
Regulation of Ferroptosis in Lung Adenocarcinoma
Abstract
:1. Introduction
2. Molecular Mechanisms of Ferroptosis
2.1. Iron Metabolism
2.2. Lipid Metabolism
2.3. Antioxidant Defenses
3. Ferroptosis in LUAD
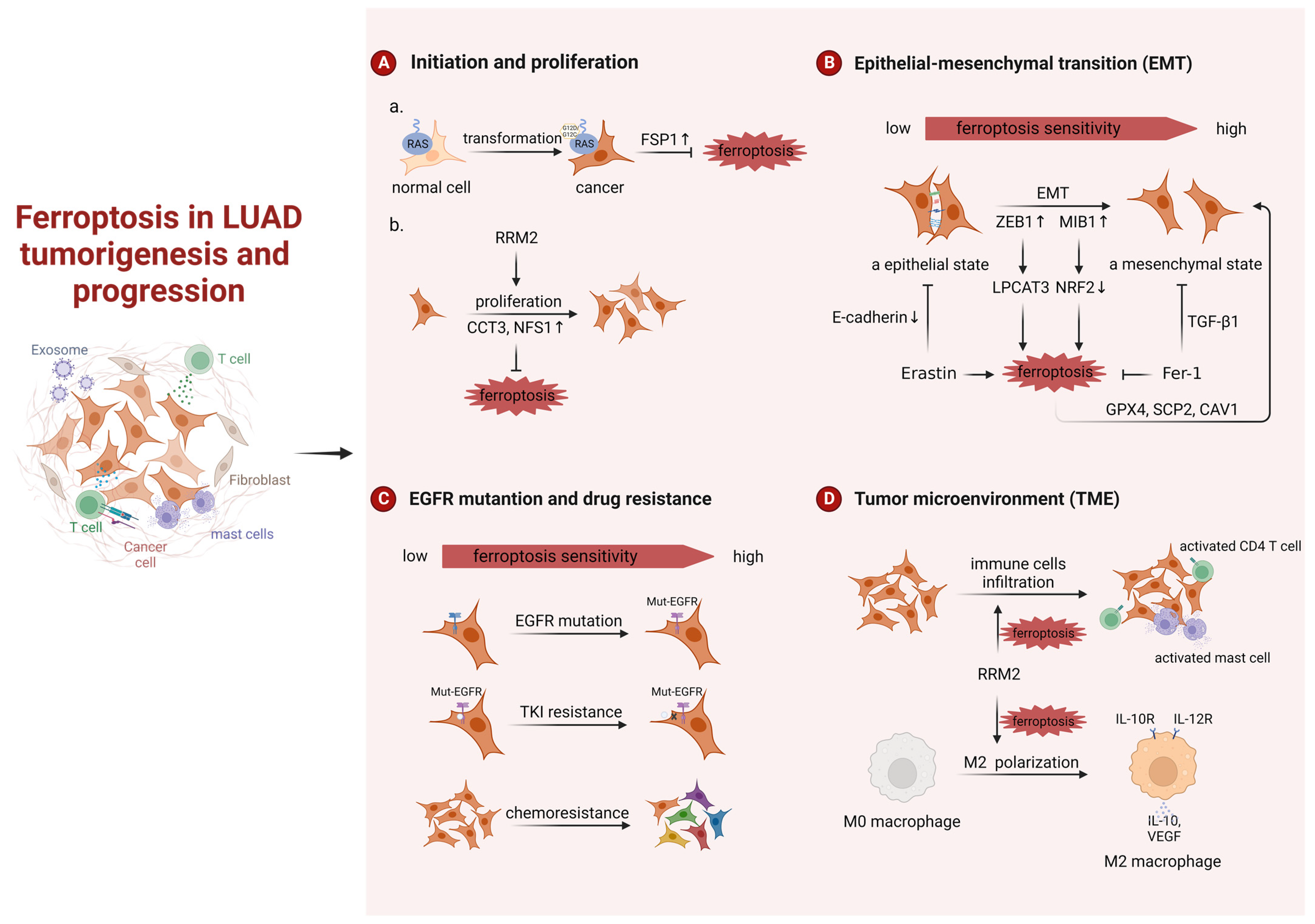
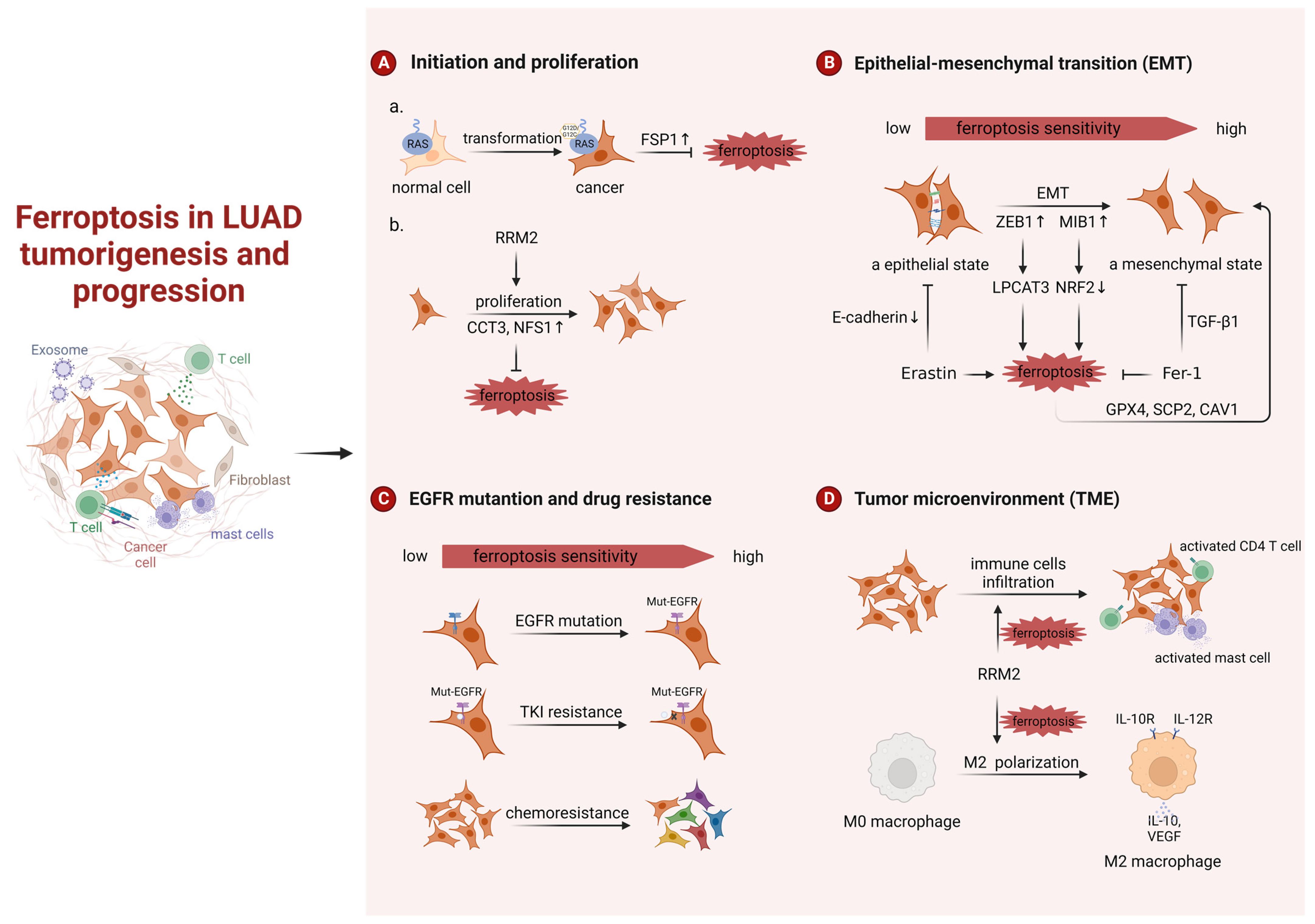
3.1. Ferroptosis in LUAD Tumorigenesis and Progression
3.1.1. Ferroptosis in LUAD Tumorigenesis
3.1.2. Ferroptosis in LUAD Progression
3.2. Regulation of Ferroptosis in LUAD
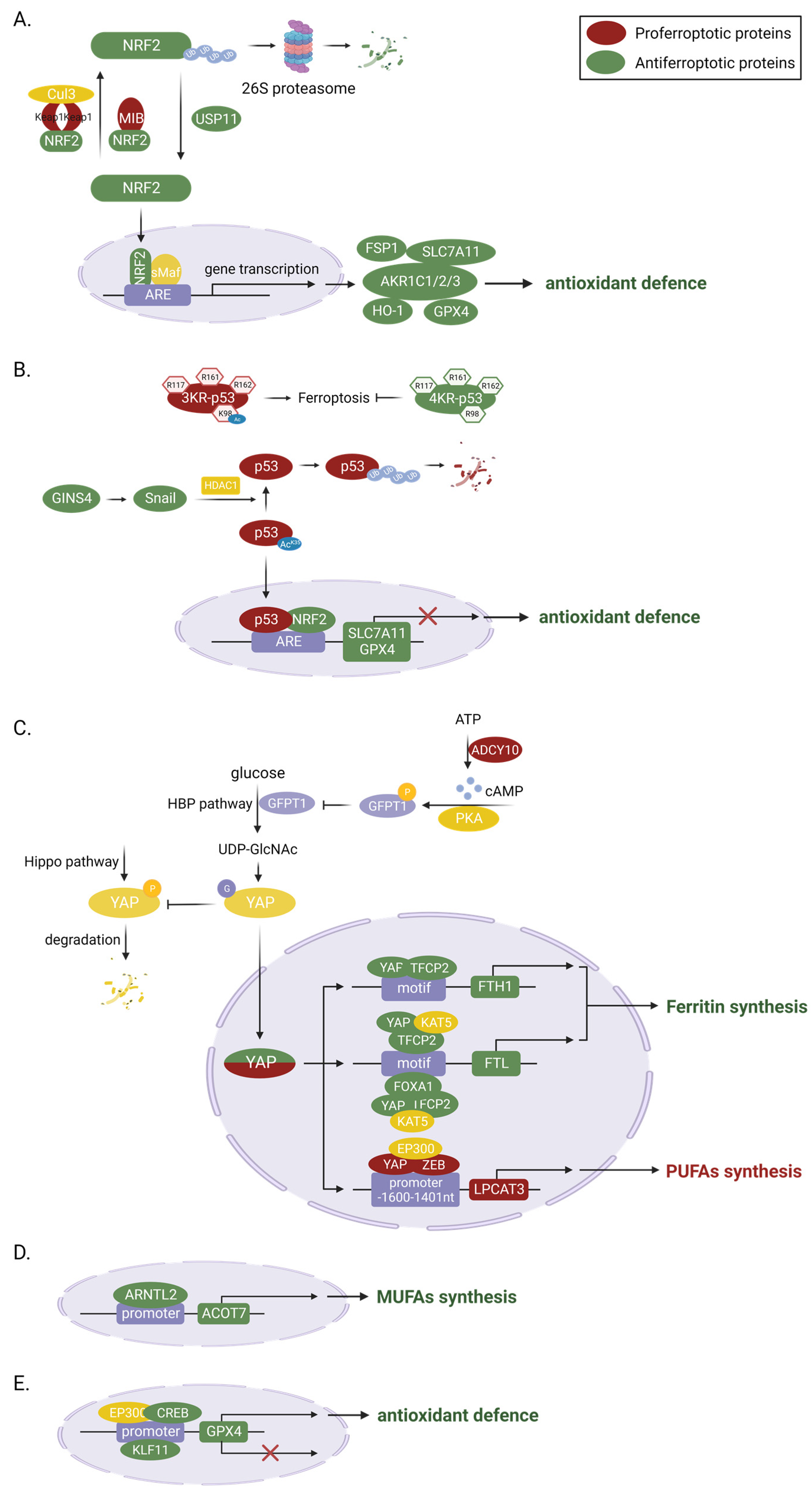
3.2.1. Transcriptional Regulation
NRF2
p53
YAP
Others
3.2.2. Epigenetic Regulation
Histone Modification
Non-Coding RNA-Mediated Processes
- miRNAs
- CircRNAs
- LncRNAs
- m6A
- modification

{kind=link}
{kind=link}
| Name | Epigenetic Mechanism | Functions in Ferroptosis | Expression in LUAD Patients | References |
|---|---|---|---|---|
| EP300 | histone modification | ① EP300/CREB complex: suppressor; increases GPX4 expression ② EP300/YAP/ZEB complex: promoter; increases LPCAT3 expression | N/A | [19,79] |
| miR-27a-3p | miRNA | Promoter; inhibits SLC7A11 expression | Downregulation | [88] |
| miR-324-3p | miRNA | Promoter; inhibits GPX4 expression | Downregulation in A549/DDP cells | [51] |
| P4HB | circRNA | Suppressor; sponge miRNA-1184 and increases SLC7A11 expression | Upregulation | [92] |
| circ_1010093 | circRNA | Suppressor; inhibits lipids (ACSL4, LPCAT3, and PLTP) | Upregulation | [94] |
| LINC00324 | lncRNA | Suppressor; sponge miR-200c-3p and promotes TFAP2A-NRF2 axis | Upregulation | [37] |
| GSEC | lncRNA | Suppressor; sponge miR-101-3p and increases CISD1 (a mitochondrial iron–sulfur protein) | Upregulation | [95] |
| Uc.339 | lncRNA | Suppressor; sponge pri-miR-339 inhibits the production of mature miR-339 and increases SLC7A11 expression | Upregulation | [96] |
| LINC00336 | lncRNA | Suppressor; sponge miR-6852 and increases cystathionine-β-synthase (CBS, involved in the transsulfuration pathway and synthesizes cysteine) expression | Upregulation | [97] |
| GMDS-AS1 and LINC01128 | lncRNA | Suppressor; sponge miR-6077 and promotes the KEAP1-NRF2-SLC7A11/NQO1 pathway | Downregulation | [59] |
| LINC00551 | lncRNA | Promotor; sponge miR-4328 and increases DDIT4 expression; DDIT4 inhibits mTOR activity and promotes autophagy-dependent ferroptosis | Downregulation | [98] |
| METTL3 | m6A | Suppressor; enhance the stability and translation of SLC7A11 | Upregulation | [104] |
| IGF2BP3 | m6A | Suppressor; enhances the stability and translation of anti-ferroptotic factors (GPX4, SLC3A2, ACSL3, and FTH1) | Upregulation | [105] |
| YTHDC2 | m6A | Promotor; inhibits system Xc (directly inhibits SLC7A11 and indirectly inhibits SLC3A5 expression) | Downregulation | [106] |
3.3. Ferroptosis in LUAD Therapy
