
Regulatory Mechanism of the IL-33–IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
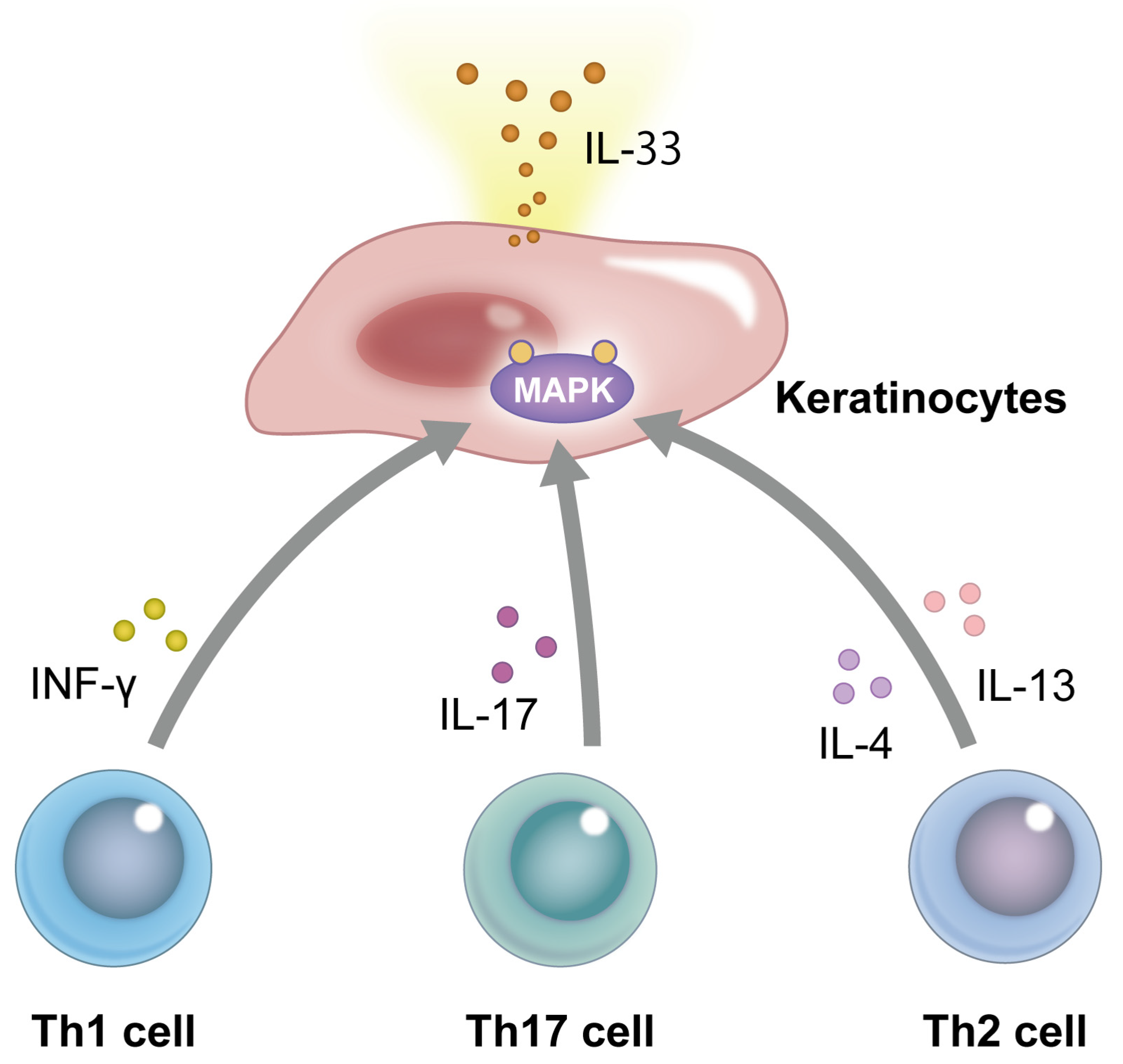
2. Structure and Signaling Pathway of IL-33
3. The Role of IL-33 in the Pathogenesis of Atopic Dermatitis (AD) and Psoriasis
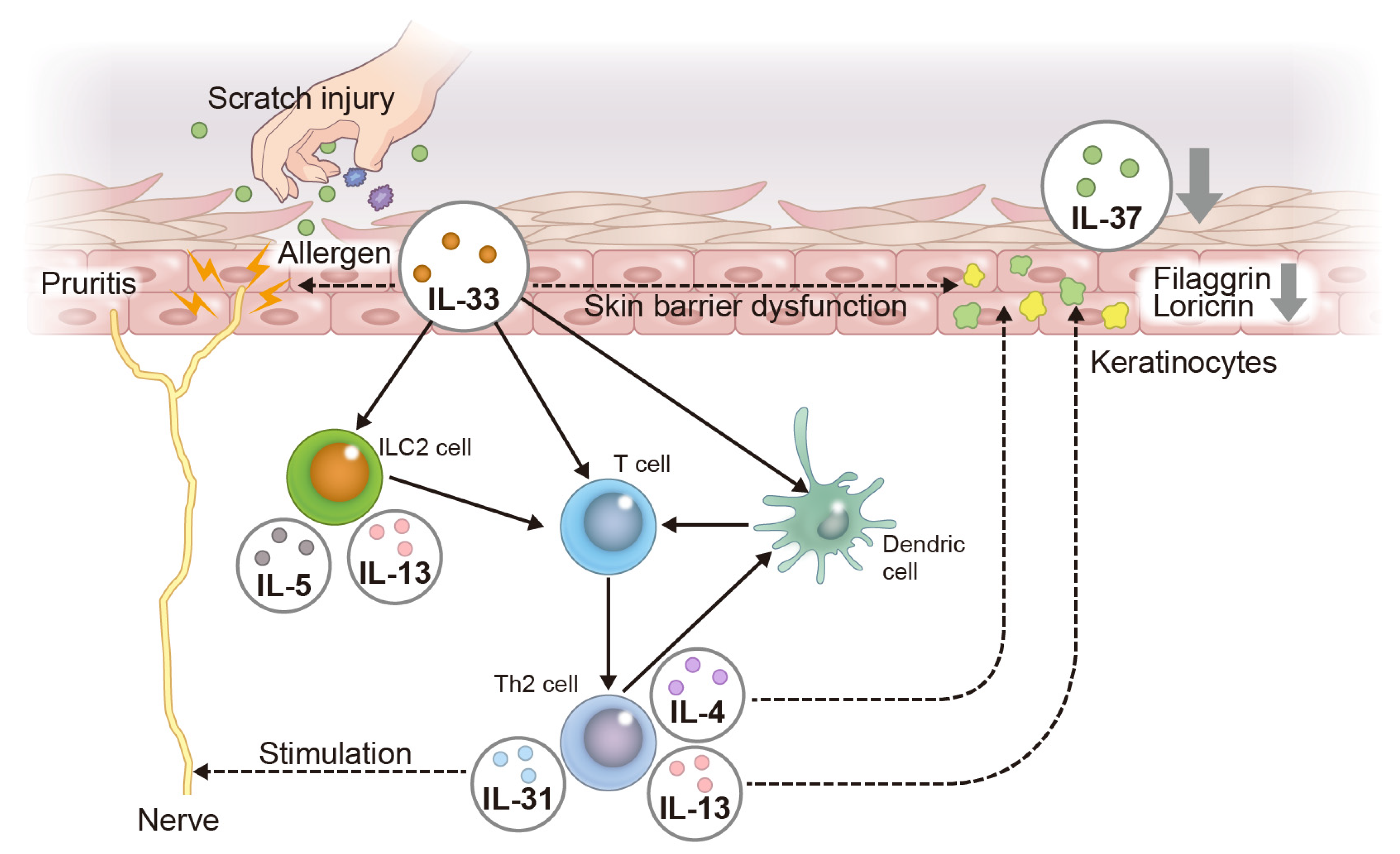
3.1. Atopic Dermatitis (AD)
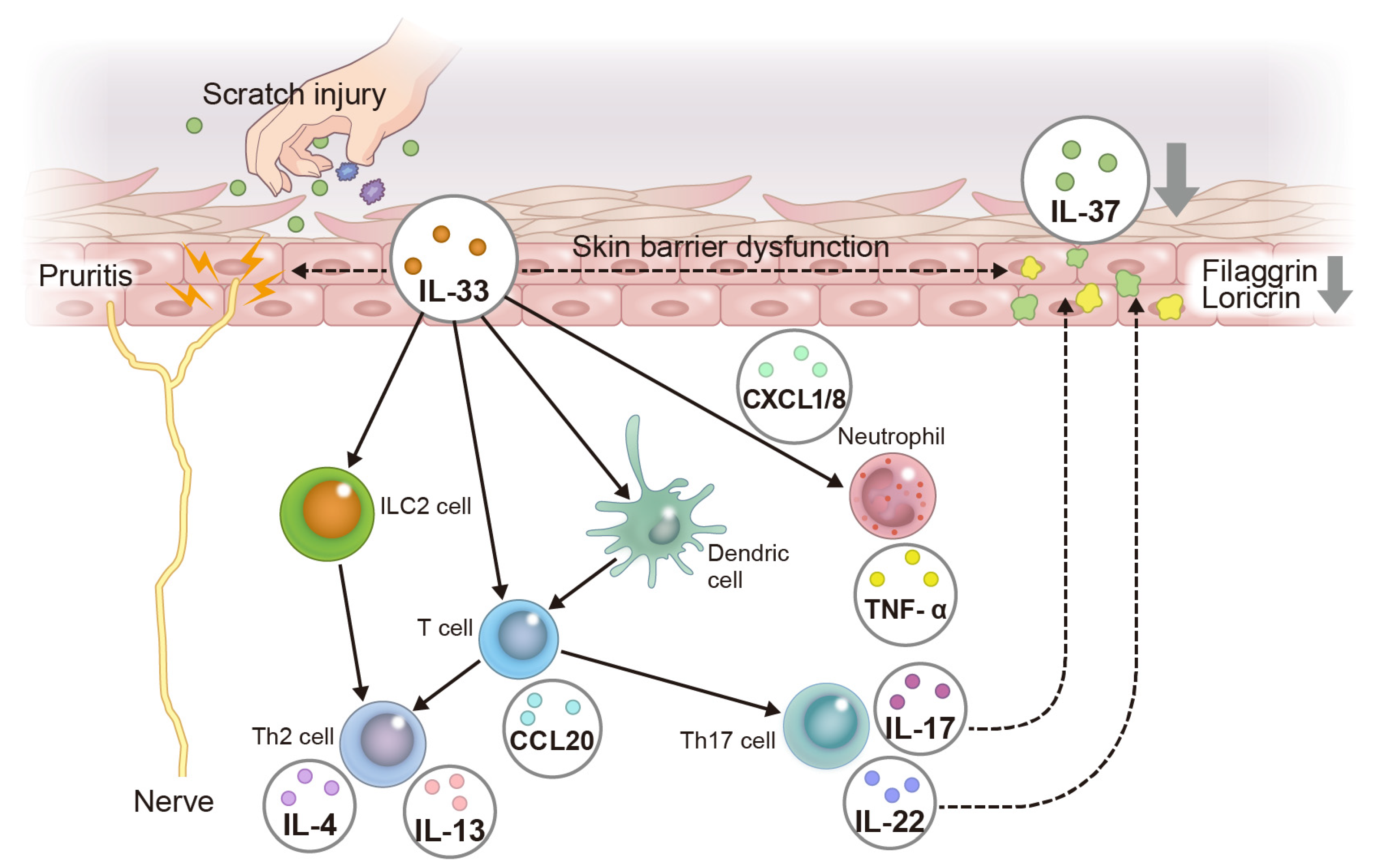
3.2. Psoriasis
4. Structure and Signaling Pathway of IL-37
5. The Role of IL-37 in the Pathogenesis of Atopic Dermatitis (AD) and Psoriasis
5.1. Atopic Dermatitis (AD)
5.2. Psoriasis
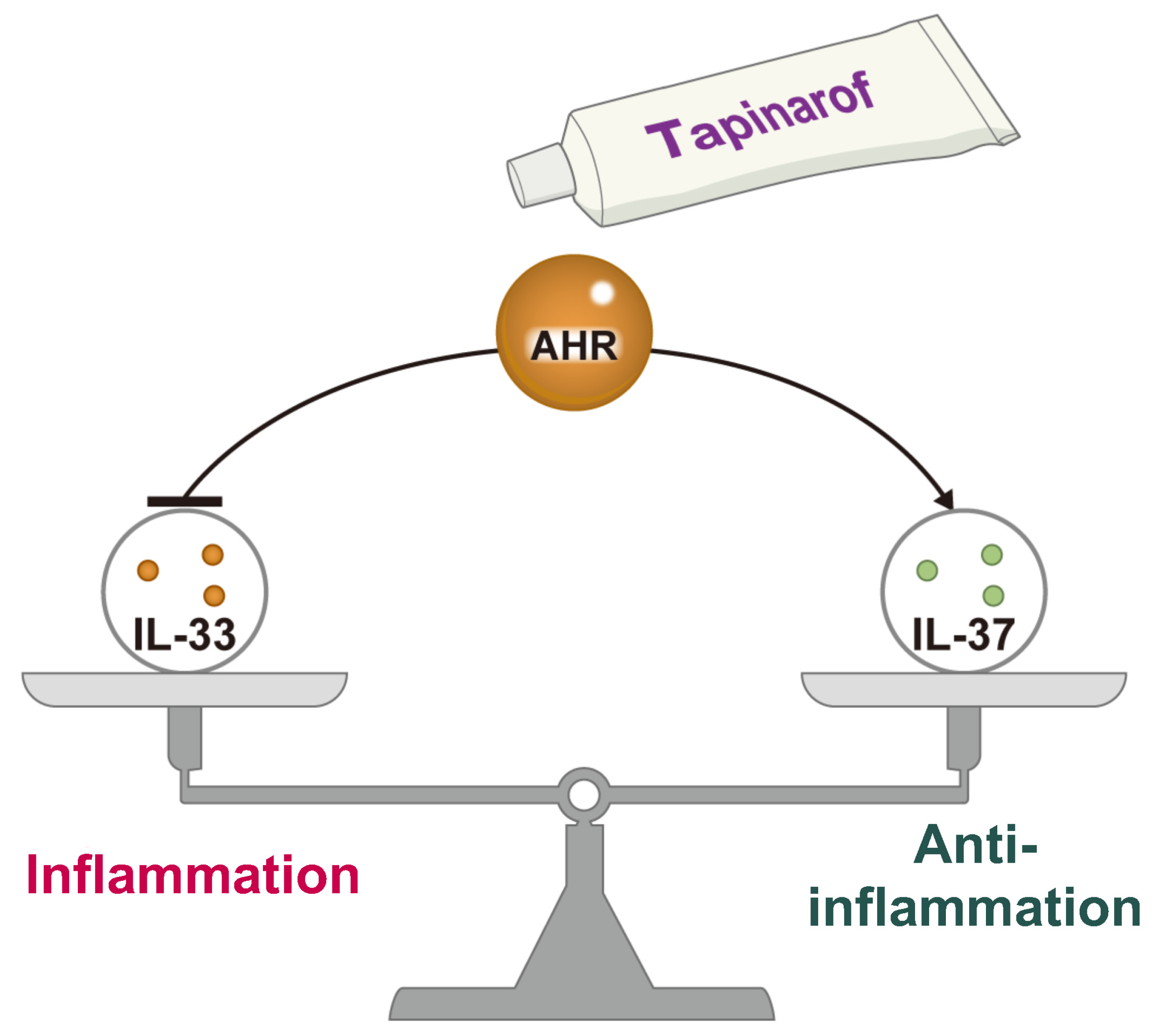
6. Regulatory Mechanism of the IL-33–IL-37 Axis via Aryl Hydrocarbon Receptor (AHR)
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Kallies, A. Interleukin (IL)-33 and the IL-1 Family of Cytokines-Regulators of Inflammation and Tissue Homeostasis. Cold Spring Harb. Perspect. Biol. 2019, 11, a028506. [Google Scholar] [CrossRef]
- Conti, P.; Pregliasco, F.E.; Bellomo, R.G.; Gallenga, C.E.; Caraffa, A.; Kritas, S.K.; Lauritano, D.; Ronconi, G. Mast Cell Cytokines IL-1, IL-33, and IL-36 Mediate Skin Inflammation in Psoriasis: A Novel Therapeutic Approach with the Anti-Inflammatory Cytokines IL-37, IL-38, and IL-1Ra. Int. J. Mol. Sci. 2021, 22, 8076. [Google Scholar] [CrossRef]
- Hou, T.; Sun, X.; Zhu, J.; Hon, K.L.; Jiang, P.; Chu, I.M.; Tsang, M.S.; Lam, C.W.; Zeng, H.; Wong, C.K. IL-37 Ameliorating Allergic Inflammation in Atopic Dermatitis Through Regulating Microbiota and AMPK-mTOR Signaling Pathway-Modulated Autophagy Mechanism. Front. Immunol. 2020, 11, 752. [Google Scholar] [CrossRef]
- Borgia, F.; Custurone, P.; Li Pomi, F.; Vaccaro, M.; Alessandrello, C.; Gangemi, S. IL-33 and IL-37: A Possible Axis in Skin and Allergic Diseases. Int. J. Mol. Sci. 2022, 24, 372. [Google Scholar] [CrossRef]
- Schröder, A.; Lunding, L.P.; Zissler, U.M.; Vock, C.; Webering, S.; Ehlers, J.C.; Orinska, Z.; Chaker, A.; Schmidt-Weber, C.B.; Lang, N.J.; et al. IL-37 Regulates Allergic Inflammation by Counterbalancing Pro-inflammatory IL-1 and IL-33. Allergy 2022, 77, 856–869. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. [Google Scholar] [CrossRef]
- van den Bogaard, E.H.; Perdew, G.H. The Enigma of AHR Activation in the Skin: Interplay among Ligands, Metabolism, and Bioavailability. J. Investig. Dermatol. 2021, 141, 1385–1388. [Google Scholar] [CrossRef]
- Napolitano, M.; Fabbrocini, G.; Martora, F.; Picone, V.; Morelli, P.; Patruno, C. Role of Aryl Hydrocarbon Receptor Activation in Inflammatory Chronic Skin Diseases. Cells 2021, 10, 3559. [Google Scholar] [CrossRef]
- Cardinali, G.; Flori, E.; Mastrofrancesco, A.; Ottaviani, M.; Dell’Anna, M.L.; Truglio, M.; Vento, A.; Zaccarini, M.; Zouboulis, C.C.; Picardo, M. Anti-Inflammatory and Pro-Differentiating Properties of the Aryl Hydrocarbon Receptor Ligands NPD-0614-13 and NPD-0614-24: Potential Therapeutic Benefits in Psoriasis. Int. J. Mol. Sci. 2021, 22, 7501. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Manetti, M.; Ibba-Manneschi, L.; Liakouli, V.; Guiducci, S.; Milia, A.F.; Benelli, G.; Marrelli, A.; Conforti, M.L.; Romano, E.; Giacomelli, R.; et al. The IL1-like Cytokine IL33 and its Receptor ST2 Are Abnormally Expressed in the Affected Skin and Visceral Organs of Patients with Systemic Sclerosis. Ann. Rheum. Dis. 2010, 69, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.N.; Beaugie, C.; O’Sullivan, C.; Leighton, S.; Halliday, G.M. The Immune-Modulating Cytokine and Endogenous Alarmin Interleukin-33 Is Upregulated in Skin Exposed to Inflammatory UVB Radiation. Am. J. Pathol. 2011, 179, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, Y.; Suzuki, M.; Matsumoto, M.; Togashi, H. Prostaglandin E(2) Enhances IL-33 Production by Dendritic Cells. Immunol. Lett. 2011, 141, 55–60. [Google Scholar] [CrossRef]
