
Applications for Deep Learning in Epilepsy Genetic Research

 ,
,  and
and
Abstract
:1. Introduction
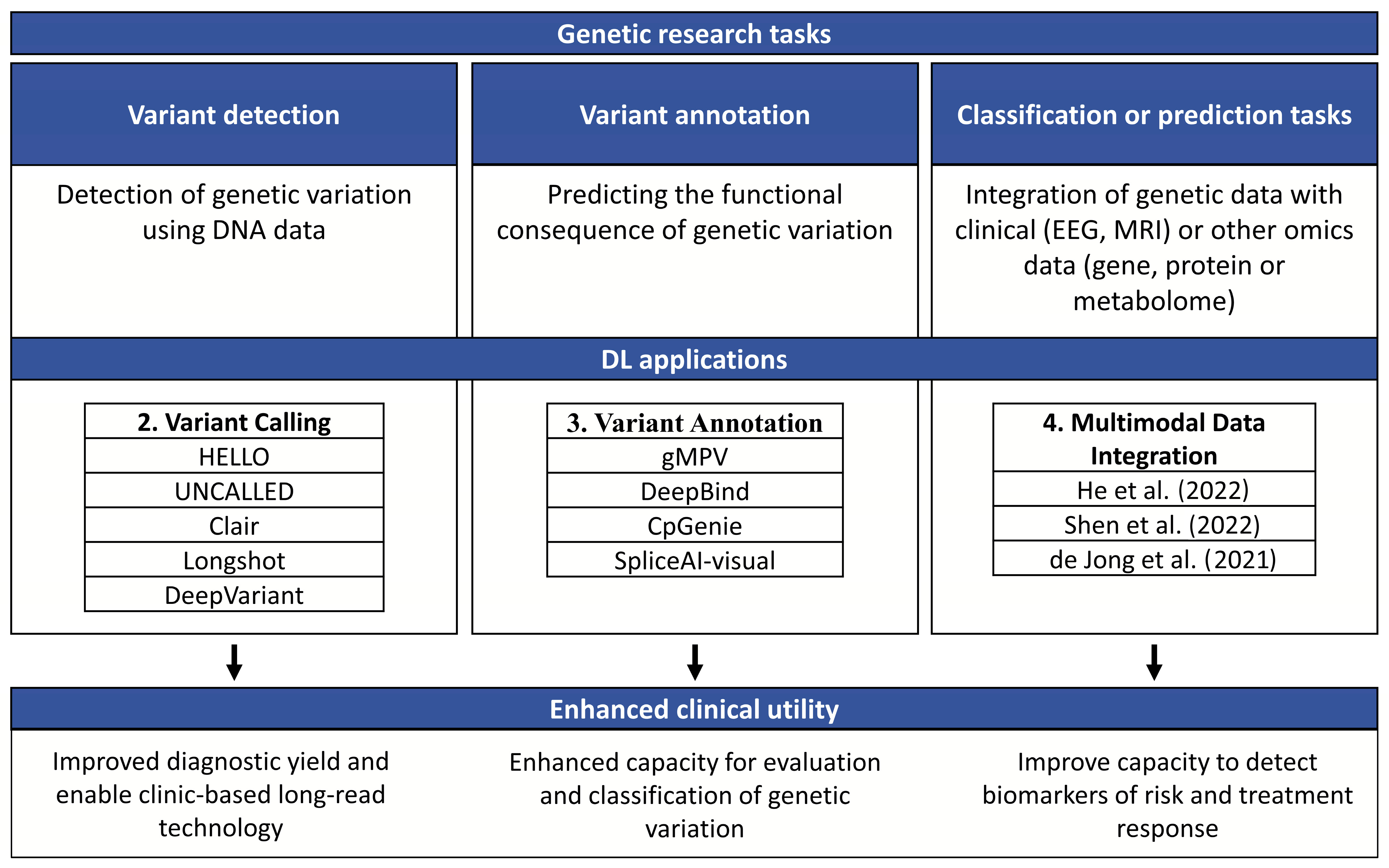
2. Variant Calling
3. Variant Annotation
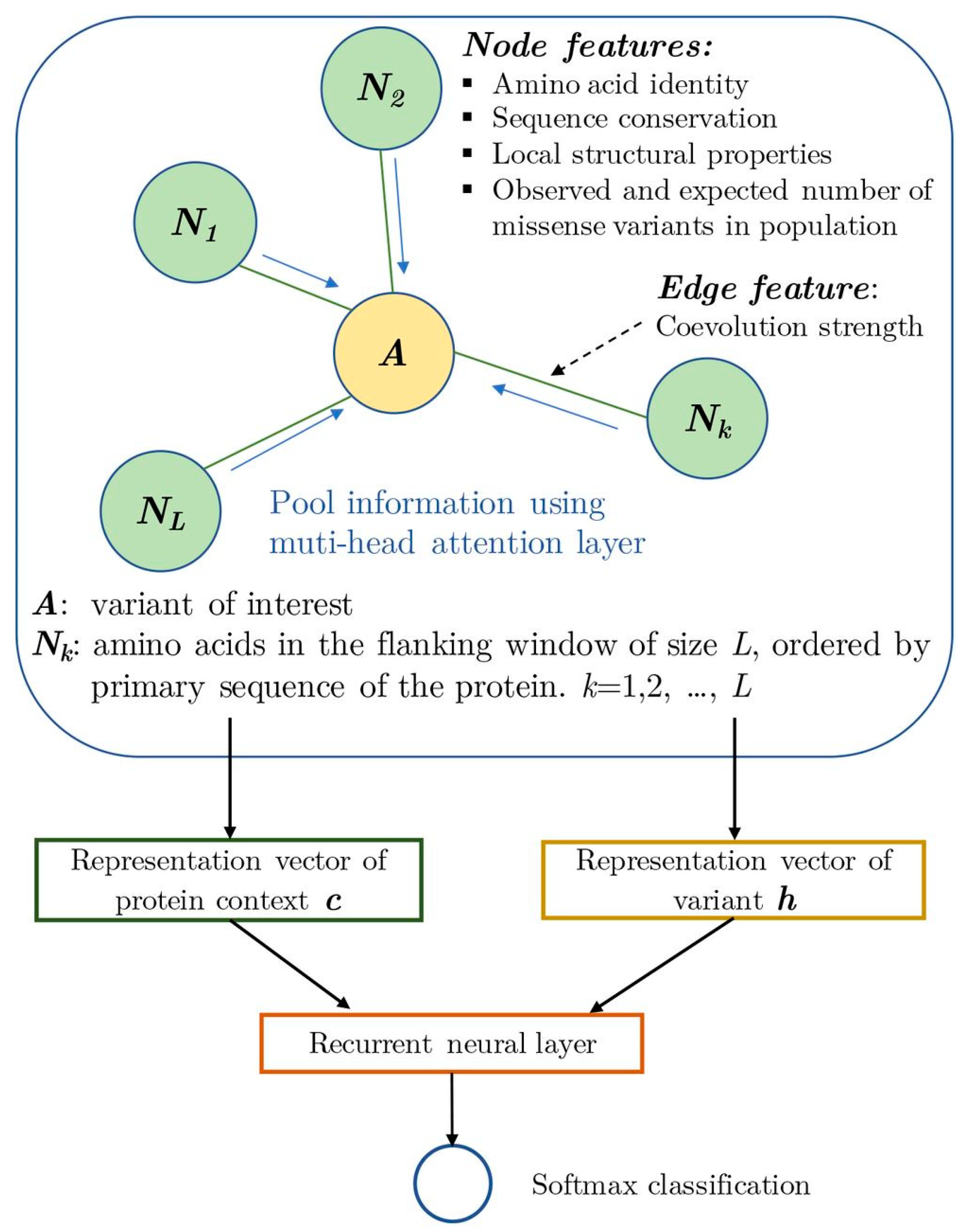
3.1. Protein-Coding Variants
3.2. Intronic and Intergenic Regions
3.2.1. Transcription Factor Binding
3.2.2. DNA Methylation
3.2.3. Splicing
4. Multimodal Data Integration
5. Challenges and Potential Solutions
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moshe, S.L.; Perucca, E.; Ryvlin, P.; Tomson, T. Epilepsy: New advances. Lancet 2015, 385, 884–898. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E. The Epidemiology of Epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, V. Molecular Genetics of Epilepsy: A Clinician’s Perspective. Ann. Indian Acad. Neurol. 2017, 20, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Mullan, K.A.; Anderson, A.; Illing, P.T.; Kwan, P.; Purcell, A.W.; Mifsud, N.A. HLA-associated antiepileptic drug-induced cutaneous adverse reactions. HLA 2019, 93, 417–435. [Google Scholar] [CrossRef]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Wirrell, E.; Tinuper, P.; Perucca, E.; Moshé, S.L. Introduction to the epilepsy syndrome papers. Epilepsia 2022, 63, 1330–1332. [Google Scholar] [CrossRef]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef]
- Sheidley, B.R.; Malinowski, J.; Bergner, A.L.; Bier, L.; Gloss, D.S.; Mu, W.; Mulhern, M.M.; Partack, E.J.; Poduri, A. Genetic testing for the epilepsies: A systematic review. Epilepsia 2022, 63, 375–387. [Google Scholar] [CrossRef]
- Perucca, P. Genetics of Focal Epilepsies: What Do We Know and Where Are We Heading? Epilepsy Curr. 2018, 18, 356–362. [Google Scholar] [CrossRef]
- Perucca, P.; Scheffer, I.E.; Harvey, A.S.; James, P.A.; Lunke, S.; Thorne, N.; Gaff, C.; Regan, B.M.; Damiano, J.A.; Hildebrand, M.S.; et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 2017, 131, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Epi4K-Consortium. Ultra-rare genetic variation in common epilepsies: A case-control sequencing study. Lancet Neurol. 2017, 16, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Gade, M.; Yang, E.; Koh, H.Y.; Lu, J.; Walley, N.M.; Buckley, A.F.; Sands, T.T.; Akman, C.I.; Mikati, M.A.; et al. Somatic variants in diverse genes leads to a spectrum of focal cortical malformations. Brain 2022, 145, 2704–2720. [Google Scholar] [CrossRef] [PubMed]