4. Conclusions and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Kris, M.G.; Gaspar, L.E.; Chaft, J.E.; Kennedy, E.B.; Azzoli, C.G.; Ellis, P.M.; Lin, S.H.; Pass, H.I.; Seth, R.; Shepherd, F.A.; et al. Adjuvant Systemic Therapy and Adjuvant Radiation Therapy for Stage I to IIIA Completely Resected Non-Small-Cell Lung Cancers: American Society of Clinical Oncology/Cancer Care Ontario Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 2960–2974. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, H.; Li, Y.; Yuan, Y.; Chen, B.; Sun, S. Elevated TRIM23 expression predicts cisplatin resistance in lung adenocarcinoma. Cancer Sci. 2020, 111, 637–646. [Google Scholar] [CrossRef]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA 2019, 322, 764–774. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Sun, H.; Zhou, J.-Y.; Jie, G.-L.; Xie, Z.; Shao, Y.; Zhang, X.; Ye, J.-Y.; Chen, C.-X.; Zhang, X.-C.; et al. Clinical characteristics and prognostic value of the KRAS G12C mutation in Chinese non-small cell lung cancer patients. Biomark. Res. 2020, 8, 22. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- van Swelm, R.P.L.; Wetzels, J.F.M.; Swinkels, D.W. The multifaceted role of iron in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Liu, J.; Kang, R.; Tang, D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem. Biophys. Res. Commun. 2020, 531, 581–587. [Google Scholar] [CrossRef]
- Song, N.; Zhang, J.; Zhai, J.; Hong, J.; Yuan, C.; Liang, M. Ferritin: A Multifunctional Nanoplatform for Biological Detection, Imaging Diagnosis, and Drug Delivery. Acc. Chem. Res. 2021, 54, 3313–3325. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, K.; Ma, L.; Qian, Z.; Tian, X.; Miao, Y.; Niu, Y.; Xu, X.; Guo, S.; Yang, Y.; et al. Endogenous glutamate determines ferroptosis sensitivity via ADCY10-dependent YAP suppression in lung adenocarcinoma. Theranostics 2021, 11, 5650–5674. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Y.; Tian, X.; Miao, Y.; Ma, L.; Zhang, C.; Xu, X.; Wang, J.; Fang, W.; Zhang, X. LPCAT3 is transcriptionally regulated by YAP/ZEB/EP300 and collaborates with ACSL4 and YAP to determine ferroptosis sensitivity. Antioxid. Redox Signal. 2023, 39, 491–511. [Google Scholar] [CrossRef]
- Wang, Y.; Qiu, S.; Wang, H.; Cui, J.; Tian, X.; Miao, Y.; Zhang, C.; Cao, L.; Ma, L.; Xu, X.; et al. Transcriptional Repression of Ferritin Light Chain Increases Ferroptosis Sensitivity in Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 719187. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc(-) cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef]
- Muller, F.; Lim, J.K.M.; Bebber, C.M.; Seidel, E.; Tishina, S.; Dahlhaus, A.; Stroh, J.; Beck, J.; Yapici, F.I.; Nakayama, K.; et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2023, 30, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Mitsushita, J.; Lambeth, J.D.; Kamata, T. The Superoxide-Generating Oxidase Nox1 Is Functionally Required for Ras Oncogene Transformation. Cancer Res. 2004, 64, 3580–3585. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Wang, K.; He, J.; Tu, C.; Xu, H.; Zhang, X.; Lv, Y.; Song, C. Upregulation of CCT3 predicts poor prognosis and promotes cell proliferation via inhibition of ferroptosis and activation of AKT signaling in lung adenocarcinoma. BMC Mol. Cell Biol. 2022, 23, 25. [Google Scholar] [CrossRef]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiang, X.; Lu, Y.; Ruan, Y.; Wang, J. Identification and integration analysis of a novel prognostic signature associated with cuproptosis-related ferroptosis genes and relevant lncRNA regulatory axis in lung adenocarcinoma. Aging 2023, 15, 1543–1563. [Google Scholar] [CrossRef]