- Chang, Y.J.; Kim, H.Y.; Albacker, L.A.; Baumgarth, N.; McKenzie, A.N.; Smith, D.E.; Dekruyff, R.H.; Umetsu, D.T. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-reactivity Independently of Adaptive Immunity. Nat. Immunol. 2011, 12, 631–638. [Google Scholar] [CrossRef]
- Hsu, C.L.; Bryce, P.J. Inducible IL-33 Expression by Mast Cells Is Regulated by a Calcium-Dependent Pathway. J. Immunol. 2012, 189, 3421–3429. [Google Scholar] [CrossRef]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like Cytokine Ligand for ST2 Receptor, Is a Chromatin-Associated Nuclear Factor In Vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef]
- Brusilovsky, M.; Rochman, M.; Rochman, Y.; Caldwell, J.M.; Mack, L.E.; Felton, J.M.; Habel, J.E.; Porollo, A.; Pasare, C.; Rothenberg, M.E. Environmental Allergens Trigger Type 2 Inflammation through Ripoptosome Activation. Nat. Immunol. 2021, 22, 1316–1326. [Google Scholar] [CrossRef]
- Fu, Z.; Thorpe, M.; Alemayehu, R.; Roy, A.; Kervinen, J.; de Garavilla, L.; Åbrink, M.; Hellman, L. Highly Selective Cleavage of Cytokines and Chemokines by the Human Mast Cell Chymase and Neutrophil Cathepsin G. J. Immunol. 2017, 198, 1474–1483. [Google Scholar] [CrossRef]
- Travers, J.; Rochman, M.; Miracle, C.E.; Habel, J.E.; Brusilovsky, M.; Caldwell, J.M.; Rymer, J.K.; Rothenberg, M.E. Chromatin Regulates IL-33 Release and Extracellular Cytokine Activity. Nat. Commun. 2018, 9, 3244. [Google Scholar] [CrossRef]
- Iwahana, H.; Yanagisawa, K.; Ito-Kosaka, A.; Kuroiwa, K.; Tago, K.; Komatsu, N.; Katashima, R.; Itakura, M.; Tominaga, S. Different Promoter Usage and Multiple Transcription Initiation Sites of the Interleukin-1 Receptor-Related Human ST2 Gene in UT-7 and TM12 Cells. Eur. J. Biochem. 1999, 264, 397–406. [Google Scholar] [CrossRef]
- Hayakawa, H.; Hayakawa, M.; Kume, A.; Tominaga, S. Soluble ST2 Blocks Interleukin-33 Signaling in Allergic Airway Inflammation. J. Biol. Chem. 2007, 282, 26369–26380. [Google Scholar] [CrossRef]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 Pathway: Therapeutic Target and Novel Biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef]
- Balato, A.; Di Caprio, R.; Canta, L.; Mattii, M.; Lembo, S.; Raimondo, A.; Schiattarella, M.; Balato, N.; Ayala, F. IL-33 is regulated by TNF-α in normal and psoriatic skin. Arch. Dermatol. Res. 2014, 306, 299–304. [Google Scholar] [CrossRef]
- Taniguchi, K.; Yamamoto, S.; Hitomi, E.; Inada, Y.; Suyama, Y.; Sugioka, T.; Hamasaki, Y. Interleukin 33 is induced by tumor necrosis factor alpha and interferon gamma in keratinocytes and contributes to allergic contact dermatitis. J. Investig. Allergol. Clin. Immunol. 2013, 23, 428–434. [Google Scholar]
- Meephansan, J.; Tsuda, H.; Komine, M.; Tominaga, S.; Ohtsuki, M. Regulation of IL-33 expression by IFN-γ and tumor necrosis factor-α in normal human epidermal keratinocytes. J. Investig. Dermatol. 2012, 132, 2593–2600. [Google Scholar] [CrossRef]
- Tsuda, H.; Komine, M.; Tominaga, S.I.; Ohtsuki, M. Identification of the promoter region of human IL-33 responsive to induction by IFNγ. J. Dermatol. Sci. 2017, 85, 137–140. [Google Scholar] [CrossRef]
- Meephansan, J.; Komine, M.; Tsuda, H.; Karakawa, M.; Tominaga, S.; Ohtsuki, M. Expression of IL-33 in the epidermis: The mechanism of induction by IL-17. J. Dermatol. Sci. 2013, 71, 107–114. [Google Scholar] [CrossRef]
- Du, H.Y.; Fu, H.Y.; Li, D.N.; Qiao, Y.; Wang, Q.W.; Liu, W. The expression and regulation of interleukin-33 in human epidermal keratinocytes: A new mediator of atopic dermatitis and its possible signaling pathway. J. Interferon Cytokine Res. 2016, 36, 552–562. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Yen, V.H.; Miake, S.; Takemura, M.; Mitamura, Y.; Ito, T.; Murata, M.; Furue, M.; Nakahara, T. Aryl Hydrocarbon Receptor Activation Downregulates IL-33 Expression in Keratinocytes via Ovo-Like 1. J. Clin. Med. 2020, 9, 891. [Google Scholar] [CrossRef]
