
A Blue Light-Responsive Strong Synthetic Promoter Based on Rational Design in Chlamydomonas reinhardtii


Abstract
:1. Introduction
2. Results
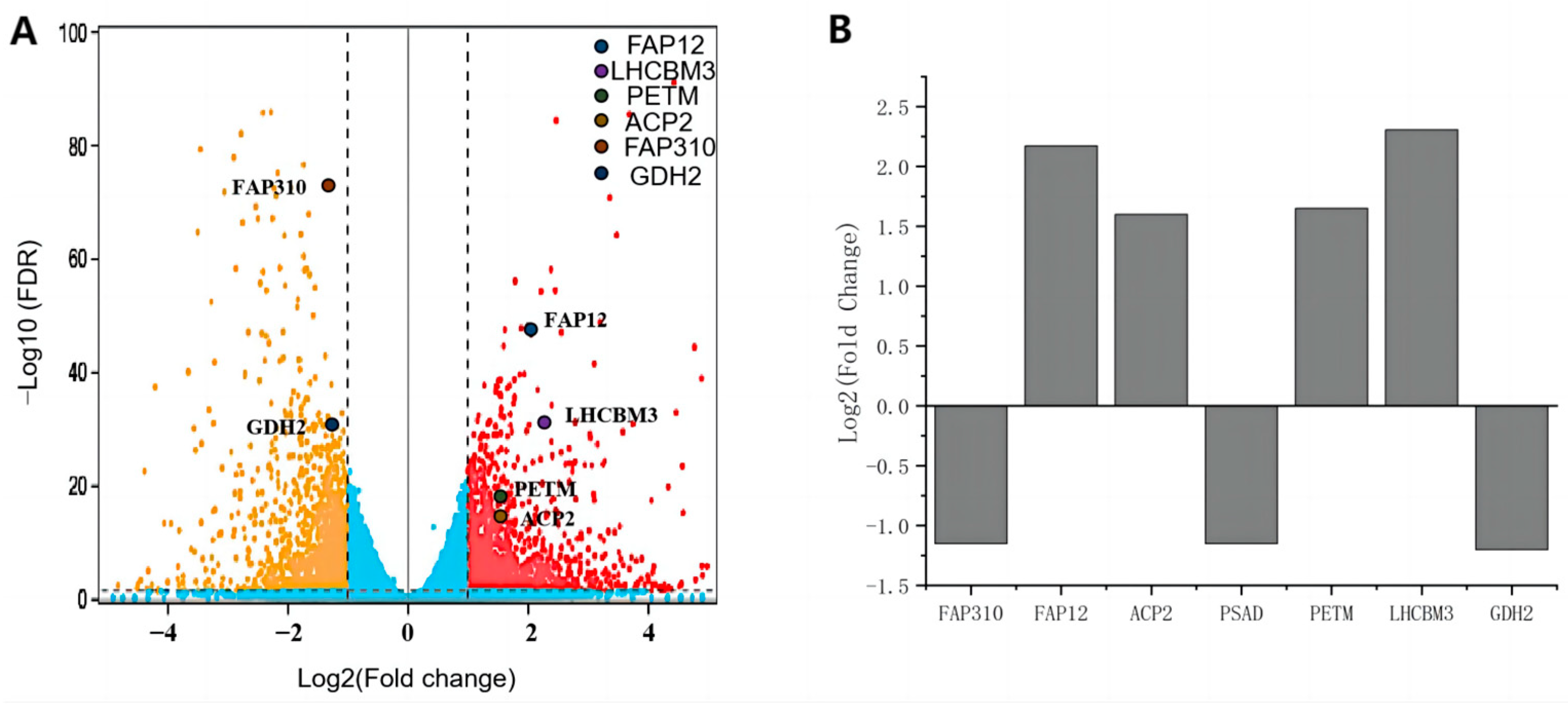
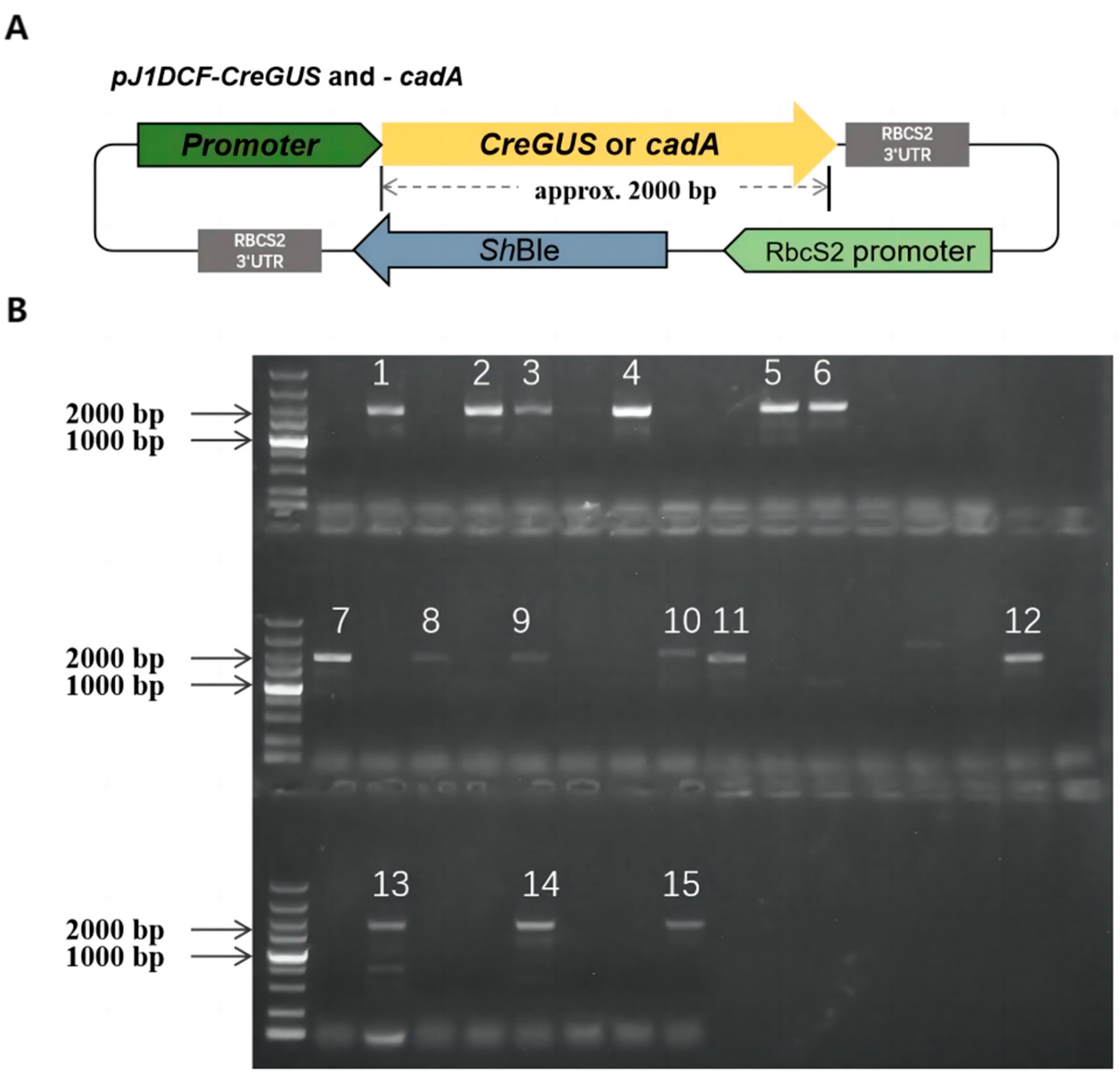
2.1. Native Promoter Identification and Construction of Transgenic Lines