- Gao, C.; Kong, N.; Zhang, F.; Tang, T.; Li, J.; Ding, H.; Sun, Z.; Wu, L.; Xu, M. Risk stratification of lung adenocarcinoma using a nomogram combined with ferroptosis-related LncRNAs and subgroup analysis with immune and N6-methyladenosine modification. BMC Med. Genom. 2022, 15, 15. [Google Scholar] [CrossRef]
- Shen, Y.; Li, D.; Liang, Q.; Yang, M.; Pan, Y.; Li, H. Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Front. Immunol. 2023, 13, 1029092. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Y.; Wu, J.; Luo, Y.; Fang, Z.; Xu, R.; Teng, W.; Chen, M.; Li, Y. A Novel Predictive Model Incorporating Ferroptosis-Related Gene Signatures for Overall Survival in Patients with Lung Adenocarcinoma. Med. Sci. Monit. 2021, 27, e934050-1. [Google Scholar] [CrossRef]
- Zhang, W.; Yao, S.; Huang, H.; Zhou, H.; Zhou, H.; Wei, Q.; Bian, T.; Sun, H.; Li, X.; Zhang, J.; et al. Molecular subtypes based on ferroptosis-related genes and tumor microenvironment infiltration characterization in lung adenocarcinoma. OncoImmunology 2021, 10, 1959977. [Google Scholar] [CrossRef]
- Zhu, G.; Huang, H.; Xu, S.; Shi, R.; Gao, Z.; Lei, X.; Zhu, S.; Zhou, N.; Zu, L.; Mello, R.A.D.; et al. Prognostic value of ferroptosis-related genes in patients with lung adenocarcinoma. Thorac. Cancer 2021, 12, 1890–1899. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Q.; Xia, J.; Cheng, S.; Pei, D.; Zhang, X.; Shu, X. The E3 Ligase MIB1 Promotes Proteasomal Degradation of NRF2 and Sensitizes Lung Cancer Cells to Ferroptosis. Mol. Cancer Res. 2022, 20, 253–264. [Google Scholar] [CrossRef]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Investig. 2016, 126, 3219–3235. [Google Scholar] [CrossRef]
- Sun, L.; Dong, H.; Zhang, W.; Wang, N.; Ni, N.; Bai, X.; Liu, N. Lipid Peroxidation, GSH Depletion, and SLC7A11 Inhibition Are Common Causes of EMT and Ferroptosis in A549 Cells, but Different in Specific Mechanisms. DNA Cell Biol. 2021, 40, 172–183. [Google Scholar] [CrossRef]
- Zhang, H.; Shan, G.; Jin, X.; Yu, X.; Bi, G.; Feng, M.; Wang, H.; Lin, M.; Zhan, C.; Wang, Q.; et al. ARNTL2 is an indicator of poor prognosis, promotes epithelial-to-mesenchymal transition and inhibits ferroptosis in lung adenocarcinoma. Transl. Oncol. 2022, 26, 101562. [Google Scholar] [CrossRef] [PubMed]
- Yatabe, Y.; Kerr, K.M.; Utomo, A.; Rajadurai, P.; Tran, V.K.; Du, X.; Chou, T.-Y.; Enriquez, M.L.D.; Lee, G.K.; Iqbal, J.; et al. EGFR Mutation Testing Practices within the Asia Pacific Region: Results of a Multicenter Diagnostic Survey. J. Thorac. Oncol. 2015, 10, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Tjulandin, S.; Hagiwara, K.; Normanno, N.; Wulandari, L.; Laktionov, K.; Hudoyo, A.; He, Y.; Zhang, Y.-P.; Wang, M.-Z.; et al. EGFR mutation prevalence in Asia-Pacific and Russian patients with advanced NSCLC of adenocarcinoma and non-adenocarcinoma histology: The IGNITE study. Lung Cancer 2017, 113, 37–44. [Google Scholar] [CrossRef]