- Furue, M. Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4–JAK–STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis. J. Clin. Med. 2020, 9, 3741. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic Dermatitis: Immune Deviation, Barrier Dysfunction, IgE Autoreactivity and New Therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and Recent Advances in the Pathophysiology of Atopic Dermatitis. J. Dermatol. 2021, 48, 130–139. [Google Scholar] [CrossRef]
- Furue, M.; Iida, K.; Imaji, M.; Nakahara, T. Microbiome Analysis of Forehead Skin in Patients with Atopic Dermatitis and Healthy Subjects: Implication of Staphylococcus and Corynebacterium. J. Dermatol. 2018, 45, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Na, C.H.; Chung, J.; Simpson, E.L. Quality of Life and Disease Impact of Atopic Dermatitis and Psoriasis on Children and Their Families. Children 2019, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Furue, K.; Ulzii, D.; Tanaka, Y.; Ito, T.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. Pathogenic Implication of Epidermal Scratch Injury in Psoriasis and Atopic Dermatitis. J. Dermatol. 2020, 47, 979–988. [Google Scholar] [CrossRef]
- Saeki, H.; Nakahara, T.; Tanaka, A.; Kabashima, K.; Sugaya, M.; Murota, H.; Ebihara, T.; Kataoka, Y.; Aihara, M.; Etoh, T.; et al. Committee for Clinical Practice Guidelines for the Management of Atopic Dermatitis of Japanese Dermatological Association. Clinical Practice Guidelines for the Management of Atopic Dermatitis 2016. J. Dermatol. 2016, 43, 1117–1145. [Google Scholar] [CrossRef]
- Martel, B.C.; Litman, T.; Hald, A.; Norsgaard, H.; Lovato, P.; Dyring-Andersen, B.; Skov, L.; Thestrup-Pedersen, K.; Skov, S.; Skak, K.; et al. Distinct Molecular Signatures of Mild Extrinsic and Intrinsic Atopic Dermatitis. Exp. Dermatol. 2016, 25, 453–459. [Google Scholar] [CrossRef]
- Weidinger, S.; Rodríguez, E.; Stahl, C.; Wagenpfeil, S.; Klopp, N.; Illig, T.; Novak, N. Filaggrin Mutations Strongly Predispose to Early-Onset and Extrinsic Atopic Dermatitis. J. Investig. Dermatol. 2007, 127, 724–726. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, L.; Gao, S.; Su, S. Guidelines for the Management of Atopic Dermatitis in Children: A Systematic Review. Int. Arch. Allergy Immunol. 2023, 184, 132–141. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-Specific Expression of IL-33 Activates Group 2 Innate Lymphoid Cells and Elicits Atopic Dermatitis-like Inflammation in Mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Maillet, I.; Mackowiak, C.; Viala, C.; Di Padova, F.; Li, M.; Togbe, D.; Quesniaux, V.; Lai, Y.; Ryffel, B. Experimental Atopic Dermatitis Depends on IL-33R Signaling via MyD88 in Dendritic Cells. Cell Death Dis. 2017, 8, e2735. [Google Scholar] [CrossRef] [PubMed]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimäki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in Atopic Dermatitis: Expression Profiles and Modulation by Triggering Factors. J. Investig. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa-Mineoka, R.; Okuzawa, Y.; Masuda, K.; Katoh, N. Increased Serum Levels of Interleukin 33 in Patients with Atopic Dermatitis. J. Am. Acad. Dermatol. 2014, 70, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y. ILC2s in Skin Disorders. Allergol. Int. 2023, 72, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Furue, M. Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. [Google Scholar] [CrossRef]
- Dai, X.; Shiraishi, K.; Muto, J.; Utsunomiya, R.; Mori, H.; Murakami, M.; Sayama, K. Nuclear IL-33 Plays an Important Role in IL-31–Mediated Downregulation of FLG, Keratin 1, and Keratin 10 by Regulating Signal Transducer and Activator of Transcription 3 Activation in Human Keratinocytes. J. Investig. Dermatol. 2022, 142, 136–144.e3. [Google Scholar] [CrossRef]