- Nilo, A.; Gelisse, P.; Crespel, A. Genetic/idiopathic generalized epilepsies: Not so good as that! Rev. Neurol. 2020, 176, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Jallon, P.; Latour, P. Epidemiology of idiopathic generalized epilepsies. Epilepsia 2005, 46 (Suppl. S9), 10–14. [Google Scholar] [CrossRef]
- Stevelink, R.; Campbell, C.; Chen, S.; Abou-Khalil, B.; Adesoji, O.M.; Afawi, Z.; Amadori, E.; Anderson, A.; Anderson, J.; Andrade, D.M.; et al. GWAS meta-analysis of over 29,000 people with epilepsy identifies 26 risk loci and subtype-specific genetic architecture. Nat. Genet. 2023, 55, 1471–1482. [Google Scholar] [CrossRef]
- Pervez, M.T.; Hasnain, M.J.U.; Abbas, S.H.; Moustafa, M.F.; Aslam, N.; Shah, S.S.M. A Comprehensive Review of Performance of Next-Generation Sequencing Platforms. BioMed Res. Int. 2022, 2022, 3457806. [Google Scholar] [CrossRef]
- McClinton, B.; Crinnion, L.A.; McKibbin, M.; Mukherjee, R.; Poulter, J.A.; Smith, C.E.L.; Ali, M.; Watson, C.M.; Inglehearn, C.F.; Toomes, C. Targeted nanopore sequencing enables complete characterisation of structural deletions initially identified using exon-based short-read sequencing strategies. Mol. Genet. Genom. Med. 2023, 11, e2164. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Goenka, S.D.; Gorzynski, J.E.; Shafin, K.; Fisk, D.G.; Pesout, T.; Jensen, T.D.; Monlong, J.; Chang, P.-C.; Baid, G.; Bernstein, J.A.; et al. Accelerated identification of disease-causing variants with ultra-rapid nanopore genome sequencing. Nat. Biotechnol. 2022, 40, 1035–1041. [Google Scholar] [CrossRef]
- He, D.; Xie, L. A cross-level information transmission network for hierarchical omics data integration and phenotype prediction from a new genotype. Bioinformatics 2022, 38, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Deng, Y.; Lin, C.; Li, J.; Lin, X.; Zou, C. Clinical Characteristics and Gene Mutation Analysis of Poststroke Epilepsy. Contrast Media Mol. Imaging 2022, 2022, 4801037. [Google Scholar] [CrossRef] [PubMed]
- de Jong, J.; Cutcutache, I.; Page, M.; Elmoufti, S.; Dilley, C.; Fröhlich, H.; Armstrong, M. Towards realizing the vision of precision medicine: AI based prediction of clinical drug response. Brain 2021, 144, 1738–1750. [Google Scholar] [CrossRef] [PubMed]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Ozdemir, M.A.; Cura, O.K.; Akan, A. Epileptic eeg classification by using time-frequency images for deep learning. Int. J. Neural Syst. 2021, 31, 2150026. [Google Scholar] [CrossRef]
- Huang, J.; Xu, J.; Kang, L.; Zhang, T. Identifying epilepsy based on deep learning using DKI images. Front. Hum. Neurosci. 2020, 14, 590815. [Google Scholar] [CrossRef]
