive Dysfunction in Amyotrophic Lateral Sclerosis: An Update


Abstract
:1. Introduction
2. Cognitive Abnormalities in ALS
3. Neuroimaging Findings in ALSci
4. Brain Perfusion and Metabolism Studies
5. Brain Network Studies
6. Neurophysiological Studies in ALSci
7. Neuropathological Findings
8. Cognitive Reserve in ALS
9. Experimental Models of ALSci
10. Putative Pathogenic Mechanisms
11. Conclusions and Outlook
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ALSci | amyotrophic lateral sclerosis with cognitive impairment |
| CI | cognitive impairment |
| CR | cognitive reserve |
| CSF | cerebrospinal fluid |
| DMN | default mode network |
| EF | executive function |
| ERP | event-related potentials |
| FA | fractional anisotropy |
| fALS | familial ALS |
| FTLD | frontotemporal lobe degeneration |
| GM | gray matter |
| MCI | mild cognitive impairment |
| rCBF | regional cerebral blood flow |
| ReHo | regional homogeneity |
| RSN | resting state network |
| sALS | sporadic ALS |
| SMN | sensorimotor network |
| SN | salience network |
| tg | transgenic |
References
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Berry, J.D.; Blanchard, M.; Bonar, K.; Drane, E.; Murton, M.; Ploug, U.; Ricchetti-Masterson, K.; Savic, N.; Worthington, E.; Heiman-Patterson, T. Epidemiology and economic burden of amyotrophic lateral sclerosis in the United States: A literature review. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 436–448. [Google Scholar] [CrossRef]
- Logroscino, G.; Piccininni, M. Amyotrophic lateral sclerosis descriptive epidemiology: The origin of geographic difference. Neuroepidemiology 2019, 52, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Marin, B.; Boumédiene, F.; Logroscino, G.; Couratier, P.; Babron, M.C.; Leutenegger, A.L.; Copetti, M.; Preux, P.M.; Beghi, E. Variation in worldwide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Hop, P.J.; Zwamborn, R.A.J.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.; Dekker, A.M.; Westeneng, H.J.; et al. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lin, F.; Ye, C.H.; Huang, H.; Li, X.; Yao, X.; Xu, Y.; Wang, C. Rare, low-frequency and common coding variants of ARHGEF28 gene and their association with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2020, 87, 138.e131–138.e136. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.D.; Khan, Y.S. Motor Neuron Disease; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560774/ (accessed on 10 May 2023).
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2023, 68, 131–152. [Google Scholar] [CrossRef]
- Megat, S.; Mora, N.; Sanogo, J.; Roman, O.; Catanese, A.; Alami, N.O.; Freischmidt, A.; Mingaj, X.; De Calbiac, H.; Muratet, F.; et al. Integrative genetic analysis illuminates ALS heritability and identifies risk genes. Nat. Commun. 2023, 14, 342. [Google Scholar] [CrossRef]
- Placek, K.; Benatar, M.; Wuu, J.; Rampersaud, E.; Hennessy, L.; Van Deerlin, V.M.; Grossman, M.; Irwin, D.J.; Elman, L.; McCluskey, L.; et al. Machine learning suggests polygenic risk for cognitive dysfunction in amyotrophic lateral sclerosis. EMBO Mol. Med. 2021, 13, e12595. [Google Scholar] [CrossRef]
- McHutchison, C.A.; Leighton, D.J.; McIntosh, A.; Cleary, E.; Warner, J.; Porteous, M.; Chandran, S.; Pal, S.; Abrahams, S. Relationship between neuropsychiatric disorders and cognitive and behavioural change in MND. J. Neurol. Neurosurg. Psychiatry 2020, 91, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron. Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Carvalho, M.D.; Swash, M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph. Lateral Scler. 2009, 10, 53–57. [Google Scholar] [CrossRef]
- Luan, W.; Wright, A.L.; Brown-Wright, H.; Le, S.; San Gil, R.; Madrid San Martin, L.; Ling, K.; Jafar-Nejad, P.; Rigo, F.; Walker, A.K. Early activation of cellular stress and death pathways caused by cytoplasmic TDP-43 in the rNLS8 mouse model of ALS and FTD. Mol. Psychiatry 2023. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular mechanisms underlying TDP-43 pathology in cellular and animal models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef] [PubMed]
- Aousji, O.; Feldengut, S.; Antonucci, S.; Schön, M.; Boeckers, T.M.; Matschke, J.; Mawrin, C.; Ludolph, A.C.; Del Tredici, K.; Roselli, F.; et al. Patterns of synaptic loss in human amyotrophic lateral sclerosis spinal cord: A clinicopathological study. Acta Neuropathol. Commun. 2023, 11, 120. [Google Scholar] [CrossRef]
- Broadhead, M.J.; Bonthron, C.; Waddington, J.; Smith, W.V.; Lopez, M.F.; Burley, S.; Valli, J.; Zhu, F.; Komiyama, N.H.; Smith, C.; et al. Selective vulnerability of tripartite synapses in amyotrophic lateral sclerosis. Acta Neuropathol. 2022, 143, 471–486. [Google Scholar] [CrossRef]
- Rusina, R.; Vandenberghe, R.; Bruffaerts, R. Cognitive and behavioral manifestations in ALS: Beyond motor system involvement. Diagnostics 2021, 11, 624. [Google Scholar] [CrossRef]
- Benatar, M.; Turner, M.R.; Wuu, J. Presymptomatic amyotrophic lateral sclerosis: From characterization to prevention. Curr. Opin. Neurol. 2023, 36, 360–364. [Google Scholar] [CrossRef]
- Corcia, P.; Couratier, P. Cognitive impairment in ALS has been known since the nineteenth century. Neurol. Sci. 2023, 44, 735–736. [Google Scholar] [CrossRef]
- Xu, Z.; Alruwaili, A.R.S.; Henderson, R.D.; McCombe, P.A. Screening for cognitive and behavioural impairment in amyotrophic lateral sclerosis: Frequency of abnormality and effect on survival. J. Neurol. Sci. 2017, 376, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Leiva, D.; Dols-Icardo, O.; Sirisi, S.; Cortés-Vicente, E.; Turon-Sans, J.; de Luna, N.; Blesa, R.; Belbin, O.; Montal, V.; Alcolea, D.; et al. Pathophysiological underpinnings of extra-motor neurodegeneration in amyotrophic lateral sclerosis: New insights from biomarker studies. Front. Neurol. 2022, 12, 750543. [Google Scholar] [CrossRef]
- Beeldman, E.; Raaphorst, J.; Klein Twennaar, M.; Govaarts, R.; Pijnenburg, Y.A.L.; de Haan, R.J.; de Visser, M.; Schmand, B.A. The cognitive profile of behavioural variant FTD and its similarities with ALS: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 995–1002. [Google Scholar] [CrossRef]
- Bersano, E.; Sarnelli, M.F.; Solara, V.; Iazzolino, B.; Peotta, L.; De Marchi, F.; Facchin, A.; Moglia, C.; Canosa, A.; Calvo, A.; et al. Decline of cognitive and behavioral functions in amyotrophic lateral sclerosis: A longitudinal study. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ceslis, A.; Argall, R.; Henderson, R.D.; McCombe, P.A.; Robinson, G.A. The spectrum of language impairments in amyotrophic lateral sclerosis. Cortex 2020, 132, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.M.; McDade, K.; Bak, T.H.; Pal, S.; Chandran, S.; Smith, C.; Abrahams, S. Executive, language and fluency dysfunction are markers of localised TDP-43 cerebral pathology in non-demented ALS. J. Neurol. Neurosurg. Psychiatry 2020, 91, 149–157. [Google Scholar] [CrossRef] [PubMed]
- McMillan, C.T.; Wuu, J.; Rascovsky, K.; Cosentino, S.; Grossman, M.; Elman, L.; Quinn, C.; Rosario, L.; Stark, J.H.; Granit, V.; et al. Defining cognitive impairment in amyotrophic lateral sclerosis: An evaluation of empirical approaches. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 517–526. [Google Scholar] [CrossRef]
