

Nicotine Exposure in a Phencyclidine-Induced Mice Model of Schizophrenia: Sex-Selective Medial Prefrontal Cortex Protein Markers of the Combined Insults in Adolescent Mice
 , , ,
, , ,
Abstract
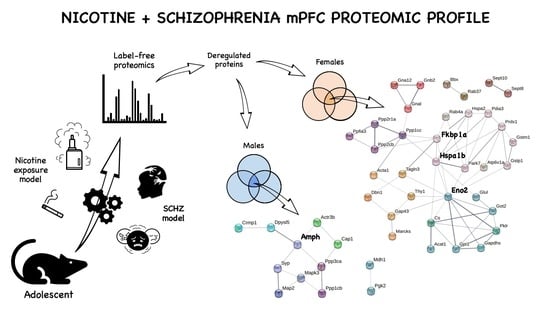
1. Introduction
2. Results
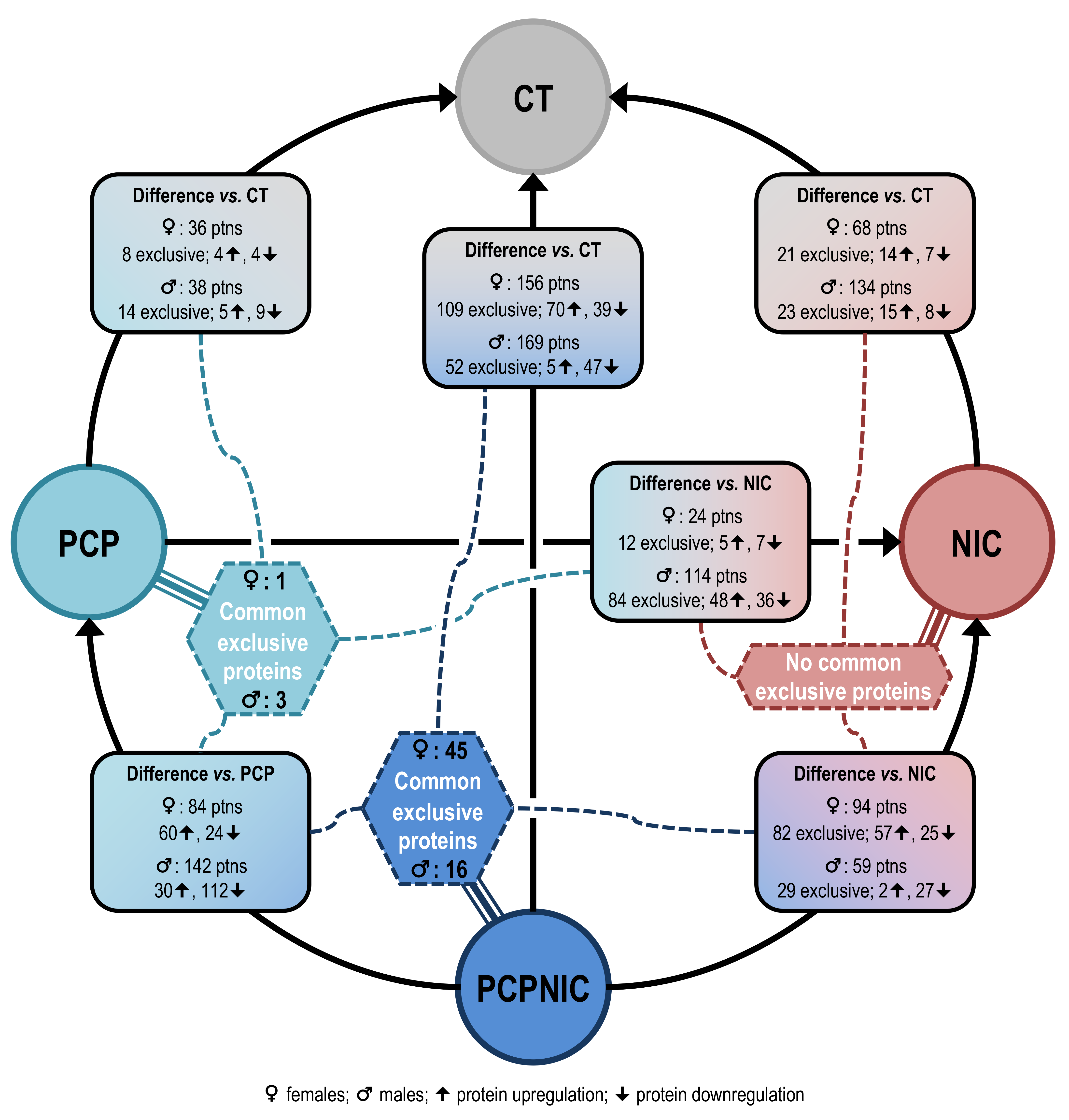
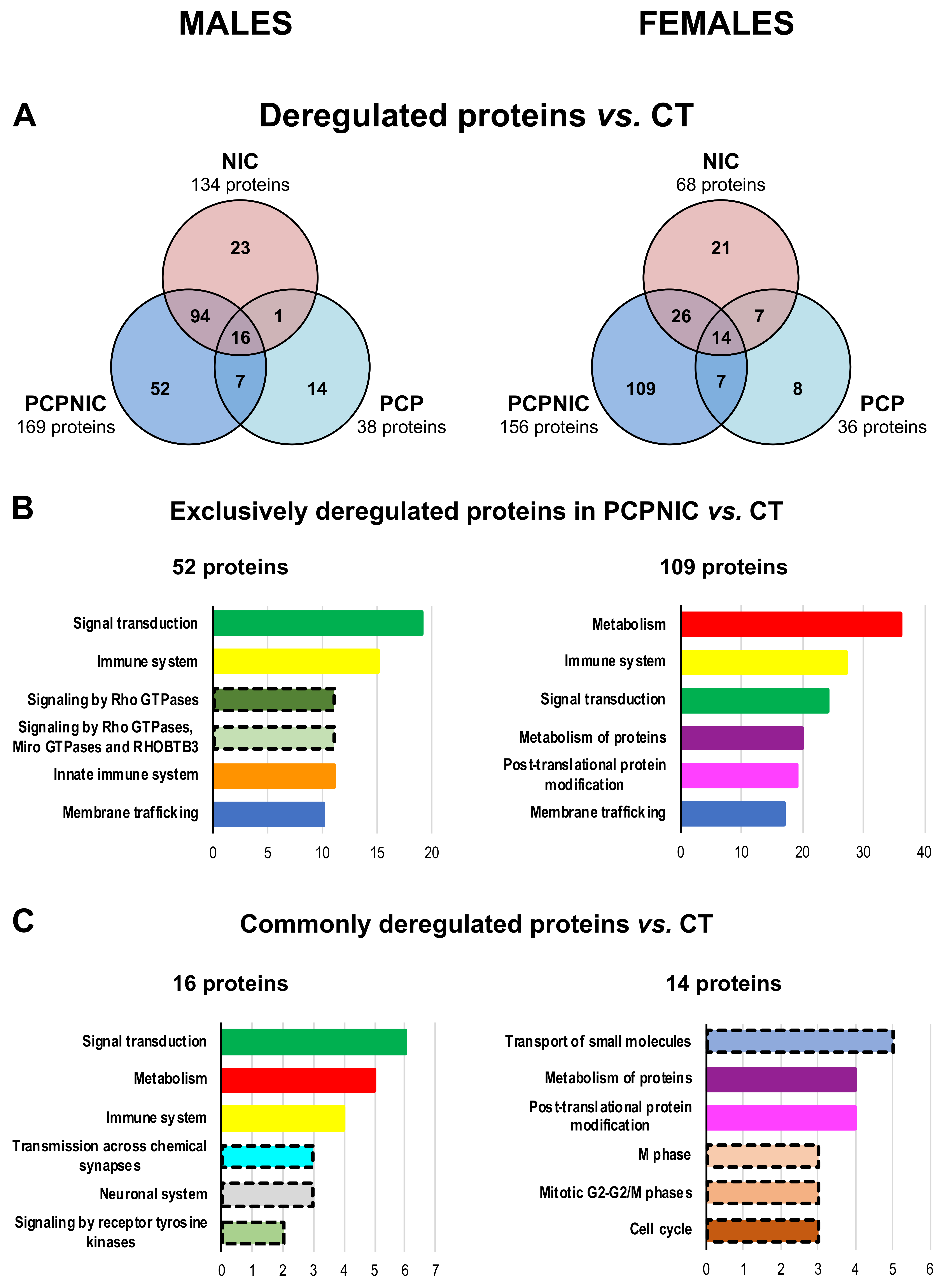
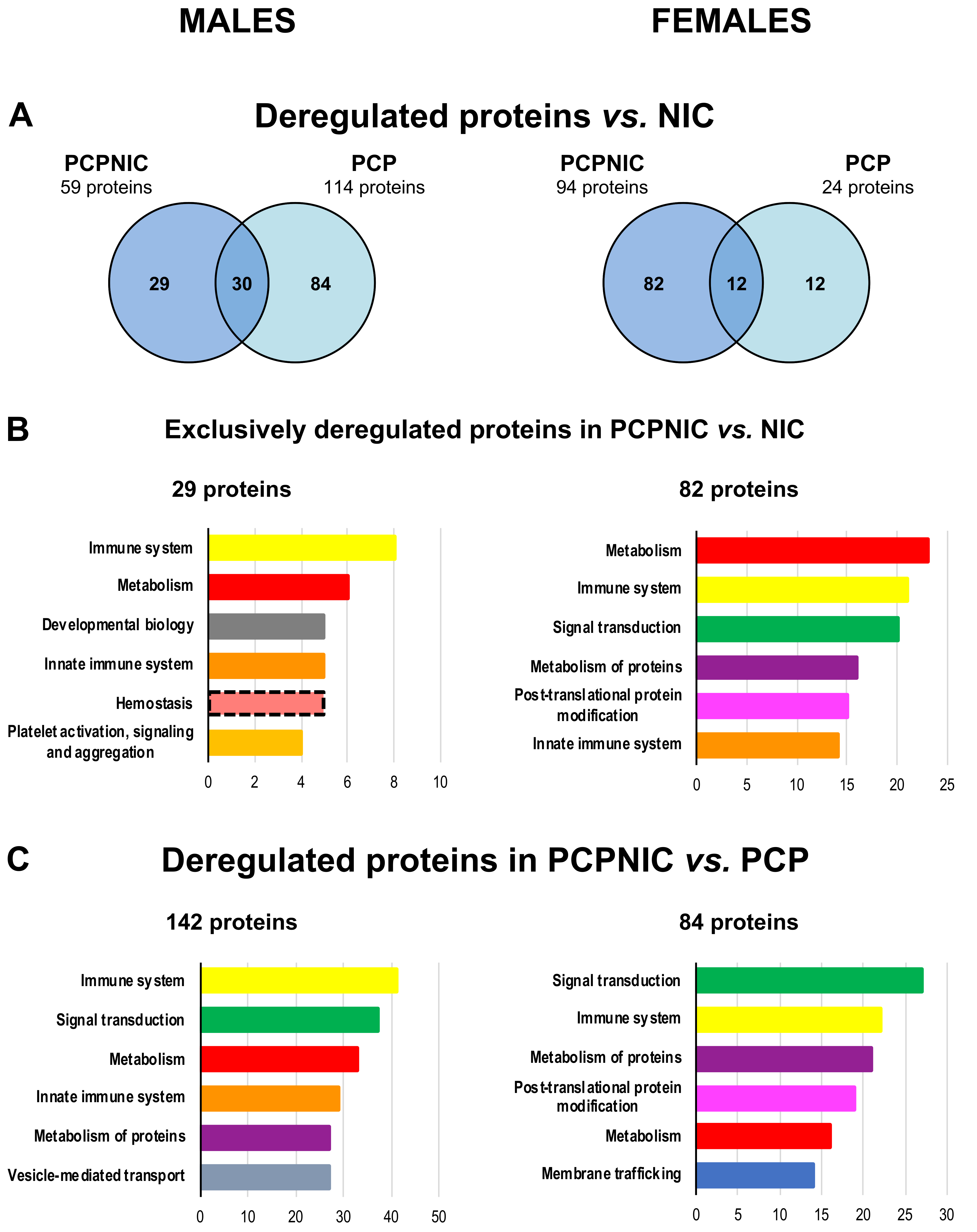
2.1. Proteomic Profile of the mPFC of PCP, NIC and PCPNIC Mice When Compared to Controls
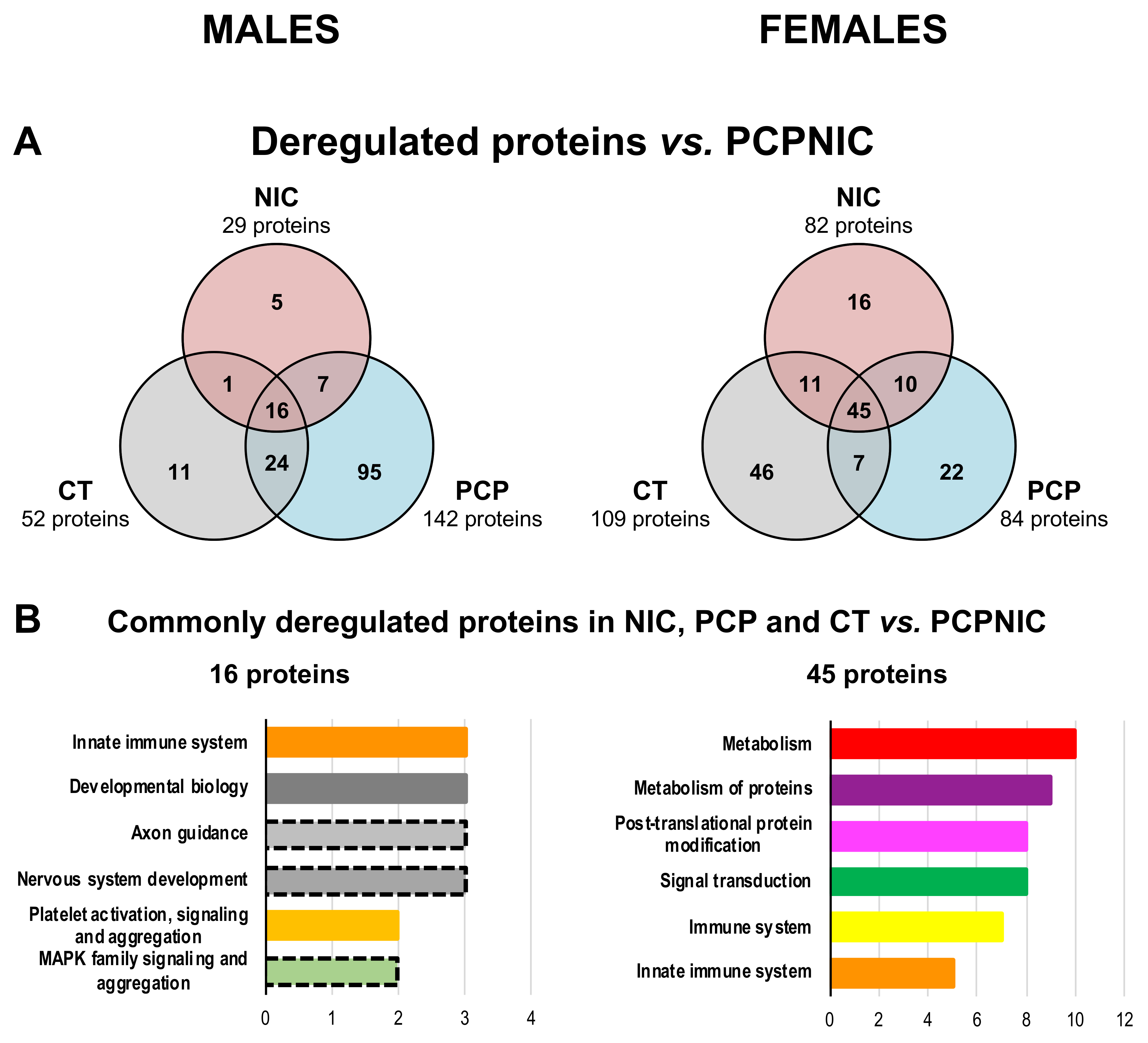
2.2. Proteomic Profile of the mPFC of PCP or PCPNIC Mice When Compared to Nicotine-Exposed Ones
2.3. Proteomic Profile of the mPFC of PCPNIC Mice When Compared to PCP-Exposed Ones
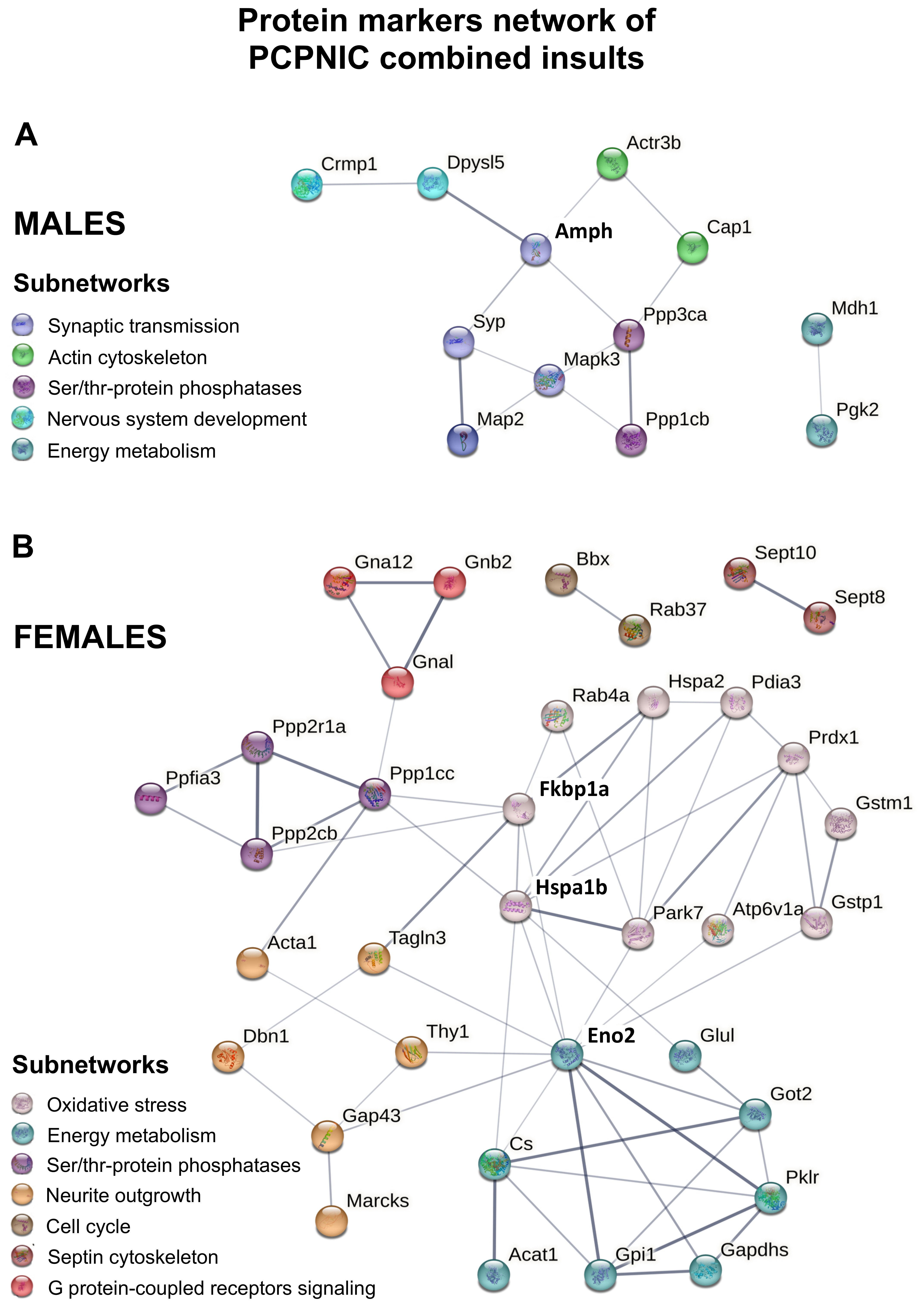
2.4. Protein Markers in the mPFC of PCPNIC Mice
3. Discussion
3.1. Males mPFC Interactome Map of PCPNIC Mice
3.2. Female mPFC Interactome Map of PCPNIC Mice
4. Materials and Methods
4.1. Materials
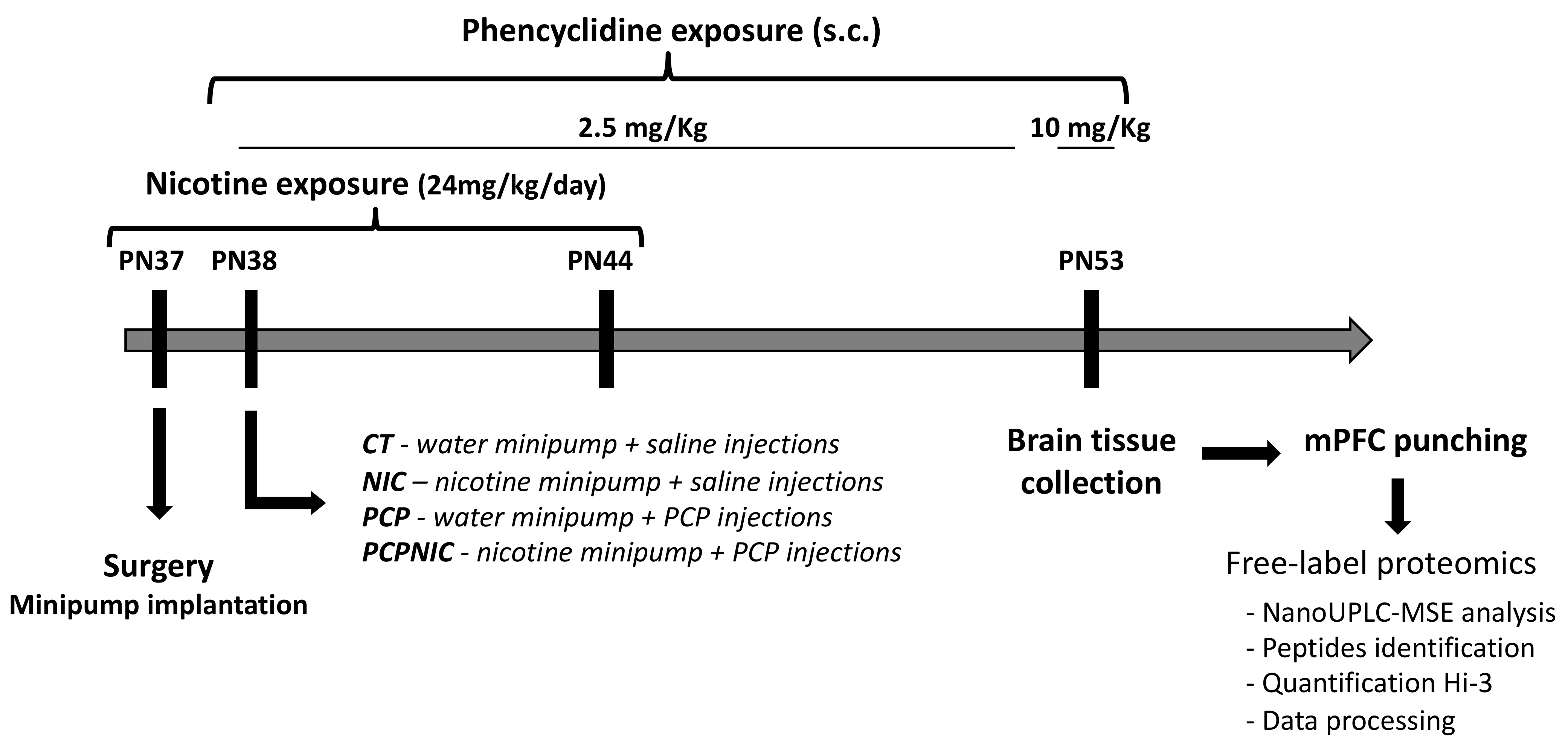
4.2. Animals and Treatment
4.3. Protein Extraction
4.4. Protein Digestion for NanoULPC-MSE Analysis
4.5. NanoUPLC-MSE Analysis
4.6. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peritogiannis, V.; Gogou, A.; Samakouri, M. Very Long-Term Outcome of Psychotic Disorders. Int. J. Soc. Psychiatry 2020, 66, 633–641. [Google Scholar] [CrossRef]
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef]
- Yang, A.; Tsai, S.-J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int. J. Mol. Sci. 2017, 18, 1689. [Google Scholar] [CrossRef]
- Buck, S.A.; Quincy Erickson-Oberg, M.; Logan, R.W.; Freyberg, Z. Relevance of Interactions between Dopamine and Glutamate Neurotransmission in Schizophrenia. Mol. Psychiatry 2022, 27, 3583–3591. [Google Scholar] [CrossRef]
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Primers 2015, 1, 15067. [Google Scholar] [CrossRef]
- Białoń, M.; Wąsik, A. Advantages and Limitations of Animal Schizophrenia Models. Int. J. Mol. Sci. 2022, 23, 5968. [Google Scholar] [CrossRef]
- Weston-Green, K. Antipsychotic Drug Development: From Historical Evidence to Fresh Perspectives. Front. Psychiatry 2022, 13, 903156. [Google Scholar] [CrossRef]
- Föcking, M.; Lopez, L.M.; English, J.A.; Dicker, P.; Wolff, A.; Brindley, E.; Wynne, K.; Cagney, G.; Cotter, D.R. Proteomic and Genomic Evidence Implicates the Postsynaptic Density in Schizophrenia. Mol. Psychiatry 2015, 20, 424–432. [Google Scholar] [CrossRef]
- Davalieva, K.; Kostovska, I.M.; Dwork, A.J. Proteomics Research in Schizophrenia. Front. Cell. Neurosci. 2016, 10, 18. [Google Scholar] [CrossRef]
- Sarnyai, Z.; Guest, P.C. Connecting Brain Proteomics with Behavioural Neuroscience in Translational Animal Models of Neuropsychiatric Disorders. Adv. Exp. Med. Biol. 2017, 974, 97–114. [Google Scholar] [CrossRef]
- Castle, D.J.; Wessely, S.; Murray, R.M. Sex and Schizophrenia: Effects of Diagnostic Stringency, and Associations with Premorbid Variables. Br. J. Psychiatry 1993, 162, 658–664. [Google Scholar] [CrossRef]
- Mcgrath, J.; Saha, S.; Chant, D.; Welham, J. Schizophrenia: A Concise Overview of Incidence, Prevalence, and Mortality. Epidemiol. Rev. 2008, 30, 67–76. [Google Scholar] [CrossRef]
- Hayes, E.; Gavrilidis, E.; Kulkarni, J. The Role of Oestrogen and Other Hormones in the Pathophysiology and Treatment of Schizophrenia. Schizophr. Res. Treat. 2012, 2012, 540273. [Google Scholar] [CrossRef]
- Mendrek, A.; Mancini-Marïe, A. Sex/Gender Differences in the Brain and Cognition in Schizophrenia. Neurosci. Biobehav. Rev. 2016, 67, 57–78. [Google Scholar] [CrossRef]
- Wickens, M.M.; Bangasser, D.A.; Briand, L.A. Sex Differences in Psychiatric Disease: A Focus on the Glutamate System. Front. Mol. Neurosci. 2018, 11, 197. [Google Scholar] [CrossRef]
- Kumari, V.; Postma, P. Nicotine Use in Schizophrenia: The Self Medication Hypotheses. Neurosci. Biobehav. Rev. 2005, 29, 1021–1034. [Google Scholar] [CrossRef]
- Ohi, K.; Shimada, T.; Kuwata, A.; Kataoka, Y.; Okubo, H.; Kimura, K.; Yasuyama, T.; Uehara, T.; Kawasaki, Y. Smoking Rates and Number of Cigarettes Smoked per Day in Schizophrenia: A Large Cohort Meta-Analysis in a Japanese Population. Int. J. Neuropsychopharmacol. 2019, 22, 19–27. [Google Scholar] [CrossRef]
- Ding, J.B.; Hu, K. Cigarette Smoking and Schizophrenia: Etiology, Clinical, Pharmacological, and Treatment Implications. Schizophr. Res. Treat. 2021, 2021, 7698030. [Google Scholar] [CrossRef]
- Danielsson, K.; Stomberg, R.; Adermark, L.; Ericson, M.; Söderpalm, B. Differential Dopamine Release by Psychosis-Generating and Non-Psychosis-Generating Addictive Substances in the Nucleus Accumbens and Dorsomedial Striatum. Transl. Psychiatry 2021, 11, 472. [Google Scholar] [CrossRef]
- Mackowick, K.M.; Barr, M.S.; Wing, V.C.; Rabin, R.A.; Ouellet-Plamondon, C.; George, T.P. Neurocognitive Endophenotypes in Schizophrenia: Modulation by Nicotinic Receptor Systems. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 52, 79–85. [Google Scholar] [CrossRef][Green Version]
- Quigley, H.; MacCabe, J.H. The Relationship between Nicotine and Psychosis. Ther. Adv. Psychopharmacol. 2019, 9, 204512531985996. [Google Scholar] [CrossRef]
- Laursen, T.M.; Munk-Olsen, T.; Vestergaard, M. Life Expectancy and Cardiovascular Mortality in Persons with Schizophrenia. Curr. Opin. Psychiatry 2012, 25, 83–88. [Google Scholar] [CrossRef]
- Marchi, M.; Grilli, M. Presynaptic Nicotinic Receptors Modulating Neurotransmitter Release in the Central Nervous System: Functional Interactions with Other Coexisting Receptors. Prog. Neurobiol. 2010, 92, 105–111. [Google Scholar] [CrossRef]
- Wonnacott, S. Presynaptic Nicotinic ACh Receptors. Trends Neurosci. 1997, 20, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, G.; Fish, K.N.; Lewis, D.A. GABA Neuron Alterations, Cortical Circuit Dysfunction and Cognitive Deficits in Schizophrenia. Neural Plast. 2011, 2011, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.M. Beyond the Dopamine Hypothesis of Schizophrenia to Three Neural Networks of Psychosis: Dopamine, Serotonin, and Glutamate. CNS Spectr. 2018, 23, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Yeom, M.; Shim, I.; Lee, H.-J.; Hahm, D.-H. Proteomic Analysis of Nicotine-Associated Protein Expression in the Striatum of Repeated Nicotine-Treated Rats. Biochem. Biophys. Res. Commun. 2005, 326, 321–328. [Google Scholar] [CrossRef]