2.2. Characterization of Native Promoters in CreGUS Expression
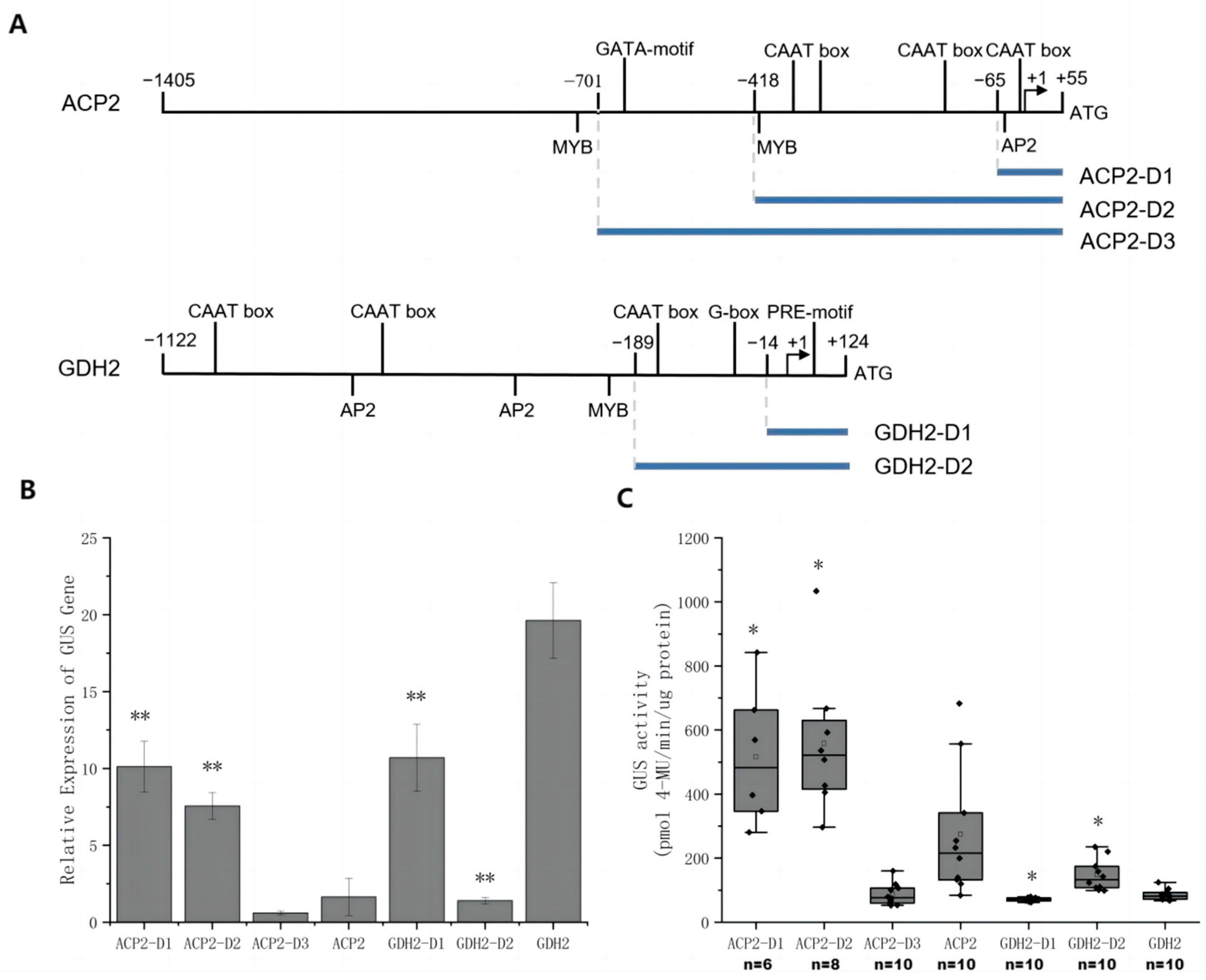
2.3. Promoter Truncation