- Rabkin, J.; Goetz, R.; Murphy, J.M.; Factor-Litvak, P.; Mitsumoto, H. Cognitive impairment, behavioral impairment, depression, and wish to die in an ALS cohort. Neurology 2016, 87, 1320–1328. [Google Scholar] [CrossRef]
- Rippon, G.A.; Scarmeas, N.; Gordon, P.H.; Murphy, P.L.; Albert, S.M.; Mitsumoto, H.; Marder, K.; Rowland, L.P.; Stern, Y. An observational study of cognitive impairment in amyotrophic lateral sclerosis. Arch. Neurol. 2006, 63, 345–352. [Google Scholar] [CrossRef]
- Ye, S.; Jin, P.P.; Zhang, N.; Wu, H.B.; Shi, L.; Zhao, Q.; Yang, K.; Yuan, H.S.; Fan, D.S. Cortical thickness and cognitive impairment in patients with amyotrophic lateral sclerosis. Beijing Da Xue Xue Bao Yi Xue Ban 2022, 54, 1158–1162. [Google Scholar]
- Huynh, W.; Ahmed, R.; Mahoney, C.J.; Nguyen, C.; Tu, S.; Caga, J.; Loh, P.; Lin, C.S.; Kiernan, M.C. The impact of cognitive and behavioral impairment in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2020, 20, 281–293. [Google Scholar] [CrossRef]
- Watanabe, Y.; Raaphorst, J.; Izumi, Y.; Yoshino, H.; Ito, S.; Adachi, T.; Takigawa, H.; Masuda, M.; Atsuta, N.; Adachi, Y.; et al. Cognitive and behavioral status in Japanese ALS patients: A multicenter study. J. Neurol. 2020, 267, 1321–1330. [Google Scholar] [CrossRef]
- Ringholz, G.M.; Appel, S.H.; Bradshaw, M.; Cooke, N.A.; Mosnik, D.M.; Schulz, P.E. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005, 65, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Phukan, J.; Elamin, M.; Bede, P.; Jordan, N.; Gallagher, L.; Byrne, S.; Lynch, C.; Pender, N.; Hardiman, O. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: A population-based study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Zhao, Q.; Liu, P.; Yuan, Y.; Liu, Z.; Hu, Y.; Li, W.; Hou, X.; Tang, X.; Jiao, B.; et al. ALS-plus related clinical and genetic study from China. Neurol. Sci. 2023, 44, 3557–3566. [Google Scholar] [CrossRef]
- Maier, P.M.; Iggena, D.; Meyer, T.; Finke, C.; Ploner, C.J. Memory-guided navigation in amyotrophic lateral sclerosis. J. Neurol. 2023, 270, 4031–4040. [Google Scholar] [CrossRef]
- Wheaton, M.W.; Salamone, A.R.; Mosnik, D.M.; McDonald, R.O.; Appel, S.H.; Schmolck, H.I.; Ringholz, G.M.; Schulz, P.E. Cognitive impairment in familial ALS. Neurology 2007, 69, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Beeldman, E.; Raaphorst, J.; Klein Twennaar, M.; de Visser, M.; Schmand, B.A.; de Haan, R.J. The cognitive profile of ALS: A systematic review and meta-analysis update. J. Neurol. Neurosurg. Psychiatry 2016, 87, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Nakamura, T.; Suzuki, M.; Ueda, M.; Hirayama, M.; Katsuno, M. Impaired pain processing and its association with attention disturbance in patients with amyotrophic lateral sclerosis. Neurol. Sci. 2021, 42, 3327–3335. [Google Scholar] [CrossRef]
- Oh, S.I.; Park, A.; Kim, H.J.; Oh, K.W.; Choi, H.; Kwon, M.J.; Ki, C.S.; Kim, H.T.; Kim, S.H. Spectrum of cognitive impairment in Korean ALS patients without known genetic mutations. PLoS ONE 2014, 9, e87163. [Google Scholar] [CrossRef]
- Panopoulou, N.; Christidi, F.; Kourtesis, P.; Ferentinos, P.; Karampetsou, P.; Tsirtsiridis, G.; Theodosiou, T.; Xirou, S.; Zouvelou, V.; Evdokimidis, I.; et al. The association of theory of mind with language and visuospatial abilities in amyotrophic lateral sclerosis: A pilot study. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.N.; Sampson, J.B. Cognitive profile of C9orf72 in frontotemporal dementia and amyotrophic lateral sclerosis. Curr. Neurol. Neurosci. Rep. 2015, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Paulus, K.S.; Magnano, I.; Piras, M.R.; Solinas, M.A.; Solinas, G.; Sau, G.F.; Aiello, I. Visual and auditory event-related potentials in sporadic amyotrophic lateral sclerosis. Clin. Neurophysiol. 2002, 113, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Machts, J.; Bittner, V.; Kasper, E.; Schuster, C.; Prudlo, J.; Abdulla, S.; Kollewe, K.; Petri, S.; Dengler, R.; Heinze, H.J.; et al. Memory deficits in amyotrophic lateral sclerosis are not exclusively caused by executive dysfunction: A comparative neuropsychological study of amnestic mild cognitive impairment. BMC Neurosci. 2014, 15, 83. [Google Scholar] [CrossRef]
- Schreiber, H.; Gaigalat, T.; Wiedemuth-Catrinescu, U.; Graf, M.; Uttner, I.; Muche, R.; Ludolph, A.C. Cognitive function in bulbar- and spinal-onset amyotrophic lateral sclerosis. A longitudinal study in 52 patients. J. Neurol. 2005, 252, 772–781. [Google Scholar] [CrossRef]
- Abrahams, S.; Leigh, P.N.; Goldstein, L.H. Cognitive change in ALS: A prospective study. Neurology 2005, 64, 1222–1226. [Google Scholar] [CrossRef]
- Finsel, J.; Uttner, I.; Vázquez Medrano, C.R.; Ludolph, A.C.; Lulé, D. Cognition in the course of ALS—A meta-analysis. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 2–13. [Google Scholar] [CrossRef]
- Pinto-Grau, M.; Donohoe, B.; O’Connor, S.; Murphy, L.; Costello, E.; Heverin, M.; Vajda, A.; Hardiman, O.; Pender, N. Patterns of language impairment in early amyotrophic lateral sclerosis. Neurol. Clin. Pract. 2021, 11, e634–e644. [Google Scholar] [CrossRef]
- Solca, F.; Aiello, E.N.; Torre, S.; Carelli, L.; Ferrucci, R.; Verde, F.; Ticozzi, N.; Silani, V.; Monti, A.; Poletti, B. Prevalence and determinants of language impairment in non-demented amyotrophic lateral sclerosis patients. Eur. J. Neurol. 2023, 30, 606–611. [Google Scholar] [CrossRef]
- Sakurai, T.; Hirano, S.; Abe, M.; Uji, Y.; Shimizu, K.; Suzuki, M.; Nakano, Y.; Ishikawa, A.; Kojima, K.; Shibuya, K.; et al. Dysfunction of the left angular gyrus may be associated with writing errors in ALS. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 267–275. [Google Scholar] [CrossRef]
- De Marchi, F.; Carrarini, C.; De Martino, A.; Diamanti, L.; Fasano, A.; Lupica, A.; Russo, M.; Salemme, S.; Spinelli, E.G.; Bombaci, A. Cognitive dysfunction in amyotrophic lateral sclerosis: Can we predict it? Neurol. Sci. 2021, 42, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Saxon, J.A.; Thompson, J.C.; Harris, J.M.; Richardson, A.M.; Langheinrich, T.; Rollinson, S.; Pickering-Brown, S.; Chaouch, A.; Ealing, J.; Hamdalla, H.; et al. Cognition and behaviour in frontotemporal dementia with and without amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.; Lippa, C.F.; Swearer, J.M. Cognition and amyotrophic lateral sclerosis (ALS). Am. J. Alzheimers Dis. Other Demen. 2007, 22, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Consonni, M.; Dalla Bella, E.; Bersano, E.; Lauria, G. Cognitive and behavioural impairment in amyotrophic lateral sclerosis: A landmark of the disease? A mini review of longitudinal studies. Neurosci. Lett 2021, 754, 135898. [Google Scholar] [CrossRef] [PubMed]
- Consonni, M.; Dalla Bella, E.; Bersano, E.; Telesca, A.; Lauria, G. Cognitive reserve is associated with altered clinical expression in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 237–247. [Google Scholar] [CrossRef]
- Crockford, C.; Newton, J.; Lonergan, K.; Chiwera, T.; Booth, T.; Chandran, S.; Colville, S.; Heverin, M.; Mays, I.; Pal, S.; et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 2018, 91, e1370–e1380. [Google Scholar] [CrossRef]
- Dubbioso, R.; Spisto, M.; Hausdorff, J.M.; Aceto, G.; Iuzzolino, V.V.; Senerchia, G.; De Marco, S.; Marcuccio, L.; Femiano, C.; Iodice, R.; et al. Cognitive impairment is associated with gait variability and fall risk in amyotrophic lateral sclerosis. Eur. J. Neurol. 2023, 30, 3056–3067. [Google Scholar] [CrossRef]