- Hwang, Y.Y.; Li, M.D. Proteins Differentially Expressed in Response to Nicotine in Five Rat Brain Regions: Identification Using a 2-DE/MS-Based Proteomics Approach. Proteomics 2006, 6, 3138–3153. [Google Scholar] [CrossRef]
- Matsuura, K.; Otani, M.; Takano, M.; Kadoyama, K.; Matsuyama, S. The Influence of Chronic Nicotine Treatment on Proteins Expressed in the Mouse Hippocampus and Cortex. Eur. J. Pharmacol. 2016, 780, 16–25. [Google Scholar] [CrossRef]
- Häfner, H. From Onset and Prodromal Stage to a Life-Long Course of Schizophrenia and Its Symptom Dimensions: How Sex, Age, and Other Risk Factors Influence Incidence and Course of Illness. Psychiatry J. 2019, 2019, 9804836. [Google Scholar] [CrossRef]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “Just the Facts” 4. Clinical Features and Conceptualization. Schizophr. Res. 2009, 110, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.J.; McGlashan, T.H.; Rosen, J.L.; Cadenhead, K.; Ventura, J.; McFarlane, W.; Perkins, D.O.; Pearlson, G.D.; Woods, S.W. Prodromal Assessment With the Structured Interview for Prodromal Syndromes and the Scale of Prodromal Symptoms: Predictive Validity, Interrater Reliability, and Training to Reliability. Schizophr. Bull. 2003, 29, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Lanza, S.T.; Vasilenko, S.A. New Methods Shed Light on Age of Onset as a Risk Factor for Nicotine Dependence. Addict. Behav. 2015, 50, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Cullen, K.A.; Ambrose, B.K.; Gentzke, A.S.; Apelberg, B.J.; Jamal, A.; King, B.A. Notes from the Field: Use of Electronic Cigarettes and Any Tobacco Product Among Middle and High School Students—United States, 2011–2018. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1276–1277. [Google Scholar] [CrossRef] [PubMed]
- Miech, R.; Johnston, L.; O’Malley, P.M.; Bachman, J.G.; Patrick, M.E. Adolescent Vaping and Nicotine Use in 2017–2018—U.S. National Estimates. N. Engl. J. Med. 2019, 380, 192–193. [Google Scholar] [CrossRef]
- Gogos, A.; Skokou, M.; Ferentinou, E.; Gourzis, P. Nicotine Consumption during the Prodromal Phase of Schizophrenia—A Review of the Literature. Neuropsychiatr. Dis. Treat. 2019, 15, 2943–2958. [Google Scholar] [CrossRef]
- Lally, J.; Spaducci, G.; Gardner-Sood, P.; Atakan, Z.; Greenwood, K.; di Forti, M.; Ismail, K.; Murphy, K.C.; Smith, S.; McNeill, A.; et al. Tobacco Smoking and Nicotine Dependence in First Episode and Established Psychosis. Asian J. Psychiatr. 2019, 43, 125–131. [Google Scholar] [CrossRef]
- Myles, N.; Newall, H.D.; Curtis, J.; Nielssen, O.; Shiers, D.; Large, M. Tobacco Use before, at, and after First-Episode Psychosis: A Systematic Meta-Analysis. J. Clin. Psychiatry 2012, 73, 468–475. [Google Scholar] [CrossRef]
- Schmidt, S.J.; Schultze-Lutter, F.; Schimmelmann, B.G.; Maric, N.P.; Salokangas, R.K.R.; Riecher-Rössler, A.; van der Gaag, M.; Meneghelli, A.; Nordentoft, M.; Marshall, M.; et al. EPA Guidance on the Early Intervention in Clinical High Risk States of Psychoses. Eur. Psychiatry 2015, 30, 388–404. [Google Scholar] [CrossRef]
- Shapiro, L.P.; Parsons, R.G.; Koleske, A.J.; Gourley, S.L. Differential Expression of Cytoskeletal Regulatory Factors in the Adolescent Prefrontal Cortex: Implications for Cortical Development. J. Neurosci. Res. 2017, 95, 1123–1143. [Google Scholar] [CrossRef]
- Jobson, D.D.; Hase, Y.; Clarkson, A.N.; Kalaria, R.N. The Role of the Medial Prefrontal Cortex in Cognition, Ageing and Dementia. Brain Commun. 2021, 3, fcab125. [Google Scholar] [CrossRef]
- Kim, I.H.; Kim, N.; Kim, S.; Toda, K.; Catavero, C.M.; Courtland, J.L.; Yin, H.H.; Soderling, S.H. Dysregulation of the Synaptic Cytoskeleton in the PFC Drives Neural Circuit Pathology, Leading to Social Dysfunction. Cell Rep. 2020, 32, 107965. [Google Scholar] [CrossRef] [PubMed]
- McFalls, A.J.; Imperio Ceasar, G.; Woodward, E.; Krikorian, C.; Brooke, S.; Wronowski, B.; Grigson, P.S.; Freeman, W.M.; Vrana, K.E. An RNA-Seq Study of the MPFC of Rats with Different Addiction Phenotypes. Brain Res. Bull. 2022, 191, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.L.; Kotzé, C. Early Deviant Behaviour as a Dimension Trait and Endophenotype in Schizophrenia. S. Afr. J. Psychiatry 2022, 28, 1747. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, S.F.; Mansvelder, H.D.; de Vries, T.J. Cholinergic Modulation of Dopamine Pathways through Nicotinic Acetylcholine Receptors. Biochem. Pharmacol. 2015, 97, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.F.; McDonald, C.G.; Bergstrom, H.C.; Ehlinger, D.G.; Brielmaier, J.M. Adolescent Nicotine Induces Persisting Changes in Development of Neural Connectivity. Neurosci. Biobehav. Rev. 2015, 55, 432–443. [Google Scholar] [CrossRef]
- Aguilar, M.C.; Gurpegui, M.; Diaz, F.J.; de Leon, J. Nicotine Dependence and Symptoms in Schizophrenia. Br. J. Psychiatry 2005, 186, 215–221. [Google Scholar] [CrossRef] [PubMed]
- de Leon, J.; Diaz, F.J. A Meta-Analysis of Worldwide Studies Demonstrates an Association between Schizophrenia and Tobacco Smoking Behaviors. Schizophr. Res. 2005, 76, 135–157. [Google Scholar] [CrossRef]
- Lodge, D.; Mercier, M.S. Ketamine and Phencyclidine: The Good, the Bad and the Unexpected. Br. J. Pharmacol. 2015, 172, 4254–4276. [Google Scholar] [CrossRef]
- Winship, I.R.; Dursun, S.M.; Baker, G.B.; Balista, P.A.; Kandratavicius, L.; Maia-de-Oliveira, J.P.; Hallak, J.; Howland, J.G. An Overview of Animal Models Related to Schizophrenia. Can. J. Psychiatry 2018, 64, 5–17. [Google Scholar] [CrossRef]
- Cadinu, D.; Grayson, B.; Podda, G.; Harte, M.K.; Doostdar, N.; Neill, J.C. NMDA Receptor Antagonist Rodent Models for Cognition in Schizophrenia and Identification of Novel Drug Treatments, an Update. Neuropharmacology 2018, 142, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Bubeníková-Valešová, V.; Horáček, J.; Vrajová, M.; Höschl, C. Models of Schizophrenia in Humans and Animals Based on Inhibition of NMDA Receptors. Neurosci. Biobehav. Rev. 2008, 32, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Yee, B.K.; Chang, D.L.T.; Feldon, J. The Effects of Dizocilpine and Phencyclidine on Prepulse Inhibition of the Acoustic Startle Reflex and on Prepulse-Elicited Reactivity in C57BL6 Mice. Neuropsychopharmacology 2004, 29, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.A.; Watson, D.J.G.; Fone, K.C.F. Animal Models of Schizophrenia. Br. J. Pharmacol. 2011, 164, 1162–1194. [Google Scholar] [CrossRef]