- Recondo, G.; Facchinetti, F.; Olaussen, K.A.; Besse, B.; Friboulet, L. Making the first move in EGFR-driven or ALK-driven NSCLC: First-generation or next-generation TKI? Nat. Rev. Clin. Oncol. 2018, 15, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-S.; Kumarakulasinghe, N.B.; Huang, Y.-Q.; Ang, Y.L.E.; Choo, J.R.-E.; Goh, B.-C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.S.; Liu, J.; Li, M.S.C.; Li, L.; Tao, Q.; Mok, T.S.K. Selenite as a dual apoptotic and ferroptotic agent synergizes with EGFR and KRAS inhibitors with epigenetic interference. Clin. Epigenetics 2023, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, B.; Chu, H.; Yao, Y. Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small-cell lung cancer with activating EGFR mutations. OncoTargets Ther. 2016, 9, 3711–3726. [Google Scholar] [CrossRef] [PubMed]
- Tumbrink, H.L.; Heimsoeth, A.; Sos, M.L. The next tier of EGFR resistance mutations in lung cancer. Oncogene 2020, 40, 1–11. [Google Scholar] [CrossRef]
- Reita, D.; Pabst, L.; Pencreach, E.; Guérin, E.; Dano, L.; Rimelen, V.; Voegeli, A.-C.; Vallat, L.; Mascaux, C.; Beau-Faller, M. Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers 2021, 13, 4926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sun, B.; Zhong, C.; Xu, K.; Wang, Z.; Hofman, P.; Nagano, T.; Legras, A.; Breadner, D.; Ricciuti, B.; et al. Targeting histone deacetylase enhances the therapeutic effect of Erastin-induced ferroptosis in EGFR-activating mutant lung adenocarcinoma. Transl. Lung Cancer Res. 2021, 10, 1857–1872. [Google Scholar] [CrossRef]
- Deng, S.H.; Wu, D.M.; Li, L.; Liu, T.; Zhang, T.; Li, J.; Yu, Y.; He, M.; Zhao, Y.Y.; Han, R.; et al. miR-324-3p reverses cisplatin resistance by inducing GPX4-mediated ferroptosis in lung adenocarcinoma cell line A549. Biochem. Biophys. Res. Commun. 2021, 549, 54–60. [Google Scholar] [CrossRef]
- Bi, G.; Liang, J.; Zhao, M.; Zhang, H.; Jin, X.; Lu, T.; Zheng, Y.; Bian, Y.; Chen, Z.; Huang, Y.; et al. miR-6077 promotes cisplatin/pemetrexed resistance in lung adenocarcinoma via CDKN1A/cell cycle arrest and KEAP1/ferroptosis pathways. Mol. Ther. Nucleic Acids 2022, 28, 366–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Y.; Wang, Y. Ferroptosis patterns modulate immunocyte communication in tumor microenvironments: Clinical value and therapeutic guidance of lung adenocarcinoma. Funct. Integr. Genom. 2023, 23, 181. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Xiang, J.; Liang, Z.; Luo, L. Identification and validation of a ferroptosis-related gene to predict survival outcomes and the immune microenvironment in lung adenocarcinoma. Cancer Cell Int. 2022, 22, 292. [Google Scholar] [CrossRef]