- Murphy, J.; Factor-Litvak, P.; Goetz, R.; Lomen-Hoerth, C.; Nagy, P.L.; Hupf, J.; Singleton, J.; Woolley, S.; Andrews, H.; Heitzman, D.; et al. Cognitive-behavioral screening reveals prevalent impairment in a large multicenter ALS cohort. Neurology 2016, 86, 813–820. [Google Scholar] [CrossRef]
- Elamin, M.; Bede, P.; Byrne, S.; Jordan, N.; Gallagher, L.; Wynne, B.; O’Brien, C.; Phukan, J.; Lynch, C.; Pender, N.; et al. Cognitive changes predict functional decline in ALS: A population-based longitudinal study. Neurology 2013, 80, 1590–1597. [Google Scholar] [CrossRef]
- Wei, Q.Q.; Ou, R.; Cao, B.; Chen, Y.; Hou, Y.; Zhang, L.; Wu, F.; Shang, H. Early weight instability is associated with cognitive decline and poor survival in amyotrophic lateral sclerosis. Brain Res. Bull. 2021, 171, 10–15. [Google Scholar] [CrossRef]
- Ye, S.; Jin, P.; Chen, L.; Zhang, N.; Fan, D. Prognosis of amyotrophic lateral sclerosis with cognitive and behavioural changes based on a sixty-month longitudinal follow-up. PLoS ONE 2021, 16, e0253279. [Google Scholar] [CrossRef]
- Stojkovic, T.; Stefanova, E.; Pekmezovic, T.; Peric, S.; Stevic, Z. Executive dysfunction and survival in patients with amyotrophic lateral sclerosis: Preliminary report from a Serbian centre for motor neuron disease. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 543–547. [Google Scholar] [CrossRef]
- Manera, U.; Peotta, L.; Iazzolino, B.; Canosa, A.; Vasta, R.; Palumbo, F.; Torrieri, M.C.; Solero, L.; Daviddi, M.; Grassano, M.; et al. The characteristics of cognitive impairment in als patients depend on the lateralization of motor damage. Brain Sci. 2020, 10, 650. [Google Scholar] [CrossRef]
- Aiello, E.N.; Pain, D.; Radici, A.; Aktipi, K.M.; Sideri, R.; Appollonio, I.; Mora, G. Cognition and motor phenotypes in ALS: A retrospective study. Neurol. Sci. 2022, 43, 5397–5402. [Google Scholar] [CrossRef] [PubMed]
- Sterling, L.E.; Jawaid, A.; Salamone, A.R.; Murthy, S.B.; Mosnik, D.M.; McDowell, E.; Wheaton, M.; Strutt, A.M.; Simpson, E.; Appel, S.; et al. Association between dysarthria and cognitive impairment in ALS: A prospective study. Amyotroph. Lateral Scler. 2010, 11, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Schrempf, T.; Finsel, J.; Uttner, I.; Ludolph, A.C.; Lulé, D. Neuropsychological deficits have only limited impact on psychological well-being in amyotrophic lateral sclerosis. J. Neurol. 2022, 269, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Elamin, M.; Bede, P.; Shatunov, A.; Walsh, C.; Corr, B.; Heverin, M.; Jordan, N.; Kenna, K.; Lynch, C.; et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: A population-based cohort study. Lancet Neurol. 2012, 11, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; Vasta, R.; Brunetti, M.; Barberis, M.; Corrado, L.; D’Alfonso, S.; Bersano, E.; et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 2019, 93, e984–e994. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Poletti, B.; Maranzano, A.; Peverelli, S.; Solca, F.; Colombrita, C.; Torre, S.; Tiloca, C.; Verde, F.; Bonetti, R.; et al. Motor, cognitive and behavioural profiles of C9orf72 expansion-related amyotrophic lateral sclerosis. J. Neurol. 2023, 270, 898–908. [Google Scholar] [CrossRef]
- Iazzolino, B.; Peotta, L.; Zucchetti, J.P.; Canosa, A.; Manera, U.; Vasta, R.; Grassano, M.; Palumbo, F.; Brunetti, M.; Barberis, M.; et al. Differential neuropsychological profile of patients with amyotrophic lateral sclerosis with and without C9orf72 mutation. Neurology 2021, 96, e141–e152. [Google Scholar] [CrossRef]
- Irwin, D.J.; McMillan, C.T.; Brettschneider, J.; Libon, D.J.; Powers, J.; Rascovsky, K.; Toledo, J.B.; Boller, A.; Bekisz, J.; Chandrasekaran, K.; et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Urso, D.; Tortelli, R. The challenge of amyotrophic lateral sclerosis descriptive epidemiology: To estimate low incidence rates across complex phenotypes in different geographic areas. Curr. Opin. Neurol. 2022, 35, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Machts, J.; Loewe, K.; Kaufmann, J.; Jakubiczka, S.; Abdulla, S.; Petri, S.; Dengler, R.; Heinze, H.J.; Vielhaber, S.; Schoenfeld, M.A.; et al. Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology 2015, 85, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Hou, Y.; Li, C.; Cao, B.; Cheng, Y.; Wei, Q.; Zhang, L.; Shang, H. Risk factors for cognitive impairment in amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 688–693. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- de Alcântara, C.; Cruzeiro, M.M.; França, M.C., Jr.; Alencar, M.A.; Jaeger, A.; de Araújo, C.M.; da Gama, N.A.S.; Camargos, S.T.; de Souza, L.C. A comparative study of cognitive and behavioral profiles between sporadic and type 8 amyotrophic lateral sclerosis. Muscle Nerve 2023, 68, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, I.; Zucchi, E.; Simonini, C.; Gianferrari, G.; Zamboni, G.; Pinti, M.; Mandrioli, J. The landscape of cognitive impairment in superoxide dismutase 1–amyotrophic lateral sclerosis. Neural Regen. Res. 2023, 18, 1427–1433. [Google Scholar] [CrossRef]
- Abe, K.; Fujimura, H.; Toyooka, K.; Sakoda, S.; Yorifuji, S.; Yanagihara, T. Cognitive function in amyotrophic lateral sclerosis. J. Neurol. Sci. 1997, 148, 95–100. [Google Scholar] [CrossRef]
- Tsermentseli, S.; Leigh, P.N.; Goldstein, L.H. The anatomy of cognitive impairment in amyotrophic lateral sclerosis: More than frontal lobe dysfunction. Cortex 2012, 48, 166–182. [Google Scholar] [CrossRef]
- Schuster, C.; Kasper, E.; Machts, J.; Bittner, D.; Kaufmann, J.; Benecke, R.; Teipel, S.; Vielhaber, S.; Prudlo, J. Longitudinal course of cortical thickness decline in amyotrophic lateral sclerosis. J. Neurol. 2014, 261, 1871–1880. [Google Scholar] [CrossRef]
- Verstraete, E.; Veldink, J.H.; Hendrikse, J.; Schelhaas, H.J.; van den Heuvel, M.P.; van den Berg, L.H. Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Christidi, F.; Karavasilis, E.; Riederer, F.; Zalonis, I.; Ferentinos, P.; Velonakis, G.; Xirou, S.; Rentzos, M.; Argiropoulos, G.; Zouvelou, V.; et al. Gray matter and white matter changes in non-demented amyotrophic lateral sclerosis patients with or without cognitive impairment: A combined voxel-based morphometry and tract-based spatial statistics whole-brain analysis. Brain Imaging Behav. 2018, 12, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Consonni, M.; Contarino, V.E.; Catricalà, E.; Dalla Bella, E.; Pensato, V.; Gellera, C.; Lauria, G.; Cappa, S.F. Cortical markers of cognitive syndromes in amyotrophic lateral sclerosis. Neuroimage Clin. 2018, 19, 675–682. [Google Scholar] [CrossRef]
- Menke, R.A.L.; Proudfoot, M.; Talbot, K.; Turner, M.R. The two-year progression of structural and functional cerebral MRI in amyotrophic lateral sclerosis. Neuroimage Clin. 2017, 17, 953–961. [Google Scholar] [CrossRef]
- Benbrika, S.; Desgranges, B.; Eustache, F.; Viader, F. Cognitive, emotional and psychological manifestations in amyotrophic lateral sclerosis at baseline and overtime: A review. Front. Neurosci. 2019, 13, 951. [Google Scholar] [CrossRef] [PubMed]
- Mioshi, E.; Lillo, P.; Yew, B.; Hsieh, S.; Savage, S.; Hodges, J.R.; Kiernan, M.C.; Hornberger, M. Cortical atrophy in ALS is critically associated with neuropsychiatric and cognitive changes. Neurology 2013, 80, 1117–1123. [Google Scholar] [CrossRef]