- Nhu, D.; Janmohamed, M.; Antonic-Baker, A.; Perucca, P.; O’Brien, T.J.; Gilligan, A.K.; Kwan, P.; Tan, C.W.; Kuhlmann, L. Deep learning for automated epileptiform discharge detection from scalp EEG: A systematic review. J. Neural Eng. 2022, 19, 051002. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Ramachandran, A.; Lumetta, S.S.; Klee, E.W.; Chen, D. HELLO: Improved neural network architectures and methodologies for small variant calling. BMC Bioinform. 2021, 22, 404. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Wong, C.-L.; Wong, Y.-S.; Tang, C.-I.; Liu, C.-M.; Leung, C.-M.; Lam, T.-W. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat. Mach. Intell. 2020, 2, 220–227. [Google Scholar] [CrossRef]
- Edge, P.; Bansal, V. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 2019, 10, 4660. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Fan, Y.; Ni, B.; Timp, W.; Schatz, M.C. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat. Biotechnol. 2021, 39, 431–441. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Conway, J.R.; Camp, S.Y.; Witkowski, L.; Kofman, E.; Reardon, B.; Han, S.; Moore, N.; Elmarakeby, H.; Salari, K.; et al. Detection of Pathogenic Variants with Germline Genetic Testing Using Deep Learning vs Standard Methods in Patients with Prostate Cancer and Melanoma. JAMA 2020, 324, 1957–1969. [Google Scholar] [CrossRef]
- Costain, G.; Cordeiro, D.; Matviychuk, D.; Mercimek-Andrews, S. Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience 2019, 418, 291–310. [Google Scholar] [CrossRef]
- Ostrander, B.E.P.; Butterfield, R.J.; Pedersen, B.S.; Farrell, A.J.; Layer, R.M.; Ward, A.; Miller, C.; DiSera, T.; Filloux, F.M.; Candee, M.S.; et al. Whole-genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. NPJ Genom. Med. 2018, 3, 22. [Google Scholar] [CrossRef]
- Smith, L.; Malinowski, J.; Ceulemans, S.; Peck, K.; Walton, N.; Sheidley, B.R.; Lippa, N. Genetic testing and counseling for the unexplained epilepsies: An evidence-based practice guideline of the National Society of Genetic Counselors. J. Genet. Couns. 2023, 32, 266–280. [Google Scholar] [CrossRef]
- Kwan, P.; Todaro, M. Genomic Sequencing for Refractory EPilepsy (GREP). 2021. Available online: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=375633&isReview (accessed on 10 August 2023).
- Kamran, M. EMPOWER-1: A Multi-Site Clinical Cohort Research Study to Reduce Health Inequality—Full Text View—ClinicalTrials.gov. 2021. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03987633?term=WGS&cond=Epilepsy&draw=2&rank=1 (accessed on 9 August 2023).