- Dutra-Tavares, A.C.; Manhães, A.C.; Semeão, K.A.; Maia, J.G.; Couto, L.A.; Filgueiras, C.C.; Ribeiro-Carvalho, A.; Abreu-Villaça, Y. Does Nicotine Exposure during Adolescence Modify the Course of Schizophrenia-like Symptoms? Behavioral Analysis in a Phencyclidine-Induced Mice Model. PLoS ONE 2021, 16, e0257986. [Google Scholar] [CrossRef]
- Hambsch, B.; Keyworth, H.; Lind, J.; Otte, D.M.; Racz, I.; Kitchen, I.; Bailey, A.; Zimmer, A. Chronic Nicotine Improves Short-Term Memory Selectively in a G72 Mouse Model of Schizophrenia. Br. J. Pharmacol. 2014, 171, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Freitas, A.L.; Ribeiro-Carvalho, A.; Lima, C.S.; Dutra-Tavares, A.C.; Manhães, A.C.; Lisboa, P.C.; Oliveira, E.; Gaspar de Moura, E.; Filgueiras, C.C.; Abreu-Villaça, Y. Nicotine Exposure during the Third Trimester Equivalent of Human Gestation: Time Course of Effects on the Central Cholinergic System of Rats. Toxicol. Sci. 2011, 123, 144–154. [Google Scholar] [CrossRef]
- Nascimento, J.M.; Garcia, S.; Saia-Cereda, V.M.; Santana, A.G.; Brandao-Teles, C.; Zuccoli, G.S.; Junqueira, D.G.; Reis-de-Oliveira, G.; Baldasso, P.A.; Cassoli, J.S.; et al. Proteomics and Molecular Tools for Unveiling Missing Links in the Biochemical Understanding of Schizophrenia. Proteom. Clin. Appl. 2016, 10, 1148–1158. [Google Scholar] [CrossRef]
- Beraki, S.; Diaz-Heijtz, R.; Tai, F.; Ögren, S.O. Effects of Repeated Treatment of Phencyclidine on Cognition and Gene Expression in C57BL/6 Mice. Int. J. Neuropsychopharmacol. 2009, 12, 243. [Google Scholar] [CrossRef]
- Barrera-Conde, M.; Ausin, K.; Lachén-Montes, M.; Fernández-Irigoyen, J.; Galindo, L.; Cuenca-Royo, A.; Fernández-Avilés, C.; Pérez, V.; de la Torre, R.; Santamaría, E.; et al. Cannabis Use Induces Distinctive Proteomic Alterations in Olfactory Neuroepithelial Cells of Schizophrenia Patients. J. Pers. Med. 2021, 11, 160. [Google Scholar] [CrossRef]
- Dutra-Tavares, A.C.; Bandeira-Martins, A.; Silva, J.O.; Couto, L.A.; Filgueiras, C.C.; Ribeiro-Carvalho, A.; Manhães, A.C.; Abreu-Villaça, Y. Adolescent Nicotine Potentiates the Inhibitory Effect of Raclopride, a D2R Antagonist, on Phencyclidine-Sensitized Psychotic-like Behavior in Mice. Toxicol. Appl. Pharmacol. 2022, 456, 116282. [Google Scholar] [CrossRef] [PubMed]
- Kanniah, G.; Kumar, R. A Selective Literature Review Exploring the Role of the Nicotinic System in Schizophrenia. Gen. Psychiatr. 2023, 36, e100756. [Google Scholar] [CrossRef] [PubMed]
- Gogos, A.; van den Buuse, M. Sex Differences in Psychosis: Focus on Animal Models. Curr. Top. Behav. Neurosci. 2023, 62, 133–163. [Google Scholar] [CrossRef]
- Dutra-Tavares, A.C.; Souza, T.P.; Silva, J.O.; Semeão, K.A.; Mello, F.F.; Filgueiras, C.C.; Ribeiro-Carvalho, A.; Manhães, A.C.; Abreu-Villaça, Y. Neonatal Phencyclidine as a Model of Sex-Biased Schizophrenia Symptomatology in Adolescent Mice. Psychopharmacology 2023, 240, 2111–2129. [Google Scholar] [CrossRef]
- English, J.A.; Dicker, P.; Föcking, M.; Dunn, M.J.; Cotter, D.R. 2-D DIGE Analysis Implicates Cytoskeletal Abnormalities in Psychiatric Disease. Proteomics 2009, 9, 3368–3382. [Google Scholar] [CrossRef] [PubMed]
- Rattay, T.W.; Martin, P.; Vittore, D.; Hengel, H.; Cebi, I.; Tünnerhoff, J.; Stefanou, M.I.; Hoffmann, J.F.; von der Ehe, K.; Klaus, J.; et al. Cerebrospinal Fluid Findings in Patients with Psychotic Symptoms—A Retrospective Analysis. Sci. Rep. 2021, 11, 7169. [Google Scholar] [CrossRef]
- Endres, D.; Leypoldt, F.; Bechter, K.; Hasan, A.; Steiner, J.; Domschke, K.; Wandinger, K.-P.; Falkai, P.; Arolt, V.; Stich, O.; et al. Autoimmune Encephalitis as a Differential Diagnosis of Schizophreniform Psychosis: Clinical Symptomatology, Pathophysiology, Diagnostic Approach, and Therapeutic Considerations. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 803–818. [Google Scholar] [CrossRef]
- Wu, Y.; Matsui, H.; Tomizawa, K. Amphiphysin I and Regulation of Synaptic Vesicle Endocytosis. Acta Med. Okayama 2009, 63, 305–323. [Google Scholar] [CrossRef]
- Egbujo, C.N.; Sinclair, D.; Hahn, C.-G. Dysregulations of Synaptic Vesicle Trafficking in Schizophrenia. Curr. Psychiatry Rep. 2016, 18, 77. [Google Scholar] [CrossRef]
- Trikash, I.; Kasatkina, L.; Lykhmus, O.; Skok, M. Nicotinic Acetylcholine Receptors Regulate Clustering, Fusion and Acidification of the Rat Brain Synaptic Vesicles. Neurochem. Int. 2020, 138, 104779. [Google Scholar] [CrossRef]
- Torres, L.H.; Garcia, R.C.T.; Blois, A.M.M.; Pacheco-Neto, M.; Camarini, R.; Britto, L.R.; Marcourakis, T. Early Postnatal Tobacco Smoke Exposure Triggers Anxiety-like Behavior and Decreases Synaptic Proteins Even after a Long Exposure-Free Period in Mice. Brain Res. 2019, 1707, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Marchisella, F.; Coffey, E.T.; Hollos, P. Microtubule and Microtubule Associated Protein Anomalies in Psychiatric Disease. Cytoskeleton 2016, 73, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Reszka, A.A.; Segert, R.; Diltz, C.D.; Krebs, E.G.; Fischer, E.H. Association of Mitogen-Activated Protein Kinase with the Microtubule Cytoskeleton. Proc. Natl. Acad. Sci. USA 1995, 92, 8881–8885. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Kosik, K.S. The Pool of MAP Kinase Associated with Microtubules Is Small but Constitutively Active. Mol. Biol. Cell 1996, 7, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Mancuso, N.; Won, H.; Kousi, M.; Finucane, H.K.; Reshef, Y.; Song, L.; Safi, A.; McCarroll, S.; Neale, B.M.; et al. Transcriptome-Wide Association Study of Schizophrenia and Chromatin Activity Yields Mechanistic Disease Insights. Nat. Genet. 2018, 50, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Loukola, A.; Gillespie, N.A.; Peterson, R.; Jia, P.; Riley, B.; Maes, H.; Dick, D.M.; Kendler, K.S.; Damaj, M.I.; et al. Genome-Wide Meta-Analyses of FTND and TTFC Phenotypes. Nicotine Tob. Res. 2020, 22, 900–909. [Google Scholar] [CrossRef]