- Tae, W.S.; Sung, J.H.; Baek, S.H.; Lee, C.N.; Kim, B.J. Shape Analysis of the Subcortical Nuclei in Amyotrophic Lateral Sclerosis without Cognitive Impairment. J. Clin. Neurol. 2020, 16, 592–598. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Bocchetta, M.; Todd, E.G.; Tse, N.Y.; Devenney, E.M.; Tu, S.; Caga, J.; Hodges, J.R.; Halliday, G.M.; Irish, M.; et al. Tackling clinical heterogeneity across the amyotrophic lateral sclerosis-frontotemporal dementia spectrum using a transdiagnostic approach. Brain Commun. 2021, 3, fcab257. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Y.; Ren, Q.; Zhang, D.; Shao, K.; Lin, P.; Yuan, Y.; Dai, T.; Zhang, Y.; Li, L.; et al. Amygdala abnormalities across disease stages in patients with sporadic amyotrophic lateral sclerosis. Hum. Brain Mapp. 2022, 43, 5421–5431. [Google Scholar] [CrossRef]
- Tse, N.Y.; Bocchetta, M.; Todd, E.G.; Devenney, E.M.; Tu, S.; Caga, J.; Hodges, J.R.; Halliday, G.M.; Irish, M.; Kiernan, M.C.; et al. Distinct hypothalamic involvement in the amyotrophic lateral sclerosis-frontotemporal dementia spectrum. Neuroimage Clin. 2023, 37, 103281. [Google Scholar] [CrossRef]
- Schuster, C.; Kasper, E.; Dyrba, M.; Machts, J.; Bittner, D.; Kaufmann, J.; Mitchell, A.J.; Benecke, R.; Teipel, S.; Vielhaber, S.; et al. Cortical thinning and its relation to cognition in amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Ash, S.; Olm, C.; McMillan, C.T.; Boller, A.; Irwin, D.J.; McCluskey, L.; Elman, L.; Grossman, M. Deficits in sentence expression in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Nevler, N.; Ash, S.; McMillan, C.; Elman, L.; McCluskey, L.; Irwin, D.J.; Cho, S.; Liberman, M.; Grossman, M. Automated analysis of natural speech in amyotrophic lateral sclerosis spectrum disorders. Neurology 2020, 95, e1629–e1639. [Google Scholar] [CrossRef]
- Buhour, M.S.; Doidy, F.; Mondou, A.; Pélerin, A.; Carluer, L.; Eustache, F.; Viader, F.; Desgranges, B. Voxel-based mapping of grey matter volume and glucose metabolism profiles in amyotrophic lateral sclerosis. EJNMMI Res. 2017, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Christidi, F.; Karavasilis, E.; Zalonis, I.; Ferentinos, P.; Giavri, Z.; Wilde, E.A.; Xirou, S.; Rentzos, M.; Zouvelou, V.; Velonakis, G.; et al. Memory-related white matter tract integrity in amyotrophic lateral sclerosis: An advanced neuroimaging and neuropsychological study. Neurobiol. Aging 2017, 49, 69–78. [Google Scholar] [CrossRef]
- Kim, H.J.; Oh, S.I.; de Leon, M.; Wang, X.; Oh, K.W.; Park, J.S.; Deshpande, A.; Buj, M.; Kim, S.H. Structural explanation of poor prognosis of amyotrophic lateral sclerosis in the non-demented state. Eur. J. Neurol. 2017, 24, 122–129. [Google Scholar] [CrossRef]
- Evans, J.; Olm, C.; McCluskey, L.; Elman, L.; Boller, A.; Moran, E.; Rascovsky, K.; Bisbing, T.; McMillan, C.T.; Grossman, M. Impaired cognitive flexibility in amyotrophic lateral sclerosis. Cogn. Behav. Neurol. 2015, 28, 17–26. [Google Scholar] [CrossRef]
- Bueno, A.P.A.; Pinaya, W.H.L.; Moura, L.M.; Bertoux, M.; Radakovic, R.; Kiernan, M.C.; Teixeira, A.L.; de Souza, L.C.; Hornberger, M.; Sato, J.R. Structural and functional Papez circuit integrity in amyotrophic lateral sclerosis. Brain Imaging Behav. 2018, 12, 1622–1630. [Google Scholar] [CrossRef]
- Tan, H.H.G.; Westeneng, H.J.; Nitert, A.D.; van Veenhuijzen, K.; Meier, J.M.; van der Burgh, H.K.; van Zandvoort, M.J.E.; van Es, M.A.; Veldink, J.H.; van den Berg, L.H. MRI clustering reveals three ALS subtypes with unique neurodegeneration patterns. Ann. Neurol. 2022, 92, 1030–1045. [Google Scholar] [CrossRef]
- Brettschneider, J.; Libon, D.J.; Toledo, J.B.; Xie, S.X.; McCluskey, L.; Elman, L.; Geser, F.; Lee, V.M.; Grossman, M.; Trojanowski, J.Q. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 123, 395–407. [Google Scholar] [CrossRef]
- Mavridis, I.N.; Pyrgelis, E.S. Nucleus accumbens changes in amyotrophic lateral sclerosis. Am. J. Neurodegener. Dis. 2023, 12, 85–88. [Google Scholar] [PubMed]
- Bede, P.; Elamin, M.; Byrne, S.; McLaughlin, R.L.; Kenna, K.; Vajda, A.; Pender, N.; Bradley, D.G.; Hardiman, O. Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology 2013, 81, 2107–2115. [Google Scholar] [CrossRef]
- Shen, D.C.; Hou, B.; Cui, B.; Li, X.L.; Peng, P.; Tai, H.F.; Zhang, K.; Liu, S.W.; Fu, H.H.; Liu, M.S.; et al. Resting-state functional MRI studies of amyotrophic lateral sclerosis patients with various levels of cognitive impairment. Zhonghua Yi Xue Za Zhi 2018, 98, 2002–2006. [Google Scholar] [PubMed]
- Hu, T.; Hou, Y.; Wei, Q.; Yang, J.; Luo, C.; Chen, Y.; Gong, Q.; Shang, H. Patterns of brain regional functional coherence in cognitive impaired ALS. Int. J. Neurosci. 2020, 130, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Kew, J.J.; Goldstein, L.H.; Leigh, P.N.; Abrahams, S.; Cosgrave, N.; Passingham, R.E.; Frackowiak, R.S.; Brooks, D.J. The relationship between abnormalities of cognitive function and cerebral activation in amyotrophic lateral sclerosis. A neuropsychological and positron emission tomography study. Brain 1993, 116, 1399–1423. [Google Scholar] [CrossRef]
- Canosa, A.; Moglia, C.; Manera, U.; Vasta, R.; Torrieri, M.C.; Arena, V.; D’Ovidio, F.; Palumbo, F.; Zucchetti, J.P.; Iazzolino, B.; et al. Metabolic brain changes across different levels of cognitive impairment in ALS: A 18F-FDG-PET study. J. Neurol. Neurosurg. Psychiatry 2021, 92, 357–363. [Google Scholar] [CrossRef]
- Carluer, L.; Mondou, A.; Buhour, M.S.; Laisney, M.; Pélerin, A.; Eustache, F.; Viader, F.; Desgranges, B. Neural substrate of cognitive theory of mind impairment in amyotrophic lateral sclerosis. Cortex 2015, 65, 19–30. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, D.; Hou, B.; Sun, X.; Yang, X.; Gao, J.; Liu, M.; Feng, F.; Cui, L. Brain structural and perfusion changes in amyotrophic lateral sclerosis-frontotemporal dementia patients with cognitive and motor onset: A preliminary study. Brain Imaging Behav. 2022, 16, 2164–2174. [Google Scholar] [CrossRef]
- Canosa, A.; Pagani, M.; Brunetti, M.; Barberis, M.; Iazzolino, B.; Ilardi, A.; Cammarosano, S.; Manera, U.; Moglia, C.; Calvo, A.; et al. Correlation between apolipoprotein E genotype and brain metabolism in amyotrophic lateral sclerosis. Eur. J. Neurol. 2019, 26, 306–312. [Google Scholar] [CrossRef]
- Saito, Y. Diffusion magnetic resonance imaging-based surrogate marker in amyotrophic lateral sclerosis. Explor. Neuroprot. Ther. 2023, 3, 186–206. [Google Scholar] [CrossRef]
- Nigri, A.; Umberto, M.; Stanziano, M.; Ferraro, S.; Fedeli, D.; Medina Carrion, J.P.; Palermo, S.; Lequio, L.; Denegri, F.; Agosta, F.; et al. C9orf72 ALS mutation carriers show extensive cortical and subcortical damage compared to matched wild-type ALS patients. Neuroimage Clin. 2023, 38, 103400. [Google Scholar] [CrossRef] [PubMed]
- Lepow, L.; Van Sweringen, J.; Strutt, A.M.; Jawaid, A.; MacAdam, C.; Harati, Y.; Schulz, P.E.; York, M.K. Frontal and temporal lobe involvement on verbal fluency measures in amyotrophic lateral sclerosis. J. Clin. Exp. Neuropsychol. 2010, 32, 913–922. [Google Scholar] [CrossRef]