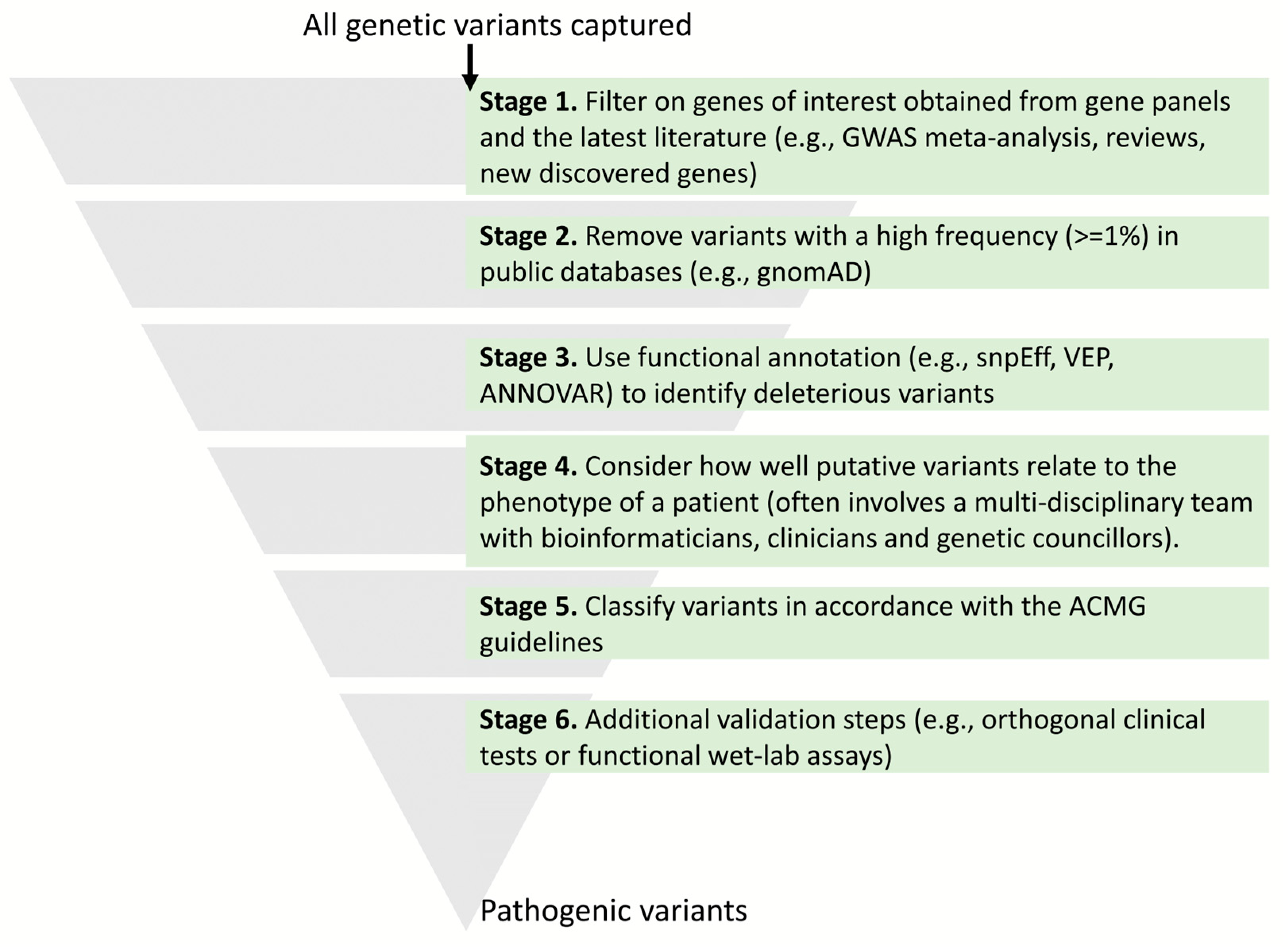
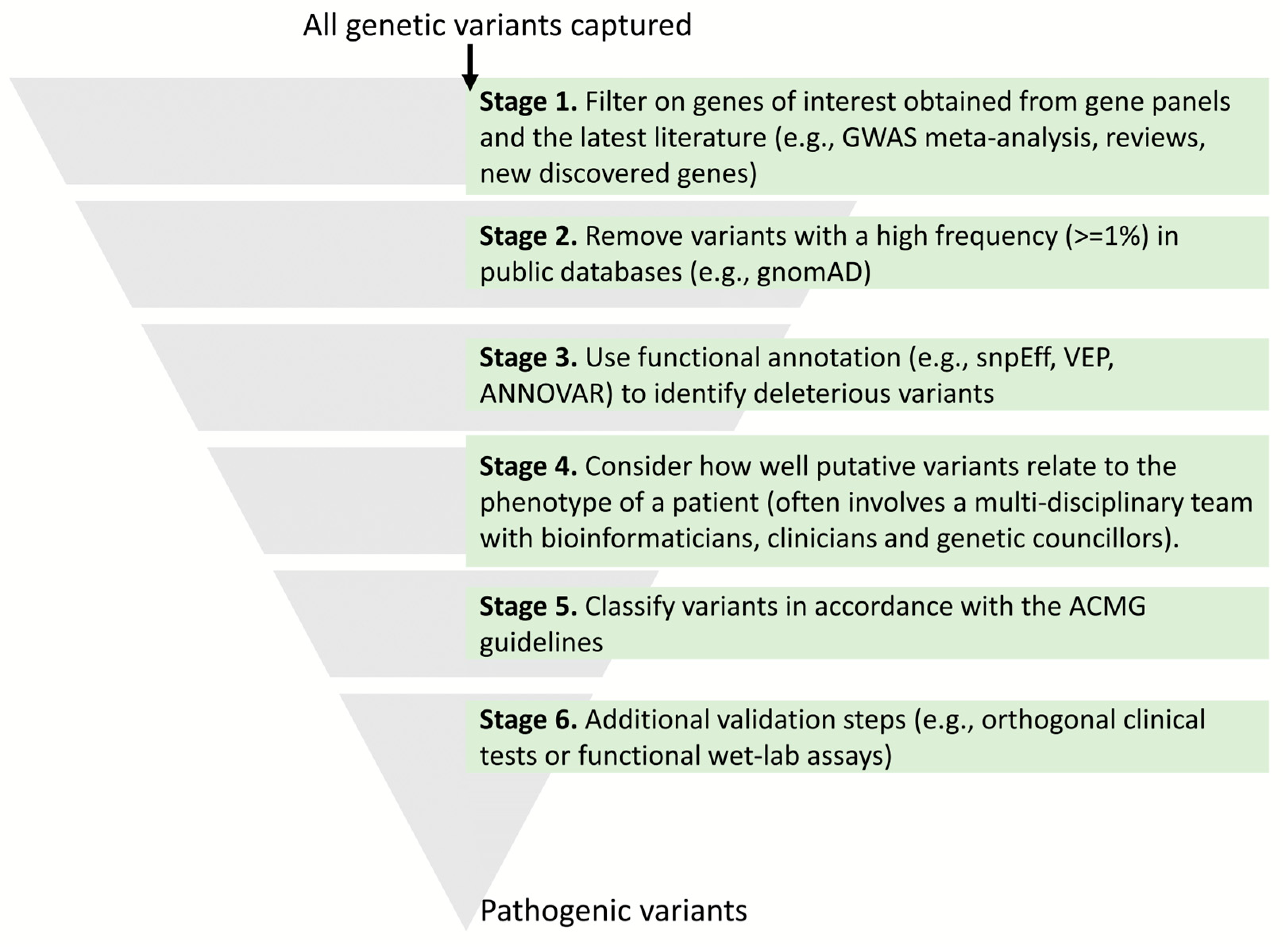
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Baux, D.; Van Goethem, C.; Ardouin, O.; Guignard, T.; Bergougnoux, A.; Koenig, M.; Roux, A.-F. MobiDetails: Online DNA variants interpretation. Eur. J. Hum. Genet. 2021, 29, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Bean, L.J.H.; Hegde, M.R. Clinical implications and considerations for evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Med. 2017, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef]
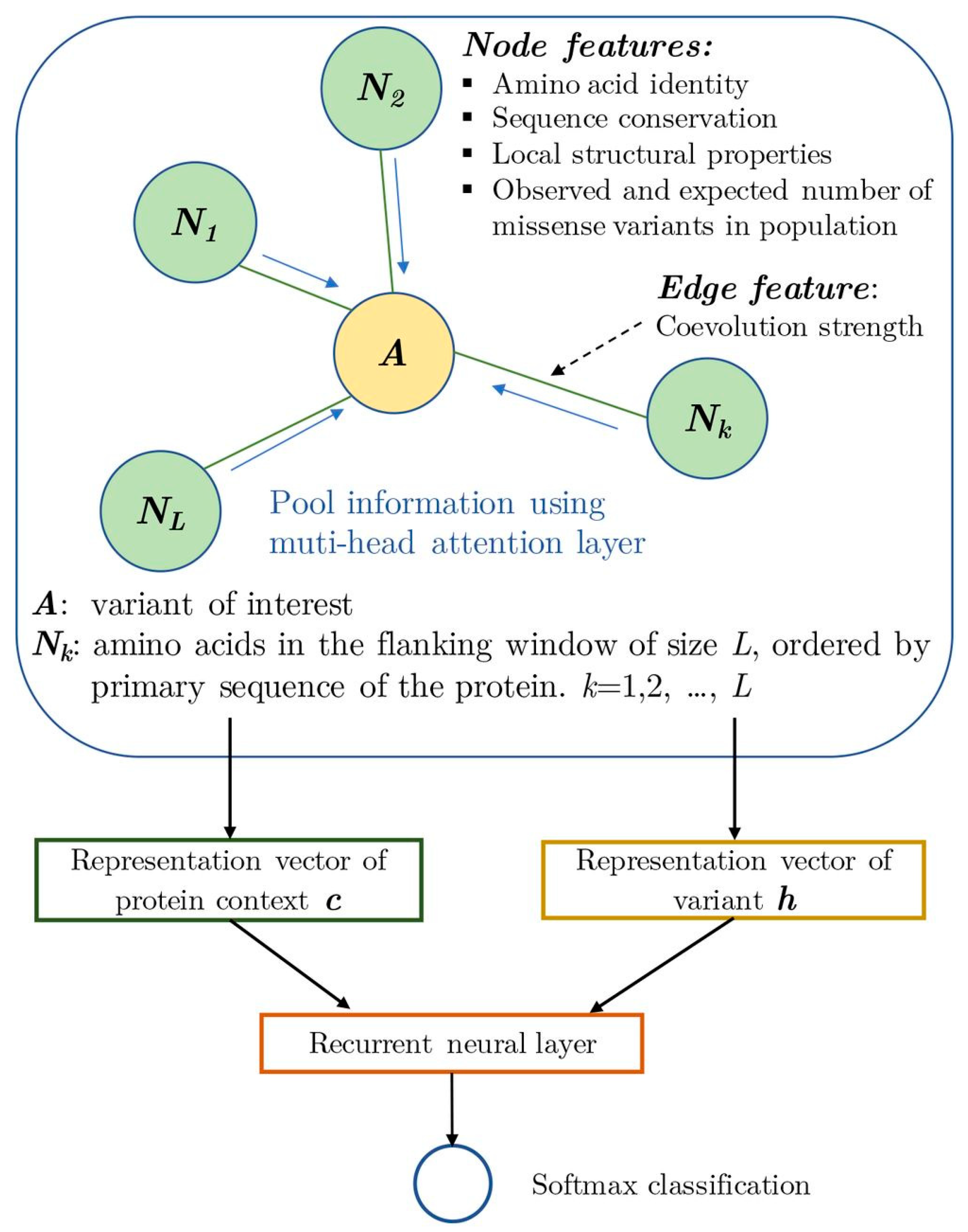
- Zhang, H.; Xu, M.S.; Fan, X.; Chung, W.K.; Shen, Y. Predicting functional effect of missense variants using graph attention neural networks. Nat. Mach. Intell. 2022, 4, 1017–1028. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef]
- Crocker, J.; Noon, E.P.; Stern, D.L. The Soft Touch: Low-Affinity Transcription Factor Binding Sites in Development and Evolution. Curr. Top. Dev. Biol. 2016, 117, 455–469. [Google Scholar] [CrossRef]
- Nishizaki, S.S.; Ng, N.; Dong, S.; Porter, R.S.; Morterud, C.; Williams, C.; Asman, C.; Switzenberg, J.A.; Boyle, A.P. Predicting the effects of SNPs on transcription factor binding affinity. Bioinformatics 2020, 36, 364–372. [Google Scholar] [CrossRef]
- Tseng, C.C.; Wong, M.C.; Liao, W.T.; Chen, C.J.; Lee, S.C.; Yen, J.H.; Chang, S.J. Genetic Variants in Transcription Factor Binding Sites in Humans: Triggered by Natural Selection and Triggers of Diseases. Int. J. Mol. Sci. 2021, 22, 4184. [Google Scholar] [CrossRef] [PubMed]
- McClelland, S.; Brennan, G.P.; Dubé, C.; Rajpara, S.; Iyer, S.; Richichi, C.; Bernard, C.; Baram, T.Z. The transcription factor NRSF contributes to epileptogenesis by selective repression of a subset of target genes. Elife 2014, 3, e01267. [Google Scholar] [CrossRef] [PubMed]
- International League Against Epilepsy Consortium on Complex Epilepsies; Berkovic, S.F.; Cavalleri, G.L.; Koeleman, B.P. Genome-wide meta-analysis of over 29,000 people with epilepsy reveals 26 loci and subtype-specific genetic architecture. medRxiv 2022. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Villicaña, S.; Bell, J.T. Genetic impacts on DNA methylation: Research findings and future perspectives. Genome Biol. 2021, 22, 127. [Google Scholar] [CrossRef]
- Belhedi, N.; Perroud, N.; Karege, F.; Vessaz, M.; Malafosse, A.; Salzmann, A. Increased CPA6 promoter methylation in focal epilepsy and in febrile seizures. Epilepsy Res. 2014, 108, 144–148. [Google Scholar] [CrossRef]
- Kobow, K.; Jeske, I.; Hildebrandt, M.; Hauke, J.; Hahnen, E.; Buslei, R.; Buchfelder, M.; Weigel, D.; Stefan, H.; Kasper, B.; et al. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 2009, 68, 356–364. [Google Scholar] [CrossRef]