- Mostowy, S.; Cossart, P. Septins: The Fourth Component of the Cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Ehlinger, D.G.; Burke, J.C.; McDonald, C.G.; Smith, R.F.; Bergstrom, H.C. Nicotine-Induced and D1-Receptor-Dependent Dendritic Remodeling in a Subset of Dorsolateral Striatum Medium Spiny Neurons. Neuroscience 2017, 356, 242–254. [Google Scholar] [CrossRef]
- Jung, Y.; Hsieh, L.S.; Lee, A.M.; Zhou, Z.; Coman, D.; Heath, C.J.; Hyder, F.; Mineur, Y.S.; Yuan, Q.; Goldman, D.; et al. An Epigenetic Mechanism Mediates Developmental Nicotine Effects on Neuronal Structure and Behavior. Nat. Neurosci. 2016, 19, 905–914. [Google Scholar] [CrossRef]
- Nestler, E.J. Is There a Common Molecular Pathway for Addiction? Nat. Neurosci. 2005, 8, 1445–1449. [Google Scholar] [CrossRef]
- King, J.R.; Kabbani, N. Alpha 7 Nicotinic Receptor Coupling to Heterotrimeric G Proteins Modulates RhoA Activation, Cytoskeletal Motility, and Structural Growth. J. Neurochem. 2016, 138, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Cerdá, J.; Chorev, D.S.; Geiger, B.; Cossart, P. The Diverse Family of Arp2/3 Complexes. Trends Cell Biol. 2017, 27, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hotulainen, P.; Llano, O.; Smirnov, S.; Tanhuanpää, K.; Faix, J.; Rivera, C.; Lappalainen, P. Defining Mechanisms of Actin Polymerization and Depolymerization during Dendritic Spine Morphogenesis. J. Cell Biol. 2009, 185, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Ganapathiraju, M.K.; Thahir, M.; Handen, A.; Sarkar, S.N.; Sweet, R.A.; Nimgaonkar, V.L.; Loscher, C.E.; Bauer, E.M.; Chaparala, S. Schizophrenia Interactome with 504 Novel Protein–Protein Interactions. NPJ Schizophr. 2016, 2, 16012. [Google Scholar] [CrossRef]
- McClure-Begley, T.D.; Esterlis, I.; Stone, K.L.; Lam, T.T.; Grady, S.R.; Colangelo, C.M.; Lindstrom, J.M.; Marks, M.J.; Picciotto, M.R. Evaluation of the Nicotinic Acetylcholine Receptor-Associated Proteome at Baseline and Following Nicotine Exposure in Human and Mouse Cortex. ENeuro 2016, 3, ENEURO.0166-16.2016. [Google Scholar] [CrossRef]
- Lippi, G.; Steinert, J.R.; Marczylo, E.L.; D’Oro, S.; Fiore, R.; Forsythe, I.D.; Schratt, G.; Zoli, M.; Nicotera, P.; Young, K.W. Targeting of the Arpc3 Actin Nucleation Factor by MiR-29a/b Regulates Dendritic Spine Morphology. J. Cell Biol. 2011, 194, 889–904. [Google Scholar] [CrossRef]
- Kakurina, G.V.; Kolegova, E.S.; Kondakova, I.V. Adenylyl Cyclase-Associated Protein 1: Structure, Regulation, and Participation in Cellular Processes. Biochemistry 2018, 83, 45–53. [Google Scholar] [CrossRef]
- Wong, A.H.C.; Lipska, B.K.; Likhodi, O.; Boffa, E.; Weinberger, D.R.; Kennedy, J.L.; Van Tol, H.H.M. Cortical Gene Expression in the Neonatal Ventral-Hippocampal Lesion Rat Model. Schizophr. Res. 2005, 77, 261–270. [Google Scholar] [CrossRef]
- Baumgärtel, K.; Mansuy, I.M. Neural Functions of Calcineurin in Synaptic Plasticity and Memory. Learn. Mem. 2012, 19, 375–384. [Google Scholar] [CrossRef]
- Penny, C.J.; Gold, M.G. Mechanisms for Localising Calcineurin and CaMKII in Dendritic Spines. Cell Signal. 2018, 49, 46–58. [Google Scholar] [CrossRef]
- Lohmann, C.; Kessels, H.W. The Developmental Stages of Synaptic Plasticity. J. Physiol. 2014, 592, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Walaas, I.S.; Caroll Hemmings, H., Jr.; Greengard, P.; Clark Nairn, A.; Mansuy, I.M.; Marin, P. Beyond the Dopamine Receptor: Regulation and Roles of Serine/Threonine Protein Phosphatases. Front. Neuroanat. 2011, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Charrier, E.; Reibel, S.; Rogemond, V.; Aguera, M.; Thomasset, N.; Honnorat, J. Collapsin Response Mediator Proteins (CRMPs): Involvement in Nervous System Development and Adult Neurodegenerative Disorders. Mol. Neurobiol. 2003, 28, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Beasley, C.L.; Pennington, K.; Behan, A.; Wait, R.; Dunn, M.J.; Cotter, D. Proteomic Analysis of the Anterior Cingulate Cortex in the Major Psychiatric Disorders: Evidence for Disease-Associated Changes. Proteomics 2006, 6, 3414–3425. [Google Scholar] [CrossRef]
- Fernando, P.; Sommer, I.E.C.; Hasan, A. Do We Need Sex-Oriented Clinical Practice Guidelines for the Treatment of Schizophrenia? Curr. Opin. Psychiatry 2020, 33, 192–199. [Google Scholar] [CrossRef]
- Bryan Dunyak, M.M.; Gestwicki, J.E. Peptidyl-Proline Isomerases (PPIases): Targets for Natural Products and Natural Product-Inspired Compounds: HHS Public Access. J. Med. Chem. 2016, 59, 9622–9644. [Google Scholar] [CrossRef]
- Harrar, Y.; Bellini, C.; Faure, J.-D. FKBPs: At the Crossroads of Folding and Transduction. Trends Plant Sci. 2001, 6, 426–431. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Tang, W.; Wong, H.; Santillan, A.; Patterson, R.J.; Martinez, L.A.; Tejada-Simon, M.V.; Paylor, R.; Hamilton, S.L.; Klann, E. Removal of FKBP12 Enhances MTOR-Raptor Interactions, LTP, Memory, and Perseverative/Repetitive Behavior. Neuron 2008, 60, 832–845. [Google Scholar] [CrossRef]
- Rosario Fernández-Fernández, M.; María Valpuesta, J. Hsp70 Chaperone: A Master Player in Protein Homeostasis. F1000Research 2018, 7, 1497. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 Chaperone Network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Arion, D.; Unger, T.; Lewis, D.A.; Levitt, P.; Mirnics, K. Molecular Evidence for Increased Expression of Genes Related to Immune and Chaperone Function in the Prefrontal Cortex in Schizophrenia. Biol. Psychiatry 2007, 62, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mandelli, L.; Lim, S.; Lim, H.K.; Kwon, O.J.; Pae, C.U.; Serretti, A.; Nimgaonkar, V.L.; Paik, I.H.; Jun, T.Y. Association Analysis of Heat Shock Protein 70 Gene Polymorphisms in Schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2008, 258, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.; Kucia, K.; Owczarek, A.; Suchanek-Raif, R.; Merk, W.; Fila-Danilow, A.; Paul-Samojedny, M.; Choreza, P.; Kowalski, J. Association of HSPA1B Polymorphisms with Paranoid Schizophrenia in a Polish Population. Neuromolecular Med. 2020, 22, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.; Owczarek, A.; Suchanek, R.; Paul-Samojedny, M.; Fila-Danilow, A.; Borkowska, P.; Kucia, K.; Kowalski, J. Heat Shock Protein 70 Gene Polymorphisms Are Associated with Paranoid Schizophrenia in the Polish Population. Cell Stress. Chaperones 2014, 19, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Pae, C.U.; Drago, A.; Kim, J.J.; Mandelli, L.; De Ronchi, D.; Serretti, A. The Impact of Heat Shock Protein 70 Gene Variations on Clinical Presentation and Outcome in Schizophrenic Inpatients. Neuropsychobiology 2009, 59, 135–141. [Google Scholar] [CrossRef]