- Dai, X.; Utsunomiya, R.; Shiraishi, K.; Mori, H.; Muto, J.; Murakami, M.; Sayama, K. Nuclear IL-33 Plays an Important Role in the Suppression of FLG, LOR, Keratin 1, and Keratin 10 by IL-4 and IL-13 in Human Keratinocytes. J. Investig. Dermatol. 2021, 141, 2646–2655.e6. [Google Scholar] [CrossRef]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef]
- Johnston, L.K.; Bryce, P.J. Understanding Interleukin 33 and Its Roles in Eosinophil Development. Front. Med. 2017, 2, 51. [Google Scholar] [CrossRef]
- Saluja, R.; Ketelaar, M.E.; Hawro, T.; Church, M.K.; Maurer, M.; Nawijn, M.C. The Role of the IL-33/IL-1RL1 Axis in Mast Cell and Basophil Activation in Allergic Disorders. Mol. Immunol. 2015, 63, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Furue, K.; Tsuji, G.; Nakahara, T. Interleukin-17A and Keratinocytes in Psoriasis. Int. J. Mol. Sci. 2020, 21, 1275. [Google Scholar] [CrossRef] [PubMed]
- Furue, K.; Ito, T.; Tsuji, G.; Kadono, T.; Furue, M. Psoriasis and the TNF/IL23/IL17 Axis. G. Ital. Dermatol. Venereol. 2019, 154, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; Picone, V.; Martora, F.; Fabbrocini, G.; Megna, M. Guselkumab, Risankizumab, and Tildrakizumab in the Management of Psoriasis: A Review of the Real-World Evidence. Clin. Cosmet. Investig. Dermatol. 2022, 15, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Vu, A.; Ulschmid, C.; Gordon, K.B. Anti-IL 23 Biologics for the Treatment of Plaque Psoriasis. Expert Opin. Biol. Ther. 2022, 12, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Ovejero-Benito, M.C.; Muñoz-Aceituno, E.; Reolid, A.; Saiz-Rodríguez, M.; Abad-Santos, F.; Daudén, E. Pharmacogenetics and Pharmacogenomics in Moderate-to-Severe Psoriasis. Am. J. Clin. Dermatol. 2018, 2, 209–222. [Google Scholar] [CrossRef]
- Zeng, F.; Chen, H.; Chen, L.; Mao, J.; Cai, S.; Xiao, Y.; Li, J.; Shi, J.; Li, B.; Xu, Y.; et al. An Autocrine Circuit of IL-33 in Keratinocytes Is Involved in the Progression of Psoriasis. J. Investig. Dermatol. 2021, 141, 596–606.e7. [Google Scholar] [CrossRef]
- Mitsui, A.; Tada, Y.; Takahashi, T.; Shibata, S.; Kamata, M.; Miyagaki, T.; Fujita, H.; Sugaya, M.; Kadono, T.; Sato, S.; et al. Serum IL-33 Levels Are Increased in Patients with Psoriasis. Clin. Exp. Dermatol. 2016, 41, 183–189. [Google Scholar] [CrossRef]
- Borsky, P.; Fiala, Z.; Andrys, C.; Beranek, M.; Hamakova, K.; Malkova, A.; Svadlakova, T.; Krejsek, J.; Palicka, V.; Borska, L.; et al. Alarmins HMGB1, IL-33, S100A7, and S100A12 in Psoriasis Vulgaris. Mediat. Inflamm. 2020, 2020, 8465083. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, M.S.; Lim, H.X.; Cho, D.; Kim, T.S. IL-33-Matured Dendritic Cells Promote Th17 Cell Responses via IL-1β and IL-6. Cytokine 2017, 99, 106–113. [Google Scholar] [CrossRef]
- Elewski, B.; Alexis, A.F.; Lebwohl, M.; Stein Gold, L.; Pariser, D.; Del Rosso, J.; Yosipovitch, G. Itch: An Under-recognized Problem in Psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Huang, Y.; Liu, T.T.; Guo, R.; Wang, B.; Wang, X.L.; Chen, L.H.; Zhou, Y.; Ji, R.R.; Liu, T. TNF-α/TNFR1 Signaling Is Required for the Full Expression of Acute and Chronic Itch in Mice via Peripheral and Central Mechanisms. Neurosci. Bull. 2018, 34, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Pavlenko, D.; Funahashi, H.; Sakai, K.; Hashimoto, T.; Lozada, T.; Yosipovitch, G.; Akiyama, T. IL-23 Modulates Histamine-Evoked Itch and Responses of Pruriceptors in Mice. Exp. Dermatol. 2020, 29, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Hu, X.; Yang, W.; Yasheng, H.; Liu, S.; Zhang, W.; Zhou, Y.; Cui, W.; Zhu, J.; Qiao, Z.; et al. Spinal IL-33/ST2 Signaling Mediates Chronic Itch in Mice through the Astrocytic JAK2-STAT3 Cascade. Glia 2019, 67, 1680–1693. [Google Scholar] [CrossRef]