- Luo, L.; Chen, X.; Huang, F. Machine learning revealed ferroptosis features and ferroptosis-related gene-based immune microenvironment in lung adenocarcinoma. Chem. Biol. Interact. 2023, 378, 110471. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xu, W.; Wang, Y.; Zhu, J.; Wang, H.; Tu, J.; Weng, Q.; Kong, C.; Yang, Y.; Qiu, R.; et al. Identification of critical ferroptosis regulators in lung adenocarcinoma that RRM2 facilitates tumor immune infiltration by inhibiting ferroptotic death. Clin. Immunol. 2021, 232, 108872. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Qu, Z.; Li, D.; Bai, F.; Xing, J.; Ding, Q.; Zhou, J.; Yao, L.; Xu, Q. Identification of a prognostic ferroptosis-related lncRNA signature in the tumor microenvironment of lung adenocarcinoma. Cell Death Discov. 2021, 7, 190. [Google Scholar] [CrossRef]
- Feng, Z.; Li, B.; Chen, Q.; Zhang, H.; Guo, Z.; Qin, J.; Zhang, Z. Identification and Validation of a GPX4-Related Immune Prognostic Signature for Lung Adenocarcinoma. J. Oncol. 2022, 2022, 9054983. [Google Scholar] [CrossRef]
- Jung, K.A.; Choi, B.H.; Nam, C.W.; Song, M.; Kim, S.T.; Lee, J.Y.; Kwak, M.K. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol. Lett. 2013, 218, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Zeh, H.J.; Kang, R.; Bai, L.; Tang, D. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat. Commun. 2020, 11, 6339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Wohlhieter, C.A.; Richards, A.L.; Uddin, F.; Hulton, C.H.; Quintanal-Villalonga, A.; Martin, A.; de Stanchina, E.; Bhanot, U.; Asher, M.; Shah, N.S.; et al. Concurrent Mutations in STK11 and KEAP1 Promote Ferroptosis Protection and SCD1 Dependence in Lung Cancer. Cell Rep. 2020, 33, 108444. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef]
- Zhang, W.; Li, X.; Xu, J.; Wang, Y.; Xing, Z.; Hu, S.; Fan, Q.; Lu, S.; Cheng, J.; Gu, J.; et al. The RSL3 Induction of KLK Lung Adenocarcinoma Cell Ferroptosis by Inhibition of USP11 Activity and the NRF2-GSH Axis. Cancers 2022, 14, 5233. [Google Scholar] [CrossRef]
- Thompson, L.R.; Oliveira, T.G.; Hermann, E.R.; Chowanadisai, W.; Clarke, S.L.; Montgomery, M.R. Distinct TP53 Mutation Types Exhibit Increased Sensitivity to Ferroptosis Independently of Changes in Iron Regulatory Protein Activity. Int. J. Mol. Sci. 2020, 21, 6751. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yu, J.; Yi, Y.; Chen, T.; Yu, L.; Zeng, W.; Ouyang, X.-k.; Huang, C.; Sun, S.; Wang, Y.; et al. Oxidative stress-amplified nanomedicine for intensified ferroptosis-apoptosis combined tumor therapy. J. Control. Release 2022, 347, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cai, Q.; Yang, R.; Wang, H.; Ling, H.; Li, T.; Liu, N.; Wang, Z.; Sun, J.; Tao, T.; et al. GINS4 suppresses ferroptosis by antagonizing p53 acetylation with Snail. Proc. Natl. Acad. Sci. USA 2023, 120, e2219585120. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef]
- Zhu, C.; Li, L.; Zhang, Z.; Bi, M.; Wang, H.; Su, W.; Hernandez, K.; Liu, P.; Chen, J.; Chen, M.; et al. A Non-canonical Role of YAP/TEAD Is Required for Activation of Estrogen-Regulated Enhancers in Breast Cancer. Mol. Cell 2019, 75, 791–806.e8. [Google Scholar] [CrossRef]