- Miller-Delaney, S.F.; Bryan, K.; Das, S.; McKiernan, R.C.; Bray, I.M.; Reynolds, J.P.; Gwinn, R.; Stallings, R.L.; Henshall, D.C. Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain 2015, 138, 616–631. [Google Scholar] [CrossRef]
- Dębski, K.J.; Pitkanen, A.; Puhakka, N.; Bot, A.M.; Khurana, I.; Harikrishnan, K.N.; Ziemann, M.; Kaspi, A.; El-Osta, A.; Lukasiuk, K.; et al. Etiology matters—Genomic DNA Methylation Patterns in Three Rat Models of Acquired Epilepsy. Sci. Rep. 2016, 6, 25668. [Google Scholar] [CrossRef]
- Kiese, K.; Jablonski, J.; Hackenbracht, J.; Wrosch, J.K.; Groemer, T.W.; Kornhuber, J.; Blümcke, I.; Kobow, K. Epigenetic control of epilepsy target genes contributes to a cellular memory of epileptogenesis in cultured rat hippocampal neurons. Acta Neuropathol. Commun. 2017, 5, 79. [Google Scholar] [CrossRef]
- Ryley Parrish, R.; Albertson, A.J.; Buckingham, S.C.; Hablitz, J.J.; Mascia, K.L.; Davis Haselden, W.; Lubin, F.D. Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus. Neuroscience 2013, 248, 602–619. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Gifford, D.K. Predicting the impact of non-coding variants on DNA methylation. Nucleic Acids Res. 2017, 45, e99. [Google Scholar] [CrossRef] [PubMed]
- López-Bigas, N.; Audit, B.; Ouzounis, C.; Parra, G.; Guigó, R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005, 579, 1900–1903. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef]
- Stamberger, H.; Crosiers, D.; Balagura, G.; Bonardi, C.M.; Basu, A.; Cantalupo, G.; Chiesa, V.; Christensen, J.; Bernardina, B.D.; Ellis, C.A.; et al. Natural History Study of STXBP1-Developmental and Epileptic Encephalopathy Into Adulthood. Neurology 2022, 99, e221–e233. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Engel, K.L.; Ramamurthy, A.; Cochran, J.N.; Roovers, J.; Stamberger, H.; Lim, N.; Schneider, A.L.; Hollingsworth, G.; Holder, D.H. Aberrant inclusion of a poison exon causes dravet syndrome and related SCN1A-associated genetic epilepsies. Am. J. Hum. Genet. 2018, 103, 1022–1029. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Ruggiero, S.M.; Gelot, A.; Soardi, F.C.; Ribeiro, B.F.; Pires, D.E.; Ascher, D.B.; Schmitt, A.; Rambaud, C.; Represa, A. A recurrent de novo splice site variant involving DNM1 exon 10a causes developmental and epileptic encephalopathy through a dominant-negative mechanism. Am. J. Hum. Genet. 2022, 109, 2253–2269. [Google Scholar] [CrossRef]
- Tsai, M.H.; Chan, C.K.; Chang, Y.C.; Yu, Y.T.; Chuang, S.T.; Fan, W.L.; Li, S.C.; Fu, T.Y.; Chang, W.N.; Liou, C.W. DEPDC5 mutations in familial and sporadic focal epilepsy. Clin. Genet. 2017, 92, 397–404. [Google Scholar] [CrossRef]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Z.; Wang, J.; Li, S. SpliceFinder: Ab initio prediction of splice sites using convolutional neural network. BMC Bioinform. 2019, 20, 652. [Google Scholar] [CrossRef]