- Bosnjak Kuharic, D.; Bozina, N.; Ganoci, L.; Makaric, P.; Kekin, I.; Prpic, N.; Bozina, T.; Rojnic Kuzman, M. Association of HSPA1B Genotypes with Psychopathology and Neurocognition in Patients with the First Episode of Psychosis: A Longitudinal 18-Month Follow-up Study. Pharmacogenomics J. 2020, 20, 638–646. [Google Scholar] [CrossRef]
- Canet-Avilé, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; Mclendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s Disease Protein DJ-1 Is Neuroprotective Due to Cysteine-Sulfinic Acid-Driven Mitochondrial Localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Krüger, R. The Genetic Architecture of Mitochondrial Dysfunction in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive Regulation of the Brain’s Antioxidant Defences by Neurons and Astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef]
- Rambaud, V.; Marzo, A.; Chaumette, B. Oxidative Stress and Emergence of Psychosis. Antioxidants 2022, 11, 1870. [Google Scholar] [CrossRef]
- Hritcu, L.; Ciobica, A.; Gorgan, L. Nicotine-Induced Memory Impairment by Increasing Brain Oxidative Stress. Open Life Sci. 2009, 4, 335–342. [Google Scholar] [CrossRef]
- Vargas, H.O.; Nunes, S.O.V.; de Castro, M.R.P.; Vargas, M.M.; Barbosa, D.S.; Bortolasci, C.C.; Venugopal, K.; Dodd, S.; Berk, M. Oxidative Stress and Inflammatory Markers Are Associated with Depression and Nicotine Dependence. Neurosci. Lett. 2013, 544, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Do, K.Q. Linking Early-Life NMDAR Hypofunction and Oxidative Stress in Schizophrenia Pathogenesis. Nat. Rev. Neurosci. 2016, 17, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.A.; Winchester, C.; Egerton, A.; Cochran, S.M.; Morris, B.J. Modelling Prefrontal Cortex Deficits in Schizophrenia: Implications for Treatment. Br. J. Pharmacol. 2008, 153, S465–S470. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An Akt/β-Arrestin 2/PP2A Signaling Complex Mediates Dopaminergic Neurotransmission and Behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef]
- Hur, E.-M.; Zhou, F.-Q. GSK3 Signalling in Neural Development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef]
- Blasi, G.; Napolitano, F.; Ursini, G.; Di Giorgio, A.; Caforio, G.; Taurisano, P.; Fazio, L.; Gelao, B.; Teresa Attrotto, M.; Colagiorgio, L.; et al. Association of GSK-3b Genetic Variation with GSK-3b Expression, Prefrontal Cortical Thickness, Prefrontal Physiology, and Schizophrenia. Am. J. Psychiatry 2013, 170, 868–876. [Google Scholar] [CrossRef]
- Xie, X.; Liang, M.; Yu, C.; Wei, Z. Liprin-α-Mediated Assemblies and Their Roles in Synapse Formation. Front. Cell Dev. Biol. 2021, 9, 653381. [Google Scholar] [CrossRef]
- Spangler, S.A.; Hoogenraad, C.C. Liprin-α Proteins: Scaffold Molecules for Synapse Maturation. Biochem. Soc. Trans. 2007, 35, 1278. [Google Scholar] [CrossRef]
- Schoch, S.; Castillo, P.E.; Jok, T.; Mukherjee, K.; Geppertk, M.; Wang, Y.; Schmitz, F.; Malenka, R.C.; Su, T.C. RIM1a Forms a Protein Scaffold for Regulating Neurotransmitter Release at the Active Zone. Nature 2002, 415, 321–326. [Google Scholar] [CrossRef]
- Ko, J.; Kim, S.; Valtschanoff, J.G.; Shin, H.; Lee, J.-R.; Sheng, M.; Premont, R.T.; Weinberg, R.J.; Kim, E. Interaction between Liprin-and GIT1 Is Required for AMPA Receptor Targeting. J. Neurosci. 2003, 23, 1667–1677. [Google Scholar] [CrossRef]
- Wyszynski, M.; Kim, E.; Dunah, A.W.; Passafaro, M.; Valtschanoff, J.G.; Serra-Pagè, C.; Streuli, M.; Weinberg, R.J. Interaction between GRIP and Liprin-Alpha/SYD2 Is Required for AMPA Receptor Targeting. Neuron 2002, 34, 39–52. [Google Scholar] [CrossRef]
- Dickinson, B.; Jo, J.; Seok, H.; Son, G.H.; Whitcomb, D.J.; Davies, C.H.; Sheng, M.; Collingridge, G.L.; Cho, K. A Novel Mechanism of Hippocampal LTD Involving Muscarinic Receptor-Triggered Interactions between AMPARs, GRIP and Liprin-Alpha. Mol. Brain 2009, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Henkel, N.D.; Wu, X.; O’Donovan, S.M.; Devine, E.A.; Jiron, J.M.; Rowland, L.M.; Sarnyai, Z.; Ramsey, A.J.; Wen, Z.; Hahn, M.K.; et al. Schizophrenia: A Disorder of Broken Brain Bioenergetics. Mol. Psychiatry 2022, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Martins-De-Souza, D.; Harris, L.W.; Guest, P.C.; Bahn, S. The Role of Energy Metabolism Dysfunction and Oxidative Stress in Schizophrenia Revealed by Proteomics. Antioxid. Redox Signal 2011, 15, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Burbaeva, G.S.; Boksha, I.S.; Tereshkina, E.B.; Savushkina, O.K.; Starodubtseva, L.I.; Turishcheva, M.S.; Mukaetova-Ladinska, E. Systemic Neurochemical Alterations in Schizophrenic Brain: Glutamate Metabolism in Focus. Neurochem. Res. 2007, 32, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Carney, J.M. Free Radical Damage to Protein and DNA: Mechanisms Involved and Relevant Observations on Brain Undergoing &dative Stress. Ann. Neurol. 1992, 32, S22–S27. [Google Scholar] [CrossRef]
- Hertz, L.; Dringen, R.; Schousboe, A.; Robinson, S.R. Astrocytes: Glutamate Producers for Neurons. J. Neurosci. Res. 1999, 57, 417–428. [Google Scholar] [CrossRef]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The Glutamine-Glutamate/GABA Cycle: Function, Regional Differences in Glutamate and GABA Production and Effects of Interference with GABA Metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef]
- Mong, J.A.; Blutstein, T. Estradiol Modulation of Astrocytic Form and Function: Implications for Hormonal Control of Synaptic Communication. Neuroscience 2006, 138, 967–975. [Google Scholar] [CrossRef]