- Yunusova, Y.; Ansari, J.; Ramirez, J.; Shellikeri, S.; Stanisz, G.J.; Black, S.E.; Gillingham, S.M.; Kiss, A.; Stuss, D.T.; Zinman, L. Frontal anatomical correlates of cognitive and speech motor deficits in amyotrophic lateral sclerosis. Behav. Neurol. 2019, 2019, 9518309. [Google Scholar] [CrossRef]
- Mao, D.; Ding, Z.; Jia, W.; Liao, W.; Li, X.; Huang, H.; Yuan, J.; Zang, Y.F.; Zhang, H. Low-frequency fluctuations of the resting brain: High magnitude does not equal high reliability. PLoS ONE 2015, 10, e0128117. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, V.; Pioro, E.P. Degeneration of gray and white matter differs between hypometabolic and hypermetabolic brain regions in a patient with ALS-FTD: A longitudinal MRI-PET multimodal study. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Canosa, A.; Palumbo, F.; Iazzolino, B.; Peotta, L.; Di Pede, F.; Manera, U.; Vasta, R.; Grassano, M.; Solero, L.; Arena, V.; et al. The interplay among education, brain metabolism, and cognitive impairment suggests a role of cognitive reserve in Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2021, 98, 205–213. [Google Scholar] [CrossRef]
- Christidi, F.; Argyropoulos, G.D.; Karavasilis, E.; Velonakis, G.; Zouvelou, V.; Kourtesis, P.; Pantoleon, V.; Tan, E.L.; Daponte, A.; Aristeidou, S.; et al. Hippocampal metabolic alterations in amyotrophic lateral sclerosis: A magnetic resonance spectroscopy study. Life 2023, 13, 571. [Google Scholar] [CrossRef]
- Ta, D.; Ishaque, A.; Srivastava, O.; Hanstock, C.; Seres, P.; Eurich, D.T.; Luk, C.; Briemberg, H.; Frayne, R.; Genge, A.L.; et al. Progressive neurochemical abnormalities in cognitive and motor subgroups of amyotrophic lateral sclerosis: A prospective multicenter study. Neurology 2021, 97, e803–e813. [Google Scholar] [CrossRef]
- Chenji, S.; Jha, S.; Lee, D.; Brown, M.; Seres, P.; Mah, D.; Kalra, S. Investigating default mode and sensorimotor network connectivity in amyotrophic lateral sclerosis. PLoS ONE 2016, 11, e0157443. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, Y.; Wang, Y.; Hu, J.; Wang, J.; Zhang, J.; Jiang, T. Disrupted effective connectivity of the sensorimotor network in amyotrophic lateral sclerosis. J. Neurol. 2016, 263, 508–516. [Google Scholar] [CrossRef]
- Dimond, D.; Ishaque, A.; Chenji, S.; Mah, D.; Chen, Z.; Seres, P.; Beaulieu, C.; Kalra, S. White matter structural network abnormalities underlie executive dysfunction in amyotrophic lateral sclerosis. Hum. Brain Mapp. 2017, 38, 1249–1268. [Google Scholar] [CrossRef] [PubMed]
- Dukic, S.; McMackin, R.; Buxo, T.; Fasano, A.; Chipika, R.; Pinto-Grau, M.; Costello, E.; Schuster, C.; Hammond, M.; Heverin, M.; et al. Patterned functional network disruption in amyotrophic lateral sclerosis. Hum. Brain Mapp. 2019, 40, 4827–4842. [Google Scholar] [CrossRef] [PubMed]
- Borgheai, S.B.; McLinden, J.; Mankodiya, K.; Shahriari, Y. Frontal functional network disruption associated with amyotrophic lateral sclerosis: An fNIRS-based minimum spanning tree analysis. Front. Neurosci. 2020, 14, 613990. [Google Scholar] [CrossRef] [PubMed]
- Cividini, C.; Basaia, S.; Spinelli, E.G.; Canu, E.; Castelnovo, V.; Riva, N.; Cecchetti, G.; Caso, F.; Magnani, G.; Falini, A.; et al. Amyotrophic lateral sclerosis-frontotemporal dementia: Shared and divergent neural correlates across the clinical spectrum. Neurology 2021, 98, e402–e415. [Google Scholar] [CrossRef]
- Bueno, A.P.A.; de Souza, L.C.; Pinaya, W.H.L.; Teixeira, A.L.; de Prado, L.G.R.; Caramelli, P.; Hornberger, M.; Sato, J.R. Papez circuit gray matter and episodic memory in amyotrophic lateral sclerosis and behavioural variant frontotemporal dementia. Brain Imaging Behav. 2021, 15, 996–1006. [Google Scholar] [CrossRef]
- Trojsi, F.; Di Nardo, F.; Caiazzo, G.; Siciliano, M.; D’Alvano, G.; Ferrantino, T.; Passaniti, C.; Ricciardi, D.; Esposito, S.; Lavorgna, L.; et al. Hippocampal connectivity in amyotrophic lateral sclerosis (ALS): More than Papez circuit impairment. Brain Imaging Behav. 2021, 15, 2126–2138. [Google Scholar] [CrossRef]
- Bede, P.; Omer, T.; Finegan, E.; Chipika, R.H.; Iyer, P.M.; Doherty, M.A.; Vajda, A.; Pender, N.; McLaughlin, R.L.; Hutchinson, S.; et al. Connectivity-based characterisation of subcortical grey matter pathology in frontotemporal dementia and ALS: A multimodal neuroimaging study. Brain Imaging Behav. 2018, 12, 1696–1707. [Google Scholar] [CrossRef]
- Bharti, K.; Graham, S.J.; Benatar, M.; Briemberg, H.; Chenji, S.; Dupré, N.; Dionne, A.; Frayne, R.; Genge, A.; Korngut, L.; et al. Functional alterations in large-scale resting-state networks of amyotrophic lateral sclerosis: A multi-site study across Canada and the United States. PLoS ONE 2022, 17, e0269154. [Google Scholar] [CrossRef]
- Chen, H.; Hu, Z.; Ke, Z.; Xu, Y.; Bai, F.; Liu, Z. Aberrant multimodal connectivity pattern involved in default mode network and limbic network in amyotrophic lateral sclerosis. Brain Sci. 2023, 13, 803. [Google Scholar] [CrossRef]
- Castelnovo, V.; Canu, E.; De Mattei, F.; Filippi, M.; Agosta, F. Basal ganglia alterations in amyotrophic lateral sclerosis. Front. Neurosci. 2023, 17, 1133758. [Google Scholar] [CrossRef]
- Crespi, C.; Cerami, C.; Dodich, A.; Canessa, N.; Iannaccone, S.; Corbo, M.; Lunetta, C.; Falini, A.; Cappa, S.F. Microstructural correlates of emotional attribution impairment in non-demented patients with amyotrophic lateral sclerosis. PLoS ONE 2016, 11, e0161034. [Google Scholar] [CrossRef] [PubMed]
- Pender, N.; Pinto-Grau, M.; Hardiman, O. Cognitive and behavioural impairment in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2020, 33, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Masuda, M.; Watanabe, H.; Ogura, A.; Ohdake, R.; Tanaka, Y.; Kato, T.; Kawabata, K.; Riku, Y.; Hara, K.; et al. The neural network basis of altered decision-making in patients with amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 2115–2126. [Google Scholar] [CrossRef]
- Abrahams, S.; Goldstein, L.H.; Simmons, A.; Brammer, M.; Williams, S.C.; Giampietro, V.; Leigh, P.N. Word retrieval in amyotrophic lateral sclerosis: A functional magnetic resonance imaging study. Brain 2004, 127, 1507–1517. [Google Scholar] [CrossRef]
- Pettit, L.D.; Bastin, M.E.; Smith, C.; Bak, T.H.; Gillingwater, T.H.; Abrahams, S. Executive deficits, not processing speed relates to abnormalities in distinct prefrontal tracts in amyotrophic lateral sclerosis. Brain 2013, 136, 3290–3304. [Google Scholar] [CrossRef]
- Castelnovo, V.; Canu, E.; Calderaro, D.; Riva, N.; Poletti, B.; Basaia, S.; Solca, F.; Silani, V.; Filippi, M.; Agosta, F. Progression of brain functional connectivity and frontal cognitive dysfunction in ALS. Neuroimage Clin. 2020, 28, 102509. [Google Scholar] [CrossRef]
- Temp, A.G.M.; Dyrba, M.; Büttner, C.; Kasper, E.; Machts, J.; Kaufmann, J.; Vielhaber, S.; Teipel, S.; Prudlo, J. Cognitive profiles of amyotrophic lateral sclerosis differ in resting-state functional connectivity: An fMRI study. Front. Neurosci. 2021, 15, 682100. [Google Scholar] [CrossRef] [PubMed]
- Sarro, L.; Agosta, F.; Canu, E.; Riva, N.; Prelle, A.; Copetti, M.; Riccitelli, G.; Comi, G.; Filippi, M. Cognitive functions and white matter tract damage in amyotrophic lateral sclerosis: A diffusion tensor tractography study. AJNR Am. J. Neuroradiol. 2011, 32, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- McMackin, R.; Dukic, S.; Costello, E.; Pinto-Grau, M.; McManus, L.; Broderick, M.; Chipika, R.; Iyer, P.M.; Heverin, M.; Bede, P.; et al. Cognitive network hyperactivation and motor cortex decline correlate with ALS prognosis. Neurobiol. Aging 2021, 104, 57–70. [Google Scholar] [CrossRef]