- Dutta, A.; Singh, K.K.; Anand, A. SpliceViNCI: Visualizing the splicing of non-canonical introns through recurrent neural networks. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alföldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv 2022. [Google Scholar] [CrossRef]
- de Sainte Agathe, J.-M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-visual: A free online tool to improve SpliceAI splicing variant interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Avsec, Ž.; Kreuzhuber, R.; Israeli, J.; Xu, N.; Cheng, J.; Shrikumar, A.; Banerjee, A.; Kim, D.S.; Beier, T.; Urban, L.; et al. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat. Biotechnol. 2019, 37, 592–600. [Google Scholar] [CrossRef]
- Nguyen, N.D.; Wang, D. Multiview learning for understanding functional multiomics. PLoS Comput. Biol. 2020, 16, e1007677. [Google Scholar] [CrossRef]
- Castanedo, F. A Review of Data Fusion Techniques. Sci. World J. 2013, 2013, 704504. [Google Scholar] [CrossRef]
- Feleke, R.; Jazayeri, D.; Abouzeid, M.; Powell, K.L.; Srivastava, P.K.; O’Brien, T.J.; Jones, N.C.; Johnson, M.R. Integrative genomics reveals pathogenic mediator of valproate-induced neurodevelopmental disability. Brain 2022, 145, 3832–3842. [Google Scholar] [CrossRef]
- Li, G.; Wang, C.; Han, D.P.; Zhang, Y.P.; Peng, P.; Calhoun, V.D.; Wang, Y.P. Deep Principal Correlated Auto-Encoders with Application to Imaging and Genomics Data Integration. IEEE Access 2020, 8, 20093–20107. [Google Scholar] [CrossRef]
- Marini, S.; Limongelli, I.; Rizzo, E.; Malovini, A.; Errichiello, E.; Vetro, A.; Da, T.; Zuffardi, O.; Bellazzi, R. A Data Fusion Approach to Enhance Association Study in Epilepsy. PLoS ONE 2016, 11, e0164940. [Google Scholar] [CrossRef]
- Wang, Z.; Gu, Y.; Zheng, S.; Yang, L.; Li, J. MGREL: A multi-graph representation learning-based ensemble learning method for gene-disease association prediction. Comput. Biol. Med. 2023, 155, 106642. [Google Scholar] [CrossRef]
- Wei, K.; Li, T.; Huang, F.; Chen, J.; He, Z. Cancer classification with data augmentation based on generative adversarial networks. Front. Comput. Sci. 2021, 16, 162601. [Google Scholar] [CrossRef]
- Iman, M.; Arabnia, H.R.; Rasheed, K. A Review of Deep Transfer Learning and Recent Advancements. Technologies 2023, 11, 40. [Google Scholar] [CrossRef]
- He, P.; Wang, L.; Cui, Y.; Wang, R.; Wu, D. Unsupervised feature learning based on autoencoder for epileptic seizures prediction. Appl. Intell. 2023, 53, 20766–20784. [Google Scholar] [CrossRef]
- Zhao, X.; Sole-Casals, J.; Sugano, H.; Tanaka, T. Seizure onset zone classification based on imbalanced iEEG with data augmentation. J. Neural Eng. 2022, 19, 065001. [Google Scholar] [CrossRef] [PubMed]