- Hertz, L.; Rothman, D.L. Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Shuying, L.; Hanrong, W.; Huirong, G.; Zheng, Z. Neuron-Specific Enclose and Myelin Basic Protein in Cerebrospinal Fluid of Patients with First Episode Schizophrenia. Huazhong Univ. Sci. Technolog. Med. Sci. 2006, 26, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Medina-Hernández, V.; Ramos-Loyo, J.; Luquin, S.; Sánchez, L.F.C.; García-Estrada, J.; Navarro-Ruiz, A. Increased Lipid Peroxidation and Neuron Specific Enolase in Treatment Refractory Schizophrenics. J. Psychiatr. Res. 2007, 41, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Li, W.; Hasan, M.; Liu, K.; Awan, U.; Saeed, Y.; Zhang, Y.; Muhammad Khan, A.; Shah, A.; Qing, H.; et al. Differential Expression of Specific Cellular Defense Proteins in Rat Hypothalamus under Simulated Microgravity Induced Conditions: Comparative Proteomics. Proteomics 2014, 14, 1424–1433. [Google Scholar] [CrossRef]
- Yang, J.; Hu, L.; Song, T.; Liu, Y.; Wu, Q.; Zhao, L.; Liu, L.; Zhao, X.; Zhang, D.; Huang, C. Proteomic Changes in Female Rat Hippocampus Following Exposure to a Terrified Sound Stress. J. Mol. Neurosci. 2014, 53, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Hattori A’b, T.; Takei A’l, N.; Mizuno, Y.; Kato, K.; Kohsaka, S. Neurotrophic and Neuroprotective Effects of Neuron-Specific Enolase on Cultured Neurons from Embryonic Rat Brain. Neurosci. Res. 1995, 21, 191–198. [Google Scholar] [CrossRef]
- Herrera-Molina, R.; Valdivia, A.; Kong, M.; Alvarez, A.; Cárdenas, A.; Quest, A.F.G.; Leyton, L. Thy-1-Interacting Molecules and Cellular Signaling in Cis and Trans. Int. Rev. Cell Mol. Biol. 2013, 305, 163–216. [Google Scholar] [CrossRef]
- Pape, M.; Doxakis, E.; Reiff, T.; Duong, C.V.; Davies, A.; Geissen, M.; Rohrer, H. A Function for the Calponin Family Member NP25 in Neurite Outgrowth. Dev. Biol. 2008, 321, 434–443. [Google Scholar] [CrossRef]
- Shirao, T.; Hanamura, K.; Koganezawa, N.; Ishizuka, Y.; Yamazaki, H.; Sekino, Y. The Role of Drebrin in Neurons. J. Neurochem. 2017, 141, 819–834. [Google Scholar] [CrossRef]
- Holahan, M.R.; Maffei, A.; Diaz, E.; Cheetham, C. A Shift from a Pivotal to Supporting Role for the Growth-Associated Protein (GAP-43) in the Coordination of Axonal Structural and Functional Plasticity. Front. Cell. Neurosci. 2017, 11, 266. [Google Scholar] [CrossRef]
- Korshunova, I.; Mosevitsky, M. Role of the Growth-Associated Protein GAP-43 in NCAM-Mediated Neurite Outgrowth. Adv. Exp. Med. Biol. 2010, 663, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Mosevitsky, M.I. Nerve Ending “Signal” Proteins GAP-43, MARCKS, and BASP1. Int. Rev. Cytol. 2005, 245, 245–325. [Google Scholar] [CrossRef]
- Brudvig, J.J.; Weimer, J.M. X MARCKS the Spot: Myristoylated Alanine-Rich C Kinase Substrate in Neuronal Function and Disease. Front. Cell Neurosci. 2015, 9, 407. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.J.; Sivagnanasundaram, S.; Weickert, C.S. Lack of Change in Markers of Presynaptic Terminal Abundance alongside Subtle Reductions in Markers of Presynaptic Terminal Plasticity in Prefrontal Cortex of Schizophrenia Patients. Biol. Psychiatry 2011, 69, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.J.; Hashimoto, T.; Lewis, D.A. Molecular Mechanisms Contributing to Dendritic Spine Alterations in the Prefrontal Cortex of Subjects with Schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Pinner, A.L.; Haroutunian, V.; Meador-Woodruff, J.H. Alterations of the Myristoylated, Alanine-Rich C Kinase Substrate (MARCKS) in Prefrontal Cortex in Schizophrenia. Schizophr. Res. 2014, 154, 36–41. [Google Scholar] [CrossRef]
- Konopaske, G.T.; Subburaju, S.; Coyle, J.T.; Benes, F.M. Altered Prefrontal Cortical MARCKS and PPP1R9A MRNA Expression in Schizophrenia and Bipolar Disorder HHS Public Access. Schizophr. Res. 2015, 164, 100–108. [Google Scholar] [CrossRef]
- Boczek, T.; Mackiewicz, J.; Sobolczyk, M.; Wawrzyniak, J.; Lisek, M.; Ferenc, B.; Guo, F.; Zylinska, L. The Role of g Protein-Coupled Receptors (Gpcrs) and Calcium Signaling in Schizophrenia. Focus on Gpcrs Activated by Neurotransmitters and Chemokines. Cells 2021, 10, 1228. [Google Scholar] [CrossRef]
- Komatsu, H.; Fukuchi, M.; Habata, Y. Potential Utility of Biased GPCR Signaling for Treatment of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 3207. [Google Scholar] [CrossRef]
- Kvajo, M.; McKellar, H.; Gogos, J. Molecules, Signaling, and Schizophrenia. Curr. Top. Behav. Neurosci. 2010, 4, 629–656. [Google Scholar] [CrossRef]
- Ishibashi, M.; Yamazaki, Y.; Miledi, R.; Sumikawa, K. Nicotinic and Muscarinic Agonists and Acetylcholinesterase Inhibitors Stimulate a Common Pathway to Enhance GluN2B-NMDAR Responses. Proc. Natl. Acad. Sci. USA 2014, 111, 12538–12543. [Google Scholar] [CrossRef] [PubMed]
- Nordman, J.C.; Kabbani, N. An Interaction between A7 Nicotinic Receptors and a G-Protein Pathway Complex Regulates Neurite Growth in Neural Cells. J. Cell Sci. 2012, 125, 5502–5513. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Belluscio, L.; Hen, R. G OLF Mediates Dopamine D 1 Receptor Signaling. J. Neurosci. 2000, 20, RC91. [Google Scholar] [CrossRef] [PubMed]
- Yu-Taeger, L.; Ott, T.; Bonsi, P.; Tomczak, C.; Wassouf, Z.; Martella, G.; Sciamanna, G.; Imbriani, P.; Ponterio, G.; Tassone, A.; et al. Impaired Dopamine- and Adenosine-Mediated Signaling and Plasticity in a Novel Rodent Model for DYT25 Dystonia. Neurobiol. Dis. 2020, 134, 104634. [Google Scholar] [CrossRef]
- Riobo, N.A.; Manning, D.R. Receptors Coupled to Heterotrimeric G Proteins of the G12 Family. Trends Pharmacol. Sci. 2005, 26, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Song, W.; Liu, M.-Y.; Jin, L.; Dykes-Hoberg, M.; Lin, C.-L.G.; Bowers, W.J.; Federoff, H.J.; Sternweis, P.C.; Rothstein, J.D. Modulation of the Neuronal Glutamate Transporter EAAT4 by Two Interacting Proteins. Nature 2001, 410, 89–93. [Google Scholar] [CrossRef]
- Chen, J.; Calhoun, V.D.; Arias-Vasquez, A.; Zwiers, M.P.; Van Hulzen, K.; En Fern, G.; Fisher, S.E.; Franke, B.; Turner, J.A.; Liu, J. G-Protein Genomic Association With Normal Variation in Gray Matter Density. Hum. Brain Mapp. 2015, 36, 4272–4286. [Google Scholar] [CrossRef]
- Jia, P.; Wang, L.; Fanous, A.H.; Pato, C.N.; Edwards, T.L. Network-Assisted Investigation of Combined Causal Signals from Genome-Wide Association Studies in Schizophrenia. PLoS Comput. Biol. 2012, 8, 1002587. [Google Scholar] [CrossRef]