- Thomas, C.; Jalil, A.; Magnani, C.; Ishibashi, M.; Quere, R.; Bourgeois, T.; Bergas, V.; Menegaut, L.; Patoli, D.; Le Guern, N.; et al. LPCAT3 deficiency in hematopoietic cells alters cholesterol and phospholipid homeostasis and promotes atherosclerosis. Atherosclerosis 2018, 275, 409–418. [Google Scholar] [CrossRef]
- Wang, T.; Wang, K.; Zhu, X.; Chen, N. ARNTL2 upregulation of ACOT7 promotes NSCLC cell proliferation through inhibition of apoptosis and ferroptosis. BMC Mol. Cell Biol. 2023, 24, 14. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Tian, X.; Yang, Y.; Ma, L.; Wang, J.; Yu, Y. CREB stimulates GPX4 transcription to inhibit ferroptosis in lung adenocarcinoma. Oncol. Rep. 2021, 45, 88. [Google Scholar] [CrossRef]
- Zhao, G.; Liang, J.; Shan, G.; Gu, J.; Xu, F.; Lu, C.; Ma, T.; Bi, G.; Zhan, C.; Ge, D. KLF11 regulates lung adenocarcinoma ferroptosis and chemosensitivity by suppressing GPX4. Commun. Biol. 2023, 6, 570. [Google Scholar] [CrossRef]
- Logie, E.; Van Puyvelde, B.; Cuypers, B.; Schepers, A.; Berghmans, H.; Verdonck, J.; Laukens, K.; Godderis, L.; Dhaenens, M.; Deforce, D.; et al. Ferroptosis Induction in Multiple Myeloma Cells Triggers DNA Methylation and Histone Modification Changes Associated with Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 12234. [Google Scholar] [CrossRef]
- Xu, Y.; Hong, M.; Kong, D.; Deng, J.; Zhong, Z.; Liang, J. Ferroptosis-associated DNA methylation signature predicts overall survival in patients with head and neck squamous cell carcinoma. BMC Genom. 2022, 23, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 2020, 160, 552–565. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.; Sevinc, K.; Gurhan Sevinc, G.; Cribbs, A.P.; Philpott, M.; Uyulur, F.; Morova, T.; Dunford, J.E.; Goklemez, S.; Ari, S.; et al. Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nat. Chem. Biol. 2019, 15, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Weaver, M.R.; Kosmacek, E.A.; O’Connor, B.P.; Harmacek, L.; Venkataraman, S.; Oberley-Deegan, R.E. MnTE-2-PyP reduces prostate cancer growth and metastasis by suppressing p300 activity and p300/HIF-1/CREB binding to the promoter region of the PAI-1 gene. Free Radic. Biol. Med. 2016, 94, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wei, K.; Yang, F.M.; Hu, L.Q.; Pan, C.F.; Pan, X.L.; Wu, W.B.; Wang, J.; Wen, W.; He, Z.C.; et al. Correction: miR-1260b, mediated by YY1, activates KIT signaling by targeting SOCS6 to regulate cell proliferation and apoptosis in NSCLC. Cell Death Dis. 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, N.; Ling, X.; Pan, M.; Du, W.; Gao, S. MiR-27a-3p Promotes Non-Small Cell Lung Cancer Through SLC7A11-Mediated-Ferroptosis. Front. Oncol. 2021, 11, 759346. [Google Scholar] [CrossRef]
- Wei, B.; Kong, W.; Mou, X.; Wang, S. Comprehensive analysis of tumor immune infiltration associated with endogenous competitive RNA networks in lung adenocarcinoma. Pathol. Res. Pract. 2019, 215, 159–170. [Google Scholar] [CrossRef]