- Habashi, A.G.; Azab, A.M.; Eldawlatly, S.; Aly, G.M. Generative adversarial networks in EEG analysis: An overview. J. NeuroEng. Rehabil. 2023, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Eraslan, G.; Avsec, Z.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Si, X.; Zhang, X.; Zhou, Y.; Sun, Y.; Jin, W.; Yin, S.; Zhao, X.; Li, Q.; Ming, D. Automated Detection of Juvenile Myoclonic Epilepsy using CNN based Transfer Learning in Diffusion MRI—Test. In Proceedings of the 2020 42nd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Montreal, QC, Canada, 20–24 July 2020; pp. 1679–1682. [Google Scholar] [CrossRef]
- Liu, L.; Meng, Q.; Weng, C.; Lu, Q.; Wang, T.; Wen, Y. Explainable deep transfer learning model for disease risk prediction using high-dimensional genomic data. PLoS Comput. Biol. 2022, 18, e1010328. [Google Scholar] [CrossRef]
- Tan, R.; Shen, Y. Accurate in silico confirmation of rare copy number variant calls from exome sequencing data using transfer learning. Nucleic Acids Res. 2022, 50, e123. [Google Scholar] [CrossRef]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-omics integration in biomedical research—A metabolomics-centric review. Anal. Chim. Acta 2021, 1141, 144–162. [Google Scholar] [CrossRef]
- Ng, A.; Jordan, M.; Weiss, Y. On spectral clustering: Analysis and an algorithm. In Proceedings of the 15th Annual Conference on Neural Information Processing Systems (NIPS 2001), Vancouver, BC, Canada, 3–8 December 2022; MIT Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Hofmann, T.; Schölkopf, B.; Smola, A.J. Kernel methods in machine learning. Ann. Stat. 2008, 36, 1171–1220. [Google Scholar] [CrossRef]
- Karim, M.R.; Beyan, O.; Zappa, A.; Costa, I.G.; Rebholz-Schuhmann, D.; Cochez, M.; Decker, S. Deep learning-based clustering approaches for bioinformatics. Brief Bioinform. 2021, 22, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Liu, H.; Hou, L.; Xu, S.; Li, H.; Chen, X.; Gao, J.; Wang, Z.; Han, B.; Liu, X.; Wan, S. Discovering Cerebral Ischemic Stroke Associated Genes Based on Network Representation Learning. Front. Genet. 2021, 12, 728333. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zeng, W.; Liu, W.; Lv, H.; Chen, T.; Jiang, R. Leveraging multiple gene networks to prioritize GWAS candidate genes via network representation learning. Methods 2018, 145, 41–50. [Google Scholar] [CrossRef]
- Xing, W.; Qi, J.; Yuan, X.; Li, L.; Zhang, X.; Fu, Y.; Xiong, S.; Hu, L.; Peng, J. A gene–phenotype relationship extraction pipeline from the biomedical literature using a representation learning approach. Bioinformatics 2018, 34, i386–i394. [Google Scholar] [CrossRef]
- Hu, W.; Meng, X.; Bai, Y.; Zhang, A.; Qu, G.; Cai, B.; Zhang, G.; Wilson, T.W.; Stephen, J.M.; Calhoun, V.D.; et al. Interpretable Multimodal Fusion Networks Reveal Mechanisms of Brain Cognition. IEEE Trans. Med. Imaging 2021, 40, 1474–1483. [Google Scholar] [CrossRef]