- Wei, L.; Baeken, C.; Liu, D.; Zhang, J.; Wu, G.R. Functional connectivity-based prediction of global cognition and motor function in riluzole-naive amyotrophic lateral sclerosis patients. Netw. Neurosci. 2022, 6, 161–174. [Google Scholar] [CrossRef]
- Trojsi, F.; Di Nardo, F.; D’Alvano, G.; Passaniti, C.; Sharbafshaaer, M.; Canale, F.; Russo, A.; Silvestro, M.; Lavorgna, L.; Cirillo, M.; et al. Cognitive, behavioral, and brain functional connectivity correlates of fatigue in amyotrophic lateral sclerosis. Brain Behav. 2023, 13, e2931. [Google Scholar] [CrossRef]
- Lange, F.; Lange, C.; Joop, M.; Seer, C.; Dengler, R.; Kopp, B.; Petri, S. Neural correlates of cognitive set shifting in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2016, 127, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Hanagasi, H.A.; Gurvit, I.H.; Ermutlu, N.; Kaptanoglu, G.; Karamursel, S.; Idrisoglu, H.A.; Emre, M.; Demiralp, T. Cognitive impairment in amyotrophic lateral sclerosis: Evidence from neuropsychological investigation and event-related potentials. Brain Res. Cogn. Brain Res. 2002, 14, 234–244. [Google Scholar] [CrossRef]
- Ogawa, T.; Tanaka, H.; Hirata, K. Cognitive deficits in amyotrophic lateral sclerosis evaluated by event-related potentials. Clin. Neurophysiol. 2009, 120, 659–664. [Google Scholar] [CrossRef]
- Geronimo, A.; Simmons, Z.; Schiff, S.J. Performance predictors of brain-computer interfaces in patients with amyotrophic lateral sclerosis. J. Neural Eng. 2016, 13, 026002. [Google Scholar] [CrossRef] [PubMed]
- Seer, C.; Joop, M.; Lange, F.; Lange, C.; Dengler, R.; Petri, S.; Kopp, B. Attenuated error-related potentials in amyotrophic lateral sclerosis with executive dysfunctions. Clin. Neurophysiol. 2017, 128, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Volpato, C.; Prats Sedano, M.A.; Silvoni, S.; Segato, N.; Cavinato, M.; Merico, A.; Piccione, F.; Palmieri, A.; Birbaumer, N. Selective attention impairment in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Dukic, S.; McMackin, R.; Costello, E.; Metzger, M.; Buxo, T.; Fasano, A.; Chipika, R.; Pinto-Grau, M.; Schuster, C.; Hammond, M.; et al. Resting-state EEG reveals four subphenotypes of amyotrophic lateral sclerosis. Brain 2022, 145, 621–631. [Google Scholar] [CrossRef]
- McMackin, R.; Dukic, S.; Broderick, M.; Iyer, P.M.; Pinto-Grau, M.; Mohr, K.; Chipika, R.; Coffey, A.; Buxo, T.; Schuster, C.; et al. Dysfunction of attention switching networks in amyotrophic lateral sclerosis. Neuroimage Clin. 2019, 22, 101707. [Google Scholar] [CrossRef]
- Hortobágyi, T.; Cairns, N.J. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration. In Neuropathology of Neurodegenerative Diseases: A Practical Guide; Kovacs, G.G., Ed.; Cambridge University Press: Cambridge, UK, 2015; pp. 209–248. [Google Scholar]
- Takeda, T.; Kitagawa, K.; Arai, K. Phenotypic variability and its pathological basis in amyotrophic lateral sclerosis. Neuropathology 2020, 40, 40–56. [Google Scholar] [CrossRef]
- Ling, S.C. Synaptic paths to neurodegeneration: The emerging role of TDP-43 and FUS in synaptic functions. Neural. Plast. 2018, 2018, 8413496. [Google Scholar] [CrossRef]
- Braak, H.; Neumann, M.; Ludolph, A.C.; Del Tredici, K. Does sporadic amyotrophic lateral sclerosis spread via axonal connectivities? Neurol. Int. Open 2017, 01, E136–E141. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Fatima, M.; Tan, R.; Halliday, G.M.; Kril, J.J. Spread of pathology in amyotrophic lateral sclerosis: Assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol. Commun. 2015, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.M.; van der Burgh, H.K.; Nitert, A.D.; Bede, P.; de Lange, S.C.; Hardiman, O.; van den Berg, L.H.; van den Heuvel, M.P. Connectome-based propagation model in amyotrophic lateral sclerosis. Ann. Neurol. 2020, 87, 725–738. [Google Scholar] [CrossRef]
- Riemenschneider, H.; Guo, Q.; Bader, J.; Frottin, F.; Farny, D.; Kleinberger, G.; Haass, C.; Mann, M.; Hartl, F.U.; Baumeister, W.; et al. Gel-like inclusions of C-terminal fragments of TDP-43 sequester stalled proteasomes in neurons. EMBO Rep. 2022, 23, e53890. [Google Scholar] [CrossRef]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Del Tredici, K.; Braak, H.; Ludolph, A. The multisystem degeneration amyotrophic lateral sclerosis—Neuropathological staging and clinical translation. Arch. Ital. Biol. 2017, 155, 118–130. [Google Scholar] [CrossRef]
- Perkins, E.M.; Burr, K.; Banerjee, P.; Mehta, A.R.; Dando, O.; Selvaraj, B.T.; Suminaite, D.; Nanda, J.; Henstridge, C.M.; Gillingwater, T.H.; et al. Altered network properties in C9ORF72 repeat expansion cortical neurons are due to synaptic dysfunction. Mol. NeuroDegener. 2021, 16, 13. [Google Scholar] [CrossRef]
- Aksman, L.M.; Wijeratne, P.A.; Oxtoby, N.P.; Eshaghi, A.; Shand, C.; Altmann, A.; Alexander, D.C.; Young, A.L. pySuStaIn: A Python implementation of the Subtype and Stage Inference algorithm. SoftwareX 2021, 16, 100811. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Vogel, J.W.; Aksman, L.M.; Wijeratne, P.A.; Eshaghi, A.; Oxtoby, N.P.; Williams, S.C.R.; Alexander, D.C. Ordinal SuStaIn: Subtype and Stage Inference for clinical scores, visual ratings, and other ordinal data. Front. Artif. Intell. 2021, 4, 613261. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Vogel, J.W.; Robinson, J.L.; McMillan, C.T.; Ossenkoppele, R.; Wolk, D.A.; Irwin, D.J.; Elman, L.; Grossman, M.; Lee, V.M.Y.; et al. Data-driven neuropathological staging and subtyping of TDP-43 proteinopathies. Brain 2023, 146, 2975–2988. [Google Scholar] [CrossRef]
- Banerjee, P.; Elliott, E.; Rifai, O.M.; O’Shaughnessy, J.; McDade, K.; Abrahams, S.; Chandran, S.; Smith, C.; Gregory, J.M. NLRP3 inflammasome as a key molecular target underlying cognitive resilience in amyotrophic lateral sclerosis. J. Pathol. 2022, 256, 262–268. [Google Scholar] [CrossRef]
- Cykowski, M.D.; Powell, S.Z.; Peterson, L.E.; Appel, J.W.; Rivera, A.L.; Takei, H.; Chang, E.; Appel, S.H. Clinical significance of TDP-43 neuropathology in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2017, 76, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tan, C.F.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. TDP-43–immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008, 115, 115–122. [Google Scholar] [CrossRef]
- Prudlo, J.; König, J.; Schuster, C.; Kasper, E.; Büttner, A.; Teipel, S.; Neumann, M. TDP-43 pathology and cognition in ALS: A prospective clinicopathologic correlation study. Neurology 2016, 87, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Koga, S.; Sekiya, H.; Oskarsson, B.; Boylan, K.; Petrucelli, L.; Josephs, K.A.; Dickson, D.W. Old age amyotrophic lateral sclerosis and limbic TDP-43 pathology. Brain Pathol 2022, 32, e13100. [Google Scholar] [CrossRef]
- Shellikeri, S.; Keith, J.; Black, S.E.; Zinman, L.; Yunusova, Y. Neuropathology of speech network distinguishes bulbar from nonbulbar amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2020, 79, 284–295. [Google Scholar] [CrossRef]