- Bodoor, K.; Al-Qarqaz, F.; Heis, L.A.; Alfaqih, M.A.; Oweis, A.O.; Almomani, R.; Obeidat, M.A. IL-33/13 Axis and IL-4/31 Axis Play Distinct Roles in Inflammatory Process and Itch in Psoriasis and Atopic Dermatitis. Clin. Cosmet. Investig. Dermatol. 2020, 13, 419–424. [Google Scholar] [CrossRef]
- Chen, J.; Li, C.; Li, H.; Yu, H.; Zhang, X.; Yan, M.; Guo, Y.; Yao, Z. Identification of a TH 2-High Psoriasis Cluster Based on Skin Biomarker Analysis in a Chinese Psoriasis Population. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 150–158. [Google Scholar] [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 Is a Fundamental Inhibitor of Innate Immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Suppression of Inflammation and Acquired Immunity by IL-37. Immunol. Rev. 2018, 281, 179–190. [Google Scholar] [CrossRef]
- Su, Z.; Tao, X. Current Understanding of IL-37 in Human Health and Disease. Front. Immunol. 2021, 12, 696605. [Google Scholar] [CrossRef]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 Requires the Receptors IL-18Rα and IL-1R8 (SIGIRR) to Carry Out Its Multifaceted Anti-inflammatory Program upon Innate Signal Transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef]
- Bulau, A.M.; Nold, M.F.; Li, S.; Nold-Petry, C.A.; Fink, M.; Mansell, A.; Schwerd, T.; Hong, J.; Rubartelli, A.; Dinarello, C.A.; et al. Role of Caspase-1 in Nuclear Translocation of IL-37, Release of the Cytokine, and IL-37 Inhibition of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2014, 111, 2650–2655. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wen, X.; Hao, D.; Wang, Y.; Wang, L.; He, G.; Jiang, X. The Role of IL-37 in Skin and Connective Tissue Diseases. Biomed. Pharmacother. 2020, 122, 109705. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Liu, H. IL-37: An Anti-inflammatory Cytokine with Antitumor Functions. Cancer Rep. 2019, 2, e1151. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Cai, X.; Liu, S.; Wang, S.; Nold-Petry, C.A.; Nold, M.F.; Bufler, P.; Norris, D.; Dinarello, C.A.; Fujita, M. Suppression of Antigen-Specific Adaptive Immunity by IL-37 via Induction of Tolerogenic Dendritic Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15178–15183. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Tsang, M.S.; Kan, L.L.; Li, P.; Chu, I.M.; Lam, C.W.; Wong, C.K. IL-37 Targets TSLP-Primed Basophils to Alleviate Atopic Dermatitis. Int. J. Mol. Sci. 2021, 22, 7393. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Inoue, Y.; Seto, K.; Komitsu, N.; Aihara, M. Interleukin-37 Is Elevated in Subjects with Atopic Dermatitis. J. Dermatol. Sci. 2013, 69, 173–175. [Google Scholar] [CrossRef]
- Hou, T.; Tsang, M.S.; Chu, I.M.; Kan, L.L.; Hon, K.L.; Leung, T.F.; Lam, C.W.; Wong, C.K. Skewed Inflammation Is Associated with Aberrant Interleukin-37 Signaling Pathway in Atopic Dermatitis. Allergy 2021, 76, 2102–2114. [Google Scholar] [CrossRef]
- Zhou, J.; Gemperline, D.C.; Turner, M.J.; Oldach, J.; Molignano, J.; Sims, J.T.; Stayrook, K.R. Transcriptomic Analysis of Healthy and Atopic Dermatitis Samples Reveals the Role of IL-37 in Human Skin. Immunohorizons. 2021, 5, 830–843. [Google Scholar] [CrossRef]
- Li, B.; Tsoi, L.C.; Swindell, W.R.; Gudjonsson, J.E.; Tejasvi, T.; Johnston, A.; Ding, J.; Stuart, P.E.; Xing, X.; Kochkodan, J.J.; et al. Transcriptome Analysis of Psoriasis in a Large Case-Control Sample: RNA-seq Provides Insights into Disease Mechanisms. J. Investig. Dermatol. 2014, 134, 1828–1838. [Google Scholar] [CrossRef]
- Teng, X.; Hu, Z.; Wei, X.; Wang, Z.; Guan, T.; Liu, N.; Liu, X.; Ye, N.; Deng, G.; Luo, C.; et al. IL-37 Ameliorates the Inflammatory Process in Psoriasis by Suppressing Proinflammatory Cytokine Production. J. Immunol. 2014, 192, 1815–1823. [Google Scholar] [CrossRef]