- Li, X.; Ding, J.; Wang, X.; Cheng, Z.; Zhu, Q. NUDT21 regulates circRNA cyclization and ceRNA crosstalk in hepatocellular carcinoma. Oncogene 2020, 39, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Duan, Y.; Sang, Y.; Li, Y.; Zhang, H.; Liang, Y.; Liu, Y.; Zhang, N.; Yang, Q. LncRNA-CDC6 promotes breast cancer progression and function as ceRNA to target CDC6 by sponging microRNA-215. J. Cell. Physiol. 2019, 234, 9105–9117. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.F.; Wei, K.; Ma, Z.J.; He, Y.Z.; Huang, J.J.; Guo, Z.Z.; Chen, Z.P.; Barr, M.P.; Shackelford, R.E.; Xia, Y.; et al. CircP4HB regulates ferroptosis via SLC7A11-mediated glutathione synthesis in lung adenocarcinoma. Transl. Lung Cancer Res. 2022, 11, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Y.; Cai, Z.R.; Liu, J.; Wang, D.S.; Ju, H.Q.; Xu, R.H. Circular RNA: Metabolism, functions and interactions with proteins. Mol. Cancer 2020, 19, 172. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, Y.; Ma, L.; Yu, K.; Niu, Y.; Xu, X.; Shi, Y.; Guo, S.; Xue, X.; Wang, Y.; et al. Essential roles of exosome and circRNA_101093 on ferroptosis desensitization in lung adenocarcinoma. Cancer Commun. 2022, 42, 287–313. [Google Scholar] [CrossRef]
- Jiang, X.; Yuan, Y.; Tang, L.; Wang, J.; Zhang, D.; Duan, L. Systematic Analysis and Validation of the Prognosis, Immunological Role and Biology Function of the Ferroptosis-Related lncRNA GSEC/miRNA-101-3p/CISD1 Axis in Lung Adenocarcinoma. Front. Mol. Biosci. 2021, 8, 793732. [Google Scholar] [CrossRef]
- Zhang, N.; Huang, J.; Xu, M.; Wang, Y. LncRNA T-UCR Uc.339/miR-339/SLC7A11 Axis Regulates the Metastasis of Ferroptosis-Induced Lung Adenocarcinoma. J. Cancer 2022, 13, 1945–1957. [Google Scholar] [CrossRef]
- Wang, M.; Mao, C.; Ouyang, L.; Liu, Y.; Lai, W.; Liu, N.; Shi, Y.; Chen, L.; Xiao, D.; Yu, F.; et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019, 26, 2329–2343. [Google Scholar] [CrossRef]
- Peng, X.; Yang, R.; Peng, W.; Zhao, Z.; Tu, G.; He, B.; Cai, Q.; Shi, S.; Yin, W.; Yu, F.; et al. Overexpression of LINC00551 promotes autophagy-dependent ferroptosis of lung adenocarcinoma via upregulating DDIT4 by sponging miR-4328. PeerJ 2022, 10, e14180. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Huttelmaier, S.; Liu, T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef]
- Song, Z.; Jia, G.; Ma, P.; Cang, S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021, 276, 119399. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lv, D.; Yan, C.; Su, H.; Zhang, X.; Shi, Y.; Ying, K. METTL3 promotes lung adenocarcinoma tumor growth and inhibits ferroptosis by stabilizing SLC7A11 m(6)A modification. Cancer Cell Int. 2022, 22, 11. [Google Scholar] [CrossRef]
- Xu, X.; Cui, J.; Wang, H.; Ma, L.; Zhang, X.; Guo, W.; Xue, X.; Wang, Y.; Qiu, S.; Tian, X.; et al. IGF2BP3 is an essential N(6)-methyladenosine biotarget for suppressing ferroptosis in lung adenocarcinoma cells. Mater. Today Bio 2022, 17, 100503. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, X.; Yu, K.; Xu, X.; Chen, T.; Shi, Y.; Wang, Y.; Qiu, S.; Guo, S.; Cui, J.; et al. Targeting SLC3A2 subunit of system X(C)(-) is essential for m(6)A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic. Biol. Med. 2021, 168, 25–43. [Google Scholar] [CrossRef]