- Borrego-Écija, S.; Turon-Sans, J.; Ximelis, T.; Aldecoa, I.; Molina-Porcel, L.; Povedano, M.; Rubio, M.A.; Gámez, J.; Cano, A.; Paré-Curell, M.; et al. Cognitive decline in amyotrophic lateral sclerosis: Neuropathological substrate and genetic determinants. Brain Pathol. 2021, 31, e12942. [Google Scholar] [CrossRef]
- Yang, T.; Wei, Q.; Li, C.; Cao, B.; Ou, R.; Hou, Y.; Zhang, L.; Chen, Y.; Shang, H. Association between vascular risk factors and cognitive impairment in amyotrophic lateral sclerosis: A case-control study. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 185–194. [Google Scholar] [CrossRef]
- Rhodes, E.; Alfa, S.; Jin, H.; Massimo, L.; Elman, L.; Amado, D.; Baer, M.; Quinn, C.; McMillan, C.T. Cognitive reserve in ALS: The role of occupational skills and requirements. medRxiv 2023. [Google Scholar] [CrossRef]
- Temp, A.G.M.; Prudlo, J.; Vielhaber, S.; Machts, J.; Hermann, A.; Teipel, S.J.; Kasper, E. Cognitive reserve and regional brain volume in amyotrophic lateral sclerosis. Cortex 2021, 139, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.; Rooney, J.; Pinto-Grau, M.; Burke, T.; Elamin, M.; Bede, P.; McMackin, R.; Dukic, S.; Vajda, A.; Heverin, M.; et al. Cognitive reserve in amyotrophic lateral sclerosis (ALS): A population-based longitudinal study. J. Neurol. Neurosurg. Psychiatry 2021, 92, 460–465. [Google Scholar] [CrossRef]
- Temp, A.G.M.; Kasper, E.; Machts, J.; Vielhaber, S.; Teipel, S.; Hermann, A.; Prudlo, J. Cognitive reserve protects ALS-typical cognitive domains: A longitudinal study. Ann. Clin. Transl. Neurol. 2022, 9, 1212–1223. [Google Scholar] [CrossRef]
- Gregory, J.M.; Elliott, E.; McDade, K.; Bak, T.; Pal, S.; Chandran, S.; Abrahams, S.; Smith, C. Neuronal clusterin expression is associated with cognitive protection in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2020, 46, 255–263. [Google Scholar] [CrossRef]
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol. Ther. 2017, 25, 127–139. [Google Scholar] [CrossRef]
- Alfieri, J.A.; Silva, P.R.; Igaz, L.M. Early cognitive/social deficits and late motor phenotype in conditional wild-type TDP-43 transgenic mice. Front. Aging Neurosci. 2016, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Ho, W.Y.; Yen, Y.C.; Sanford, E.; Ling, S.C. The vulnerability of motor and frontal cortex-dependent behaviors in mice expressing ALS-linked mutation in TDP-43. Neurobiol. Aging 2020, 92, 43–60. [Google Scholar] [CrossRef]
- Jury, N.; Abarzua, S.; Diaz, I.; Guerra, M.V.; Ampuero, E.; Cubillos, P.; Martinez, P.; Herrera-Soto, A.; Arredondo, C.; Rojas, F.; et al. Widespread loss of the silencing epigenetic mark H3K9me3 in astrocytes and neurons along with hippocampal-dependent cognitive impairment in C9orf72 BAC transgenic mice. Clin. Epigenetics 2020, 12, 32. [Google Scholar] [CrossRef]
- Sgobio, C.; Trabalza, A.; Spalloni, A.; Zona, C.; Carunchio, I.; Longone, P.; Ammassari-Teule, M. Abnormal medial prefrontal cortex connectivity and defective fear extinction in the presymptomatic G93A SOD1 mouse model of ALS. Genes Brain Behav. 2008, 7, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Spalloni, A.; Longone, P. Cognitive impairment in amyotrophic lateral sclerosis, clues from the SOD1 mouse. Neurosci. Biobehav. Rev. 2016, 60, 12–25. [Google Scholar] [CrossRef]
- Ho, W.Y.; Chang, J.C.; Tyan, S.H.; Yen, Y.C.; Lim, K.; Tan, B.S.Y.; Ong, J.; Tucker-Kellogg, G.; Wong, P.; Koo, E.; et al. FUS-mediated dysregulation of Sema5a, an autism-related gene, in FUS mice with hippocampus-dependent cognitive deficits. Hum. Mol. Genet. 2019, 28, 3777–3791. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.Y.; Agrawal, I.; Tyan, S.H.; Sanford, E.; Chang, W.T.; Lim, K.; Ong, J.; Tan, B.S.Y.; Moe, A.A.K.; Yu, R.; et al. Dysfunction in nonsense-mediated decay, protein homeostasis, mitochondrial function, and brain connectivity in ALS-FUS mice with cognitive deficits. Acta Neuropathol. Commun. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and cellular mechanisms affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Rifai, O.M.; Longden, J.; O’Shaughnessy, J.; Sewell, M.; Pate, J.; McDade, K.; Daniels, M.J.; Abrahams, S.; Chandran, S.; McColl, B.W.; et al. Random forest modelling demonstrates microglial and protein misfolding features to be key phenotypic markers in C9orf72–ALS. J. Pathol. 2022, 258, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Sassani, M.; Alix, J.J.; McDermott, C.J.; Baster, K.; Hoggard, N.; Wild, J.M.; Mortiboys, H.J.; Shaw, P.J.; Wilkinson, I.D.; Jenkins, T.M. Magnetic resonance spectroscopy reveals mitochondrial dysfunction in amyotrophic lateral sclerosis. Brain 2020, 143, 3603–3618. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Duranti, E.; Villa, C. Molecular investigations of protein aggregation in the pathogenesis of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2022, 24, 704. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Jeon, Y.M.; Kwon, Y.; Lee, S.; Kim, H.J. Potential roles of the endoplasmic reticulum stress pathway in amyotrophic lateral sclerosis. Front. Aging Neurosci. 2023, 15, 1047897. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Prater, K.E.; Latimer, C.S.; Jayadev, S. Glial TDP-43 and TDP-43 induced glial pathology, focus on neurodegenerative proteinopathy syndromes. Glia 2022, 70, 239–255. [Google Scholar] [CrossRef]
- Verde, F.; Milone, I.; Maranzano, A.; Colombo, E.; Torre, S.; Solca, F.; Doretti, A.; Gentile, F.; Manini, A.; Bonetti, R.; et al. Serum levels of glial fibrillary acidic protein in patients with amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2023, 10, 118–129. [Google Scholar] [CrossRef]
- Rifai, O.M.; O’Shaughnessy, J.; Dando, O.R.; Munro, A.F.; Sewell, M.D.E.; Abrahams, S.; Waldron, F.M.; Sibley, C.R.; Gregory, J.M. Distinct neuroinflammatory signatures exist across genetic and sporadic amyotrophic lateral sclerosis cohorts. Brain 2023, awad243. [Google Scholar] [CrossRef] [PubMed]
- Higashihara, M.; Pavey, N.; van den Bos, M.; Menon, P.; Kiernan, M.C.; Vucic, S. Association of cortical hyperexcitability and cognitive impairment in patients with amyotrophic lateral sclerosis. Neurology 2021, 96, e2090–e2097. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Sideris, D.I.; Carroll, E.; Rotariu, S.; Salomonsson, S.; Tzioras, M.; McKenzie, C.A.; Smith, C.; von Arnim, C.A.F.; Ludolph, A.C.; et al. Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Ince, P.G.; Slade, J.; Chinnery, R.M.; McKenzie, J.; Royston, C.; Roberts, G.W.; Shaw, P.J. Quantitative study of synaptophysin immunoreactivity of cerebral cortex and spinal cord in motor neuron disease. J. Neuropathol. Exp. Neurol. 1995, 54, 673–679. [Google Scholar] [CrossRef]
- Sasaki, S.; Maruyama, S. Synapse loss in anterior horn neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1994, 88, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y.; Seilhean, D.; Duyckaerts, C.; Boluda, S.; Iguchi, Y.; Ishigaki, S.; Iwasaki, Y.; Yoshida, M.; Sobue, G.; Katsuno, M. Pathway from TDP-43-related pathology to neuronal dysfunction in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Int. J. Mol. Sci. 2021, 22, 3843. [Google Scholar] [CrossRef]
- Yang, W.; Strong, M.J. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph. Lateral Scler. 2012, 13, 178–193. [Google Scholar] [CrossRef]