- Rønholt, K.; Nielsen, A.L.; Johansen, C.; Vestergaard, C.; Fauerbye, A.; López-Vales, R.; Dinarello, C.A.; Iversen, L. IL-37 Expression Is Downregulated in Lesional Psoriasis Skin. Immunohorizons 2020, 4, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Sehat, M.; Talaei, R.; Dadgostar, E.; Nikoueinejad, H.; Akbari, H. Evaluating Serum Levels of IL-33, IL-36, IL-37 and Gene Expression of IL-37 in Patients with Psoriasis Vulgaris. Iran. J. Allergy Asthma Immunol. 2018, 17, 179–187. [Google Scholar] [PubMed]
- Krueger, J.; Clark, J.D.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Cueto, I.; Wang, C.Q.; Tan, H.; Wolk, R.; Rottinghaus, S.T.; Whitley, M.Z.; et al. Tofacitinib Attenuates Pathologic Immune Pathways in Patients with Psoriasis: A Randomized Phase 2 Study. J. Allergy Clin. Immunol. 2016, 137, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Howell, M.D.; Guttman-Yassky, E.; Gilleaudeau, P.M.; Cardinale, I.R.; Boguniewicz, M.; Krueger, J.G.; Leung, D.Y. TNF-α downregulates filaggrin and loricrin through c-Jun N-terminal kinase: Role for TNF-α antagonists to improve skin barrier. J. Investig. Dermatol. 2011, 131, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Smithrithee, R.; Niyonsaba, F.; Kiatsurayanon, C.; Ushio, H.; Ikeda, S.; Okumura, K.; Ogawa, H. Human β-defensin-3 increases the expression of interleukin-37 through CCR6 in human keratinocytes. J. Dermatol. Sci. 2015, 77, 46–53. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: A novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 2011, 186, 2613–2622. [Google Scholar] [CrossRef]
- Furue, M.; Takahara, M.; Nakahara, T.; Uchi, H. Role of AhR/ARNT systemin skin homeostasis. Arch. Dermatol. Res. 2014, 306, 769–779. [Google Scholar] [CrossRef]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Chiba, T.; Ito, T.; Nakahara, T.; Tsuji, G. Antioxidants for Healthy Skin: The emerging role of aryl hydrocarbon receptors and nuclear factor-erythroid 2-related factor-2. Nutrients 2017, 9, 223. [Google Scholar] [CrossRef]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Tsuji, G.; Takahara, M.; Uchi, H.; Takeuchi, S.; Mitoma, C.; Moroi, Y.; Furue, M. An environmental contaminant, benzo(a)pyrene, induces oxidative stress-mediated interleukin-8 production in human keratinocytes via the aryl hydrocarbon receptor signaling pathway. J. Dermatol. Sci. 2011, 62, 42–49. [Google Scholar] [CrossRef]
- Blunder, S.; Kõks, S.; Kõks, G.; Reimann, E.; Hackl, H.; Gruber, R.; Moosbrugger-Martinz, V.; Schmuth, M.; Dubrac, S. Enhanced Expression of Genes Related to Xenobiotic Metabolism in the Skin of Patients with Atopic Dermatitis but Not with Ichthyosis Vulgaris. J. Investig. Dermatol. 2018, 138, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Takahara, M.; Uchi, H.; Matsuda, T.; Chiba, T.; Takeuchi, S.; Yasukawa, F.; Moroi, Y.; Furue, M. Identification of ketoconazole as an AhR-Nrf2 activator in cultured human keratinocytes: The basis of its anti-inflammatory effect. J. Investig. Dermatol. 2012, 132, 59–68. [Google Scholar] [CrossRef] [PubMed]
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef]
- Nukaya, M.; Lin, B.C.; Glover, E.; Moran, S.M.; Kennedy, G.D.; Bradfield, C.A. The aryl hydrocarbon receptor-interacting protein (AIP) is required for dioxin-induced hepatotoxicity but not for the induction of the Cyp1a1 and Cyp1a2 genes. J. Biol. Chem. 2010, 285, 35599–35605. [Google Scholar] [CrossRef]
- Habano, W.; Miura, T.; Terashima, J.; Ozawa, S. Aryl Hydrocarbon Receptor as a DNA Methylation Reader in the Stress Response Pathway. Toxicology 2022, 470, 153154. [Google Scholar] [CrossRef]
- Ishihara, Y.; Haarmann-Stemmann, T.; Kado, N.Y.; Vogel, C.F.A. Interleukin 33 Expression Induced by Aryl Hydrocarbon Receptor in Macrophages. Toxicol. Sci. 2019, 170, 404–414. [Google Scholar] [CrossRef]