- Liu, Y.; Qu, H.-Q.; Chang, X.; Tian, L.; Glessner, J.; Sleiman, P.A.M.; Hakonarson, H. Expansion of Schizophrenia Gene Network Knowledge Using Machine Learning Selected Signals From Dorsolateral Prefrontal Cortex and Amygdala RNA-Seq Data. Front. Psychiatry 2022, 13, 797329. [Google Scholar] [CrossRef]
- Schwab, S.G.; Hallmayer, J.; Lerer, B.; Albus, M.; Borrmann, M.; Hönig, S.; Strauß, M.; Segman, R.; Lichtermann, D.; Knapp, M.; et al. Support for a Chromosome 18p Locus Conferring Susceptibility to Functional Psychoses in Families with Schizophrenia, by Association and Linkage Analysis. Am. J. Hum. Genet. 1998, 63, 1139–1152. [Google Scholar] [CrossRef][Green Version]
- Cullen, K.A.; Liu, S.T.; Bernat, J.K.; Slavit, W.I.; Tynan, M.A.; King, B.A.; Neff, L.J. Flavored Tobacco Product Use Among Middle and High School Students—United States, 2014–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Arrazola, R.A.; Corey, C.G.; Husten, C.G.; Neff, L.J.; Homa, D.M.; King, B.A. Tobacco Use Among Middle and High School Students—United States, 2011–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 361–367. [Google Scholar] [CrossRef]
- Massadeh, A.M.; Gharaibeh, A.A.; Omari, K.W. A Single-Step Extraction Method for the Determination of Nicotine and Cotinine in Jordanian Smokers’ Blood and Urine Samples by RP-HPLC and GC-MS. J. Chromatogr. Sci. 2009, 47, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.; Ellen Wewers, M.; Groner, J.; Ahijevych, K. Smoke Constituent Exposure and Smoking Topography of Adolescent Daily Cigarette Smokers. Nicotine Tob. Res. 2004, 6, 853–862. [Google Scholar] [CrossRef]
- Engel, M.; Snikeris, P.; Jenner, A.; Karl, T.; Huang, X.-F.; Frank, E. Neuregulin 1 Prevents Phencyclidine-Induced Behavioral Impairments and Disruptions to GABAergic Signaling in Mice. Int. J. Neuropsychopharmacol. 2015, 18, pyu114. [Google Scholar] [CrossRef] [PubMed]
- Spielewoy, C.; Markou, A. Strain-Specificity in Nicotine Attenuation of Phencyclidine-Induced Disruption of Prepulse Inhibition in Mice: Relevance to Smoking in Schizophrenia Patients. Behav. Genet. 2004, 34, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Selvendra, A.; Stewart, A.; Castle, D. Risk Factors in Early and Late Onset Schizophrenia. Compr. Psychiatry 2018, 80, 155–162. [Google Scholar] [CrossRef]
- Abreu-Villaça, Y.; Nunes, F.; do E Queiroz-Gomes, F.; Manhães, A.C.; Filgueiras, C.C. Combined Exposure to Nicotine and Ethanol in Adolescent Mice Differentially Affects Anxiety Levels during Exposure, Short-Term, and Long-Term Withdrawal. Neuropsychopharmacology 2008, 33, 599–610. [Google Scholar] [CrossRef]
- Abreu-Villaça, Y.; Medeiros, A.H.; Lima, C.S.; Faria, F.P.; Filgueiras, C.C.; Manhães, A.C. Combined Exposure to Nicotine and Ethanol in Adolescent Mice Differentially Affects Memory and Learning during Exposure and Withdrawal. Behav. Brain Res. 2007, 181, 136–146. [Google Scholar] [CrossRef]
- Ribeiro-Carvalho, A.; Lima, C.S.; Nunes-Freitas, A.L.; Filgueiras, C.C.; Manhães, A.C.; Abreu-Villaça, Y. Exposure to Nicotine and Ethanol in Adolescent Mice: Effects on Depressive-like Behavior during Exposure and Withdrawal. Behav. Brain Res. 2011, 221, 282–289. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Rodríguez-Vega, A.; Losada-Barragán, M.; Berbert, L.R.; Mesquita-Rodrigues, C.; Bombaça, A.C.S.; Menna-Barreto, R.; Aquino, P.; Carvalho, P.C.; Padrón, G.; de Jesus, J.B.; et al. Quantitative Analysis of Proteins Secreted by Leishmania (Viannia) Braziliensis Strains Associated to Distinct Clinical Manifestations of American Tegumentary Leishmaniasis. J. Proteom. 2021, 232, 104077. [Google Scholar] [CrossRef]
- Silva, J.C.; Gorenstein, M.V.; Li, G.Z.; Vissers, J.P.C.; Geromanos, S.J. Absolute Quantification of Proteins by LCMSE: A Virtue of Parallel MS Acquisition. Mol. Cell. Proteom. 2006, 5, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER Version 11: Expanded Annotation Data from Gene Ontology and Reactome Pathways, and Data Analysis Tool Enhancements. Nucleic Acids Res. 2017, 45, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An Interactive Venn Diagram Viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein-Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2014, 43, 447–452. [Google Scholar] [CrossRef]
- Ageta-Ishihara, N.; Kinoshita, M. Developmental and postdevelopmental roles of septins in the brain. Neurosci. Res. 2021, 170, 6–12. [Google Scholar] [CrossRef]
- Benoit, B.; Poüs, C.; Baillet, A. Septins as membrane influencers: Direct play or in association with other cytoskeleton partners. Front. Cell Dev. Biol. 2023, 11, 1112319. [Google Scholar] [CrossRef]
- Chen, T.; Zhou, L.; Yuan , Y.; Fang, Y.; Guo, Y.; Huang, H.; Zhou, Q.; Lv, X. Characterization of Bbx, a member of a novel subfamily of the HMG-box superfamily together with Cic. Dev. Genes Evol. 2014, 224, 261–268. [Google Scholar] [CrossRef]
- Dixon, C.; Harvey, T.J.; Smith, A.G.; Gronostajski, R.M.; Bailey, T.L.; Piper, M. Nuclear Factor One X Regulates Bobby Sox During Development of the Mouse Forebrain. Cell. Mol. Neurobiol. 2013, 33, 867–873. [Google Scholar] [CrossRef]
- Guest, P.C.; Iwata, K.; Kato, T.A.; Steiner, J.; Schmitt, A.; Turck, C.W.; Martins-De-Souza, D. MK-801 treatment affects glycolysis in oligodendrocytes more than in astrocytes and neuronal cells: Insights for schizophrenia. Front. Cell. Neurosci. 2015, 9, 180. [Google Scholar] [CrossRef]
- Hagag, N.; Halegoua, S.; Viola, M. Inhibition of growth factor-induced differentiation of PC12 cells by microinjection of antibody to ras p21. Nature 1986, 319, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Martins-de-Souza, D.; Gattaz, W.F.; Schmitt, A.; Maccarrone, G.; Hunyadi-Gulyás, E.; Eberlin, M.N.; Souza, G.H.M.F.; Marangoni, S.; Novello, J.C.; Turck, C.W.; et al. Proteomic analysis of dorsolateral prefrontal cortex indicates the involvement of cytoskeleton, oligodendrocyte, energy metabolism and new potential markers in schizophrenia. J. Psychiatr. Res. 2009, 43, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.A.; Mirnics, K.; Pierri, J.N.; Lewis, D.A.; Levitt, P. Gene Expression Profiling Reveals Alterations of Specific Metabolic Pathways in Schizophrenia. J. Neurosci. 2002, 22, 2718–2729. [Google Scholar] [CrossRef]
- Noda, M.; Ko, M.; Ogura, A.; Liu, D.; Amano, T.; Takano, T.; Ikawa, Y. Sarcoma viruses carrying ras oncogenes induce differentiation-associated properties in a neuronal cell line. Nature 1985, 318, 73–75. [Google Scholar] [CrossRef]