- Stahlschmidt, S.R.; Ulfenborg, B.; Synnergren, J. Multimodal deep learning for biomedical data fusion: A review. Brief. Bioinform. 2022, 23, bbab569. [Google Scholar] [CrossRef]
- Bengio, Y.; Goodfellow, I.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2017; Volume 1. [Google Scholar]
- Wilcox, E.H.; Sarmady, M.; Wulf, B.; Wright, M.W.; Rehm, H.L.; Biesecker, L.G.; Abou Tayoun, A.N. Evaluating the impact of in silico predictors on clinical variant classification. Genet. Med. 2022, 24, 924–930. [Google Scholar] [CrossRef]
- Deignan, J.L.; Chung, W.K.; Kearney, H.M.; Monaghan, K.G.; Rehder, C.W.; Chao, E.C.; ACMG Laboratory Quality Assurance Committee. Points to consider in the reevaluation and reanalysis of genomictest results: A statement of the American College of Medical Genetics and Genomics(ACMG). Genet. Med. 2019, 21, 1267–1270. [Google Scholar] [CrossRef]
- Chen, K.M.; Cofer, E.M.; Zhou, J.; Troyanskaya, O.G. Selene: A PyTorch-based deep learning library for sequence data. Nat. Methods 2019, 16, 315–318. [Google Scholar] [CrossRef]
- Budach, S.; Marsico, A. Pysster: Classification of biological sequences by learning sequence and structure motifs with convolutional neural networks. Bioinformatics 2018, 34, 3035–3037. [Google Scholar] [CrossRef] [PubMed]
- Kopp, W.; Monti, R.; Tamburrini, A.; Ohler, U.; Akalin, A. Deep learning for genomics using Janggu. Nat. Commun. 2020, 11, 3488. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Lai, B.; Qian, S.; Zhang, H.; Zhang, S.; Kozlova, A.; Duan, J.; Xu, J.; He, X. Annotating functional effects of non-coding variants in neuropsychiatric cell types by deep transfer learning. PLoS Comput. Biol. 2022, 18, e1010011. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeibich, R.; Kwan, P.; J. O’Brien, T.; Perucca, P.; Ge, Z.; Anderson, A. Applications for Deep Learning in Epilepsy Genetic Research. Int. J. Mol. Sci. 2023, 24, 14645. https://doi.org/10.3390/ijms241914645
Zeibich R, Kwan P, J. O’Brien T, Perucca P, Ge Z, Anderson A. Applications for Deep Learning in Epilepsy Genetic Research. International Journal of Molecular Sciences. 2023; 24(19):14645. https://doi.org/10.3390/ijms241914645
Chicago/Turabian StyleZeibich, Robert, Patrick Kwan, Terence J. O’Brien, Piero Perucca, Zongyuan Ge, and Alison Anderson. 2023. "Applications for Deep Learning in Epilepsy Genetic Research" International Journal of Molecular Sciences 24, no. 19: 14645. https://doi.org/10.3390/ijms241914645
APA StyleZeibich, R., Kwan, P., J. O’Brien, T., Perucca, P., Ge, Z., & Anderson, A. (2023). Applications for Deep Learning in Epilepsy Genetic Research. International Journal of Molecular Sciences, 24(19), 14645. https://doi.org/10.3390/ijms241914645