- Ishihara, N.; Okuda, T.; Hagino, H.; Oguro, A.; Tani, Y.; Okochi, H.; Tokoro, C.; Fujii-Kuriyama, Y.; Itoh, K.; Vogel, C.F.A.; et al. Involvement of polycyclic aromatic hydrocarbons and endotoxin in macrophage expression of interleukin-33 induced by exposure to particulate matter. J. Toxicol. Sci. 2022, 47, 201–210. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhu, Z.; Liu, X.; Chen, B.; Yin, H.; Gu, C.; Fang, X.; Zhu, R.; Yu, T.; Mi, W.; et al. A dysregulated sebum-microbial metabolite-IL-33 axis initiates skin inflammation in atopic dermatitis. J. Exp. Med. 2022, 219, e20212397. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Matsuda-Taniguchi, T.; Takai-Yumine, A.; Takemura, M.; Yan, X.; Furue, M.; Nakahara, T. Natural Compounds Tapinarof and Galactomyces Ferment Filtrate Downregulate IL-33 Expression via the AHR/IL-37 Axis in Human Keratinocytes. Front. Immunol. 2022, 13, 745997. [Google Scholar] [CrossRef]
- Peppers, J.; Paller, A.S.; Maeda-Chubachi, T.; Wu, S.; Robbins, K.; Gallagher, K.; Kraus, J.E. A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 89–98.e3. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.; Stein Gold, L.; Bissonnette, R.; Armstrong, A.W.; Kircik, L.; Tyring, S.K.; Piscitelli, S.C.; Brown, P.M.; Rubenstein, D.S.; Tallman, A.M.; et al. One-year safety and efficacy of tapinarof cream for the treatment of plaque psoriasis: Results from the PSOARING 3 trial. J. Am. Acad. Dermatol. 2022, 87, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Vu, Y.H.; Hashimoto-Hachiya, A.; Takemura, M.; Yumine, A.; Mitamura, Y.; Nakahara, T.; Furue, M.; Tsuji, G. IL-24 Negatively Regulates Keratinocyte Differentiation Induced by Tapinarof, an Aryl Hydrocarbon Receptor Modulator: Implication in the Treatment of Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 9412. [Google Scholar] [CrossRef]
- Lebwohl, M.G.; Stein Gold, L.; Strober, B.; Papp, K.A.; Armstrong, A.W.; Bagel, J.; Kircik, L.; Ehst, B.; Hong, H.C.; Soung, J.; et al. Phase 3 Trials of Tapinarof Cream for Plaque Psoriasis. N. Engl. J. Med. 2021, 385, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef]
- Hirota, T.; Takahashi, A.; Kubo, M.; Tsunoda, T.; Tomita, K.; Sakashita, M.; Yamada, T.; Fujieda, S.; Tanaka, S.; Doi, S.; et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 2012, 44, 1222–1226. [Google Scholar] [CrossRef]
- Paternoster, L.; Standl, M.; Chen, C.M.; Ramasamy, A.; Bønnelykke, K.; Duijts, L.; Ferreira, M.A.; Alves, A.C.; Thyssen, J.P.; Albrecht, E.; et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat. Genet. 2011, 44, 187–192. [Google Scholar] [CrossRef]
- Dragan, M.; Sun, P.; Chen, Z.; Ma, X.; Vu, R.; Shi, Y.; Villalta, S.A.; Dai, X. Epidermis-Intrinsic Transcription Factor Ovol1 Coordinately Regulates Barrier Maintenance and Neutrophil Accumulation in Psoriasis-Like Inflammation. J. Investig. Dermatol. 2022, 142 Pt A, 583–593.e5. [Google Scholar] [CrossRef]
- Chan, B.C.L.; Lam, C.W.K.; Tam, L.S.; Wong, C.K. IL33: Roles in Allergic Inflammation and Therapeutic Perspectives. Front. Immunol. 2019, 10, 364. [Google Scholar] [CrossRef]
- Jeon, W.Y.; Shin, H.K.; Shin, I.S.; Kim, S.K.; Lee, M.Y. Soshiho-tang Water Extract Inhibits Ovalbumin-Induced Airway Inflammation via the Regulation of Heme Oxygenase-1. BMC Complement. Altern. Med. 2015, 15, 329. [Google Scholar] [CrossRef]
- Kasai, A.; Hiramatsu, N.; Hayakawa, K.; Yao, J.; Kitamura, M. Blockade of the Dioxin Pathway by Herbal Medicine Formula Bupleuri Minor: Identification of Active Entities for Suppression of AhR Activation. Biol. Pharm. Bull. 2008, 31, 838–846. [Google Scholar] [CrossRef]
- Harada, A.; Sugihara, K.; Watanabe, Y.; Yamaji, S.; Kitamura, S.; Ohta, S. Aryl Hydrocarbon Receptor Ligand Activity of Extracts from 62 Herbal Medicines and Effect on Cytochrome P450 Activity. Yakugaku Zasshi 2015, 135, 1185–1196. [Google Scholar] [CrossRef]