- Peterson, E.A.; Petty, E.M. Conquering the complex world of human septins: Implications for health and disease. Clin. Genet. 2010, 77, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Dias Az, A.; Blanco, M.A.; Jones, N.; Moreno, S. HBP2: A new mammalian protein that complements the ®ssion yeast MBF transcription complex. Curr. Genet. 2001, 40, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H.; Olkkonen, V.M. The Rab GTPase family. Genome Biol. 2001, 2, REVIEWS3007. [Google Scholar] [CrossRef]
- Werner, B.; Yadav, S. Phosphoregulation of the septin cytoskeleton in neuronal development and disease. Cytoskeleton 2022, 80, 275–289. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subnetwork | Protein Name | Gene Name | PCPNIC vs. CT | PCPNIC vs. NIC | PCPNIC vs. PCP |
|---|---|---|---|---|---|
| Synaptic transmission | |||||
| Amphiphysin | Amph | Down | Down | Down | |
| Synaptophysin | Syp | Down | Down | Down | |
| Microtubule-associated protein 2 | Map2 | Down | Down | Down | |
| Mitogen-activated protein kinase 3 | Mapk3 | Down | Down | Down | |
| Actin cytoskeleton | |||||
| Actin-related protein 3B | Actr3b | Down | Down | Down | |
| Adenylyl cyclase-associated protein 1 | Cap1 | Down | Down | Down | |
| Serine/threonine-protein phosphatases | |||||
| Serine/threonine-protein phosphatase 2B catalytic subunit alpha isoform | Ppp3ca | Down | Down | Down | |
| Serine/threonine-protein phosphatase PP1-beta catalytic subunit | Ppp1cb | Down | Down | Down | |
| Nervous system development | |||||
| Dihydropyrimidinase-related protein 5 | Dpysl5 | Down | Down | Down | |
| Dihydropyrimidinase-related protein 1 | Crmp1 | Down | Down | Down | |
| Energy metabolism | |||||
| Malate dehydrogenase_cytoplasmic | Mdh1 | Down | Down | Down | |
| Phosphoglycerate kinase 2 | Pgk2 | Down | Down | Down | |
| Subnetwork | Protein Name | Gene Name | PCPNIC vs. CT | PCPNIC vs. NIC | PCPNIC vs. PCP |
|---|---|---|---|---|---|
| Oxidative stress | |||||
| Peptidyl-prolyl cis-trans isomerase FKBP1A | Fkbp1a | Up | Up | Up | |
| Heat shock 70 kDa protein 1B | Hspa1b | Down | Down | Down | |
| Heat shock-related 70 kDa protein 2 | Hspa2 | Up | Up | Up | |
| Ras-related protein Rab-4A | Rab4a | Up | Up | Up | |
| Peroxiredoxin-1 | Prdx1 | Up | Up | Up | |
| V-type proton ATPase catalytic subunit A | Atp6v1a | Up | Up | Up | |
| Parkinson disease protein 7 homolog | Park7 | Up | Up | Up | |
| Glutathione S-transferase P 1 | Gstp1 | Up | Up | Up | |
| Glutathione S-transferase Mu 1 | Gstm1 | Up | Up | Up | |
| Protein disulfide-isomerase A3 | Pdia3 | Up | Up | Up | |
| Energy metabolism | |||||
| Gamma-enolase | Eno2 | Up | Up | Up | |
| Glyceraldehyde-3-phosphate dehydrogenase_testis-specific | Gapdhs | Down | Down | Down | |
| Glucose-6-phosphate isomerase | Gpi | Up | Up | Up | |
| Pyruvate kinase PKLR | Pklr | Up | Up | Up | |
| Aspartate aminotransferase_mitochondrial | Got2 | Up | Up | Up | |
| Citrate synthase_mitochondrial | Cs | Down | Down | Down | |
| Acetyl-CoA acetyltransferase_mitochondrial | Acat1 | Down | Down | Down | |
| Glutamine synthetase | Glul | Up | Up | Up | |
| Serine/threonine-protein phosphatases | |||||
| Serine/threonine-protein phosphatase PP1-gamma catalytic subunit | Ppp1cc | Up | Up | Up | |
| Serine/threonine-protein phosphatase 2A catalytic subunit beta isoform | Ppp2cb | Up | Up | Up | |
| Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform | Ppp2r1a | Up | Up | Up | |
| Isoform 2 of Liprin-alpha-3 | Ppfia3 | Up | Up | Up | |
| Neurite outgrowth | |||||
| Thy-1 membrane glycoprotein | Thy1 | Down | Down | Down | |
| Transgelin-3 | Tagln3 | Up | Up | Up | |
| Drebrin | Dbn1 | Up | Up | Up | |
| Neuromodulin | Gap43 | Down | Down | Down | |
| Myristoylated alanine-rich C-kinase substrate | Marcks | Down | Down | Down | |
| Actin_alpha skeletal muscle | Acta1 | Up | Up | Up | |
| G protein-coupled receptors signaling | |||||
| Guanine nucleotide-binding protein subunit alpha-12 | Gna12 | Up | Up | Up | |
| Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2 | Gnb2 | Up | Up | Up | |
| Guanine nucleotide-binding protein G(olf) subunit alpha | Gnal | Up | Up | Up | |
| Septin cytoskeleton | |||||
| Septin-8 | Septin8 | Up | Up | Up | |
| Septin-10 | Septin10 | Down | Down | Down | |
| Cell cycle | |||||
| Ras-related protein Rab-37 | Rab37 | Up | Up | Up | |
| HMG box transcription factor BBX | Bbx | Up | Up | Up | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Vega, A.; Dutra-Tavares, A.C.; Souza, T.P.; Semeão, K.A.; Filgueiras, C.C.; Ribeiro-Carvalho, A.; Manhães, A.C.; Abreu-Villaça, Y. Nicotine Exposure in a Phencyclidine-Induced Mice Model of Schizophrenia: Sex-Selective Medial Prefrontal Cortex Protein Markers of the Combined Insults in Adolescent Mice. Int. J. Mol. Sci. 2023, 24, 14634. https://doi.org/10.3390/ijms241914634
Rodríguez-Vega A, Dutra-Tavares AC, Souza TP, Semeão KA, Filgueiras CC, Ribeiro-Carvalho A, Manhães AC, Abreu-Villaça Y. Nicotine Exposure in a Phencyclidine-Induced Mice Model of Schizophrenia: Sex-Selective Medial Prefrontal Cortex Protein Markers of the Combined Insults in Adolescent Mice. International Journal of Molecular Sciences. 2023; 24(19):14634. https://doi.org/10.3390/ijms241914634
Chicago/Turabian StyleRodríguez-Vega, Andrés, Ana Carolina Dutra-Tavares, Thainá P. Souza, Keila A. Semeão, Claudio C. Filgueiras, Anderson Ribeiro-Carvalho, Alex C. Manhães, and Yael Abreu-Villaça. 2023. "Nicotine Exposure in a Phencyclidine-Induced Mice Model of Schizophrenia: Sex-Selective Medial Prefrontal Cortex Protein Markers of the Combined Insults in Adolescent Mice" International Journal of Molecular Sciences 24, no. 19: 14634. https://doi.org/10.3390/ijms241914634
APA StyleRodríguez-Vega, A., Dutra-Tavares, A. C., Souza, T. P., Semeão, K. A., Filgueiras, C. C., Ribeiro-Carvalho, A., Manhães, A. C., & Abreu-Villaça, Y. (2023). Nicotine Exposure in a Phencyclidine-Induced Mice Model of Schizophrenia: Sex-Selective Medial Prefrontal Cortex Protein Markers of the Combined Insults in Adolescent Mice. International Journal of Molecular Sciences, 24(19), 14634. https://doi.org/10.3390/ijms241914634