- Latimer, C.S.; Burke, B.T.; Liachko, N.F.; Currey, H.N.; Kilgore, M.D.; Gibbons, L.E.; Henriksen, J.; Darvas, M.; Domoto-Reilly, K.; Jayadev, S.; et al. Resistance and resilience to Alzheimer’s disease pathology are associated with reduced cortical pTau and absence of limbic-predominant age-related TDP-43 encephalopathy in a community-based cohort. Acta Neuropathol. Commun. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Latimer, C.S.; Stair, J.G.; Hincks, J.C.; Currey, H.N.; Bird, T.D.; Keene, C.D.; Kraemer, B.C.; Liachko, N.F. TDP-43 promotes tau accumulation and selective neurotoxicity in bigenic Caenorhabditis elegans. Dis. Model Mech. 2022, 15, dmm049323. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.A.; Gan, K.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 expression influences amyloidβ plaque deposition and tau aggregation. Neurobiol. Dis. 2017, 103, 154–162. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and tau oligomers in Alzheimer’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Moszczynski, A.J.; Hintermayer, M.A.; Strong, M.J. Phosphorylation of threonine 175 tau in the induction of tau pathology in amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD). A review. Front. Neurosci. 2018, 12, 259. [Google Scholar] [CrossRef]
- Strong, M.J.; Donison, N.S.; Volkening, K. Alterations in tau metabolism in ALS and ALS-FTSD. Front. Neurol. 2020, 11, 598907. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, P.; Li, W.; Liu, Z.; Zhou, M.; Li, J.; Yuan, Y.; Fang, L.; Wang, M.; Shen, L.; et al. Detection of changes in synaptic density in amyotrophic lateral sclerosis patients using 18F-SynVesT-1 positron emission tomography. Eur. J. Neurol. 2022, 29, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, Z.I.; Hindley, N.; Sanchez Avila, A.; Kline, R.A.; Eaton, S.L.; Lamont, D.J.; Smith, C.; Spires-Jones, T.L.; Wishart, T.M.; Henstridge, C.M. Synaptic proteomics reveal distinct molecular signatures of cognitive change and C9ORF72 repeat expansion in the human ALS cortex. Acta Neuropathol. Commun. 2022, 10, 156. [Google Scholar] [CrossRef]
- Gulino, R. Synaptic dysfunction and plasticity in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2023, 24, 4613. [Google Scholar] [CrossRef]
- Mora, S.; Allodi, I. Neural circuit and synaptic dysfunctions in ALS-FTD pathology. Front. Neural Circuits 2023, 17, 1208876. [Google Scholar] [CrossRef]
- Bacioglu, M.; Maia, L.F.; Preische, O.; Schelle, J.; Apel, A.; Kaeser, S.A.; Schweighauser, M.; Eninger, T.; Lambert, M.; Pilotto, A.; et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 2016, 91, 56–66. [Google Scholar] [CrossRef]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Verde, F.; Milone, I.; Colombo, E.; Maranzano, A.; Solca, F.; Torre, S.; Doretti, A.; Gentile, F.; Manini, A.; Bonetti, R.; et al. Phenotypic correlates of serum neurofilament light chain levels in amyotrophic lateral sclerosis. Front. Aging Neurosci. 2023, 15, 1132808. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Portelius, E.; Cullen, N.C.; Sandelius, Å.; Zetterberg, H.; Andreasson, U.; Höglund, K.; Irwin, D.J.; Grossman, M.; Weintraub, D.; et al. Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol. 2019, 76, 318–325. [Google Scholar] [CrossRef]
- Meyer, T.; Salkic, E.; Grehl, T.; Weyen, U.; Kettemann, D.; Weydt, P.; Günther, R.; Lingor, P.; Koch, J.C.; Petri, S.; et al. Performance of serum neurofilament light chain in a wide spectrum of clinical courses of amyotrophic lateral sclerosis—A cross-sectional multicenter study. Eur. J. Neurol. 2023, 30, 1600–1610. [Google Scholar] [CrossRef] [PubMed]
- Vidovic, M.; Müschen, L.H.; Brakemeier, S.; Machetanz, G.; Naumann, M.; Castro-Gomez, S. Current state and future directions in the diagnosis of amyotrophic lateral sclerosis. Cells 2023, 12, 736. [Google Scholar] [CrossRef] [PubMed]
- Trojsi, F.; Di Nardo, F.; D’Alvano, G.; Caiazzo, G.; Passaniti, C.; Mangione, A.; Sharbafshaaer, M.; Russo, A.; Silvestro, M.; Siciliano, M.; et al. Resting state fMRI analysis of pseudobulbar affect in Amyotrophic Lateral Sclerosis (ALS): Motor dysfunction of emotional expression. Brain Imaging Behav. 2023, 17, 77–89. [Google Scholar] [CrossRef]
- Tedeschi, G.; Trojsi, F.; Tessitore, A.; Corbo, D.; Sagnelli, A.; Paccone, A.; D’Ambrosio, A.; Piccirillo, G.; Cirillo, M.; Cirillo, S.; et al. Interaction between aging and neurodegeneration in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 886–898. [Google Scholar] [CrossRef]
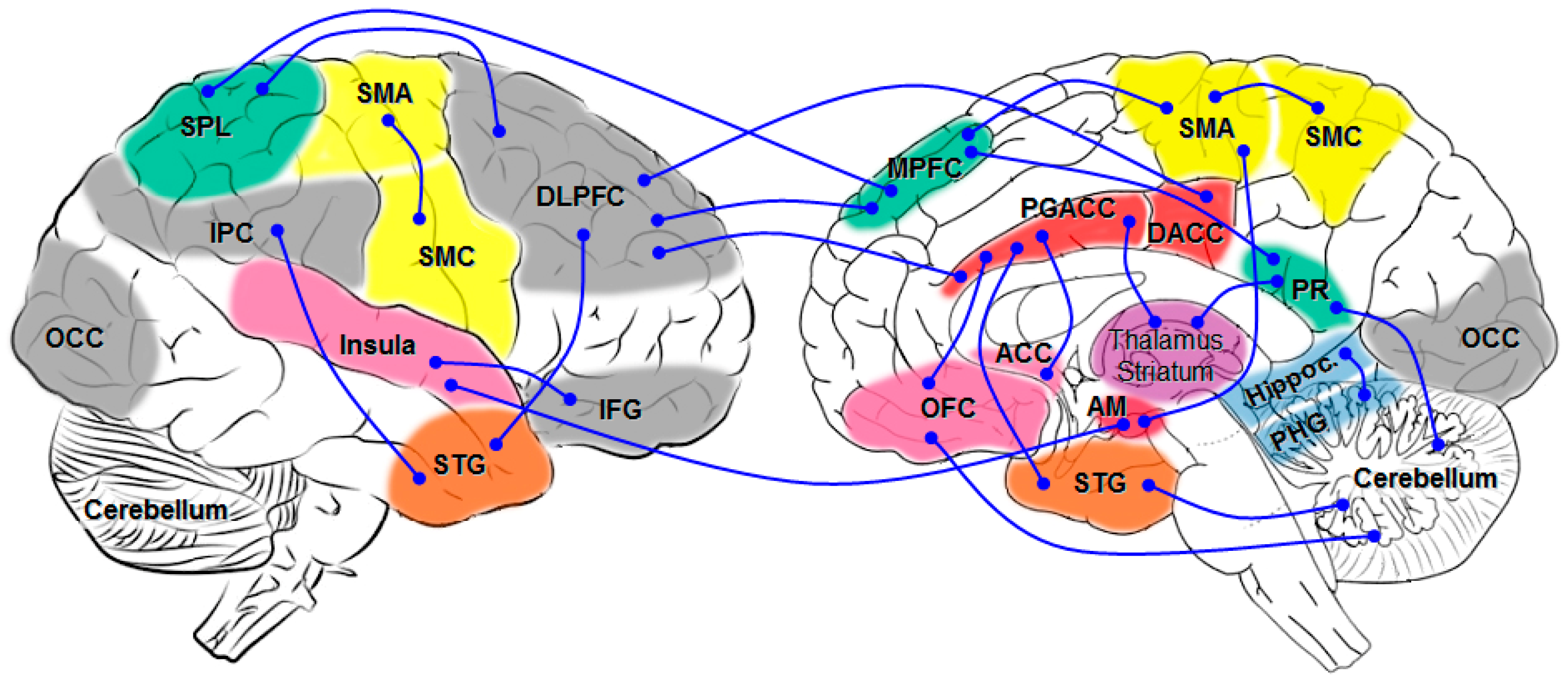


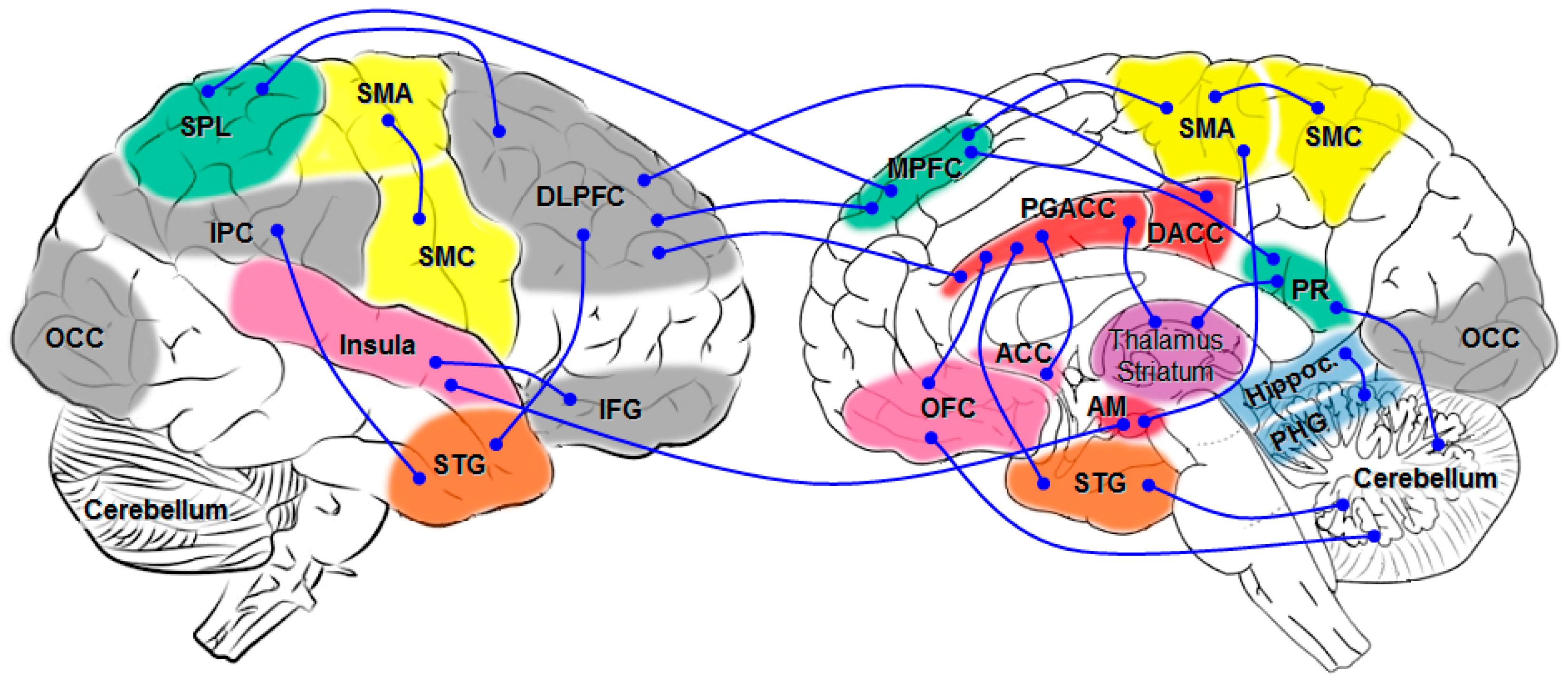{kind=link}
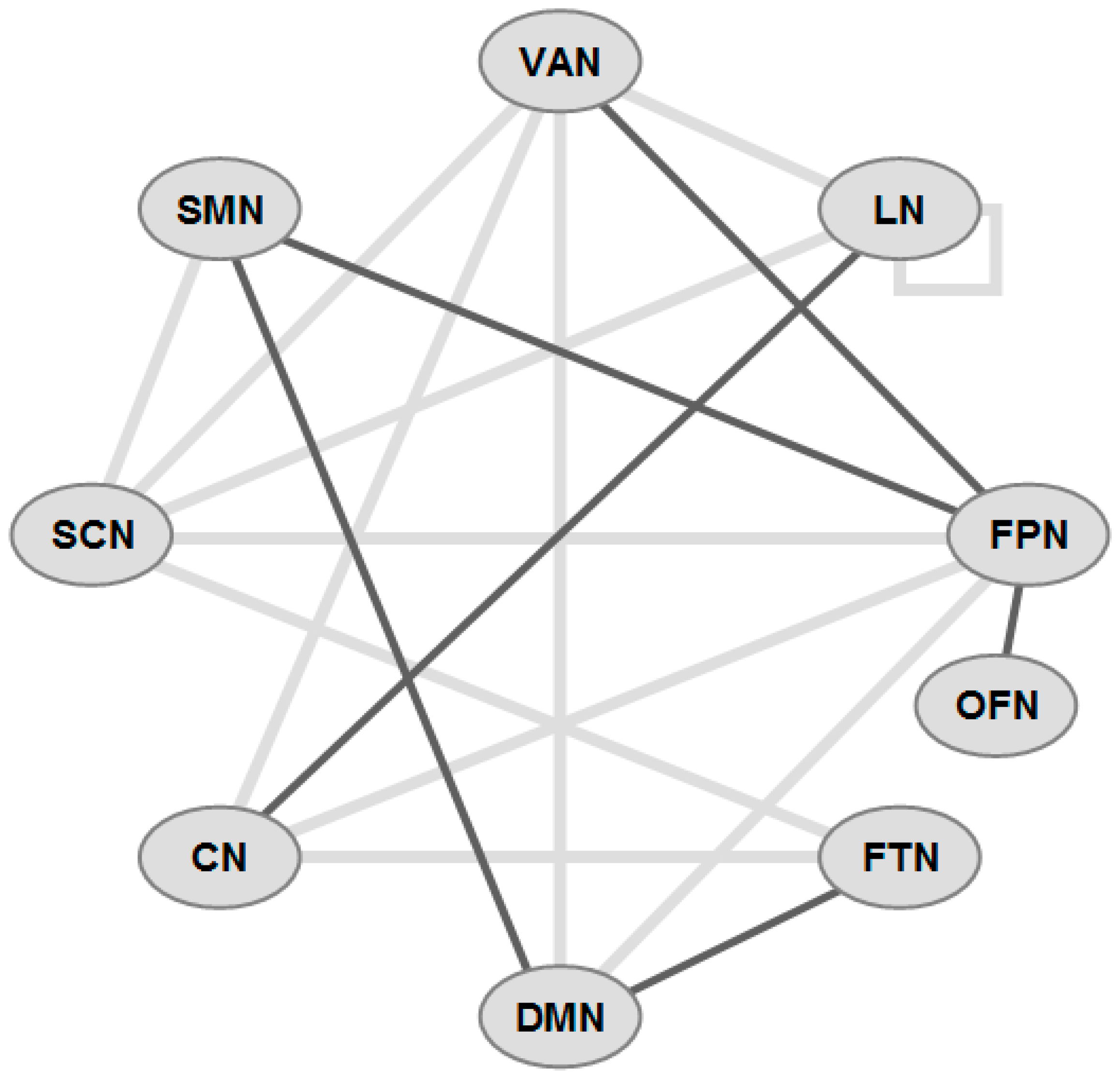{kind=link}
| Type of Lesion, Location | References |
|---|---|
| Cortical atrophy | |
| Frontoparietal cortex, somatosensory area | [87] |
| Frontal, temporal, and limbic cortex | [80] |
| Frontal, temporal and parietal cortex | [82,92] |
| Precentral gyri, posterior cingulate cortex, thalamus, striatum, pallidum, hippocampus | [85] |
| Left inferior temporal and anterior temporal cortex | [93] |
| Primary motor cortex and perisylvian regions | [94] (speech deficits) |
| Temporal poles | [95] |
| Inferior frontal, temporal, cingulate, and insular cortices, frontoparietal cortex | [84] |
| Frontoparietal cortex, right insula, striatum, precentral cortex, right frontal and temporal cortex, precuneus | [89] |
| Primary motor cortex, frontotemporal cortex, basal ganglia, cerebellum | [96,97] |
| Inferior frontal cortex, insular region | [98] |
| Left hippocampus, entorhinal cortex, posterior cingulate cortex | [99] |
| Precentral gyrus, orbitofrontal and temporal cortex, posterior cingulate cortex, temporal operculum, parietal white matter, cerebellum | [100] |
| Left entorhinal cortex -”- medial orbitofrontal lobe -”- inferior temporal gyrus -”- insular lobe | [31] |
| Amygdala | [90] |
| Nucleus accumbens | [101,102] |
| Right nucleus accumbens, left caudate nucleus and hippocampus | [103] |
| Reduced fractional anisotropy/altered regional homogeneity (ReHo) | |
| Corticospinal tract bilaterally Corpus callosum, extramotor tracts | [83] |
| Increased ReHo Left prefrontal lobe, anterior cingulate cortex | [104] |
| Increased ReHo: bilateral inferior parietal lobules, precuneus, right parietal and inferior cerebellar area Decreased ReHo: bilateral sensorimotor cortices | [105] |
| Reduced brain perfusion, hypometabolism | |
| Prefrontal cortex, anterior cingulate cortex, right hippocampus, anterior thalamic nucleus | [106] |
| Frontal and prefrontal area | [107] |
| Dorsomedial/dorsolateral prefrontal cortex | [108] |
| Left parahippocampal gyrus | [109] |
| Prefrontal, orbitofrontal, anterior cigulate cortex | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jellinger, K.A. The Spectrum of Cognitive Dysfunction in Amyotrophic Lateral Sclerosis: An Update. Int. J. Mol. Sci. 2023, 24, 14647. https://doi.org/10.3390/ijms241914647
Jellinger KA. The Spectrum of Cognitive Dysfunction in Amyotrophic Lateral Sclerosis: An Update. International Journal of Molecular Sciences. 2023; 24(19):14647. https://doi.org/10.3390/ijms241914647
Chicago/Turabian StyleJellinger, Kurt A. 2023. "The Spectrum of Cognitive Dysfunction in Amyotrophic Lateral Sclerosis: An Update" International Journal of Molecular Sciences 24, no. 19: 14647. https://doi.org/10.3390/ijms241914647
APA StyleJellinger, K. A. (2023). The Spectrum of Cognitive Dysfunction in Amyotrophic Lateral Sclerosis: An Update. International Journal of Molecular Sciences, 24(19), 14647. https://doi.org/10.3390/ijms241914647