- Gum, S.I.; Jo, S.J.; Ahn, S.H.; Kim, S.G.; Kim, J.T.; Shin, H.M.; Cho, M.K. The Potent Protective Effect of Wild Ginseng (Panax ginseng C.A. Meyer) against Benzo[alpha]pyrene-Induced Toxicity through Metabolic Regulation of CYP1A1 and GSTs. J. Ethnopharmacol. 2007, 112, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Che, D.N.; Cho, B.O.; Kim, J.S.; Shin, J.Y.; Kang, H.J.; Jang, S.I. Effect of Luteolin and Apigenin on the Production of Il-31 and Il-33 in Lipopolysaccharides-Activated Microglia Cells and Their Mechanism of Action. Nutrients 2020, 12, 811. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zheng, T.; Hou, Z.; Lv, C.; Xue, A.; Han, T.; Han, B.; Sun, X.; Wei, Y. Luteolin, an Aryl Hydrocarbon Receptor Ligand, Suppresses Tumor Metastasis In Vitro and In Vivo. Oncol. Rep. 2020, 44, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Park, H.; Li, X.; Davidson, L.A.; Allred, C.; Patil, B.; Jayaprakasha, G.; Orr, A.A.; Mao, L.; Chapkin, R.S.; et al. Structure-Dependent Modulation of Aryl Hydrocarbon Receptor-Mediated Activities by Flavonoids. Toxicol. Sci. 2018, 164, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Lim, S.; Bae, M.J.; Lee, W.; Kim, S. PG102 Upregulates IL-37 through p38, ERK, and Smad3 Pathways in HaCaT Keratinocytes. Mediat. Inflamm. 2019, 2019, 6085801. [Google Scholar] [CrossRef] [PubMed]
- Matich, A.J.; Young, H.; Allen, J.M.; Wang, M.Y.; Fielder, S.; McNeilage, M.A.; MacRae, E.A. Actinidia arguta: Volatile Compounds in Fruit and Flowers. Phytochemistry 2003, 63, 285–301. [Google Scholar] [CrossRef]
- Wojdyło, A.; Nowicka, P. Anticholinergic Effects of Actinidia arguta Fruits and their Polyphenol Content Determined by Liquid Chromatography-Photodiode Array Detector-Quadrupole/Time of Flight-Mass Spectrometry (LC-MS-PDA-Q/TOF). Food Chem. 2019, 271, 216–223. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Daschner, P.J.; Yeh, G.C. Dietary Flavonols Quercetin and Kaempferol Are Ligands of the Aryl Hydrocarbon Receptor that Affect CYP1A1 Transcription Differentially. Biochem. J. 1999, 340 Pt 3, 715–722. [Google Scholar] [CrossRef]
- Lossius, A.H.; Berents, T.L.; Saetre, F.; Nilsen, H.R.; Bradley, M.; Asad, S.; Haraldsen, G.; Sundnes, O.; Holm, J.Ø. Early Transcriptional Changes after UVB Treatment in Atopic Dermatitis Include Inverse Regulation of IL-36γ and IL-37. Exp. Dermatol. 2021, 30, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Bergander, L.; Wincent, E.; Rannug, A.; Foroozesh, M.; Alworth, W.; Rannug, U. Metabolic Fate of the Ah Receptor Ligand 6-Formylindolo[3,2-b]carbazole. Chem. Biol. Interact. 2004, 149, 151–164. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuji, G.; Yamamura, K.; Kawamura, K.; Kido-Nakahara, M.; Ito, T.; Nakahara, T. Regulatory Mechanism of the IL-33–IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2023, 24, 14633. https://doi.org/10.3390/ijms241914633
Tsuji G, Yamamura K, Kawamura K, Kido-Nakahara M, Ito T, Nakahara T. Regulatory Mechanism of the IL-33–IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. International Journal of Molecular Sciences. 2023; 24(19):14633. https://doi.org/10.3390/ijms241914633
Chicago/Turabian StyleTsuji, Gaku, Kazuhiko Yamamura, Koji Kawamura, Makiko Kido-Nakahara, Takamichi Ito, and Takeshi Nakahara. 2023. "Regulatory Mechanism of the IL-33–IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis" International Journal of Molecular Sciences 24, no. 19: 14633. https://doi.org/10.3390/ijms241914633
APA StyleTsuji, G., Yamamura, K., Kawamura, K., Kido-Nakahara, M., Ito, T., & Nakahara, T. (2023). Regulatory Mechanism of the IL-33–IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. International Journal of Molecular Sciences, 24(19), 14633. https://doi.org/10.3390/ijms241914633