- He, H.; Liang, L.; Huang, J.; Jiang, S.; Liu, Y.; Sun, X.; Li, Y.; Cong, L.; Jiang, Y. KIF20A is associated with clinical prognosis and synergistic effect of gemcitabine combined with ferroptosis inducer in lung adenocarcinoma. Front. Pharmacol. 2022, 13, 1007429. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib Induces Ferroptosis in Human Cancer Cell Lines Originating from Different Solid Tumors. Anticancer Res. 2014, 34, 6417. [Google Scholar]
- Mo, X.; Hu, D.; Yuan, K.; Luo, J.; Huang, C.; Xu, M. Tetrandrine citrate suppresses lung adenocarcinoma growth via SLC7A11/GPX4-mediated ferroptosis. Discov. Oncol. 2023, 14, 85. [Google Scholar] [CrossRef]
- Huang, F.; Pang, J.; Xu, L.; Niu, W.; Zhang, Y.; Li, S.; Li, X. Hedyotis diffusa injection induces ferroptosis via the Bax/Bcl2/VDAC2/3 axis in lung adenocarcinoma. Phytomedicine 2022, 104, 154319. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.S.; Zhao, L.P.; Huang, Z.H.; Chen, X.Y.; Xu, J.T.; Tai, W.C.; Tsim, K.W.K.; Chen, Y.T.; Xie, T. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine 2021, 80, 153370. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fu, F.; Huang, Z.; Wang, W.; Chen, M.; Yue, X.; Fu, J.; Feng, X.; Huang, Y.; Wu, C.; et al. Inhalable Biomimetic Protein Corona-Mediated Nanoreactor for Self-Amplified Lung Adenocarcinoma Ferroptosis Therapy. ACS Nano 2022, 16, 8370–8387. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.; He, X.; Zeng, N. Tetrandrine: A review of its anticancer potentials, clinical settings, pharmacokinetics and drug delivery systems. J. Pharm. Pharmacol. 2020, 72, 1491–1512. [Google Scholar] [CrossRef] [PubMed]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lipper, C.H.; Stofleth, J.T.; Bai, F.; Sohn, Y.S.; Roy, S.; Mittler, R.; Nechushtai, R.; Onuchic, J.N.; Jennings, P.A. Redox-dependent gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. USA 2019, 116, 19924–19929. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.; Li, X.; Hu, S.; Cheng, J.; Cai, R. Regulation of Ferroptosis in Lung Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 14614. https://doi.org/10.3390/ijms241914614
Wei X, Li X, Hu S, Cheng J, Cai R. Regulation of Ferroptosis in Lung Adenocarcinoma. International Journal of Molecular Sciences. 2023; 24(19):14614. https://doi.org/10.3390/ijms241914614
Chicago/Turabian StyleWei, Xiangyun, Xiaohe Li, Shuming Hu, Jinke Cheng, and Rong Cai. 2023. "Regulation of Ferroptosis in Lung Adenocarcinoma" International Journal of Molecular Sciences 24, no. 19: 14614. https://doi.org/10.3390/ijms241914614
APA StyleWei, X., Li, X., Hu, S., Cheng, J., & Cai, R. (2023). Regulation of Ferroptosis in Lung Adenocarcinoma. International Journal of Molecular Sciences, 24(19), 14614. https://doi.org/10.3390/ijms241914614