and Characterization of Promoter Fragments
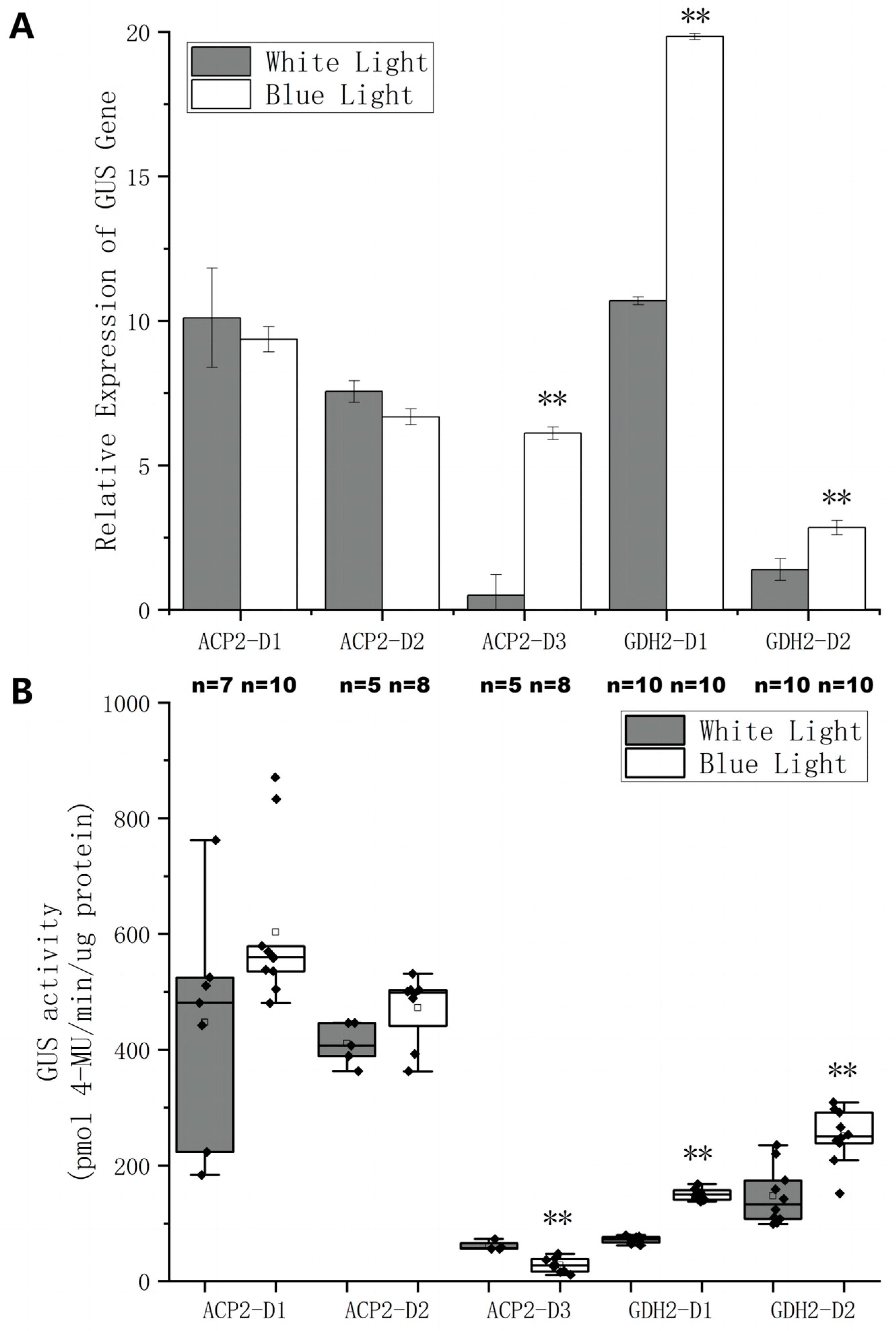
2.4. Construction and Characterization of Chimeric Promoters
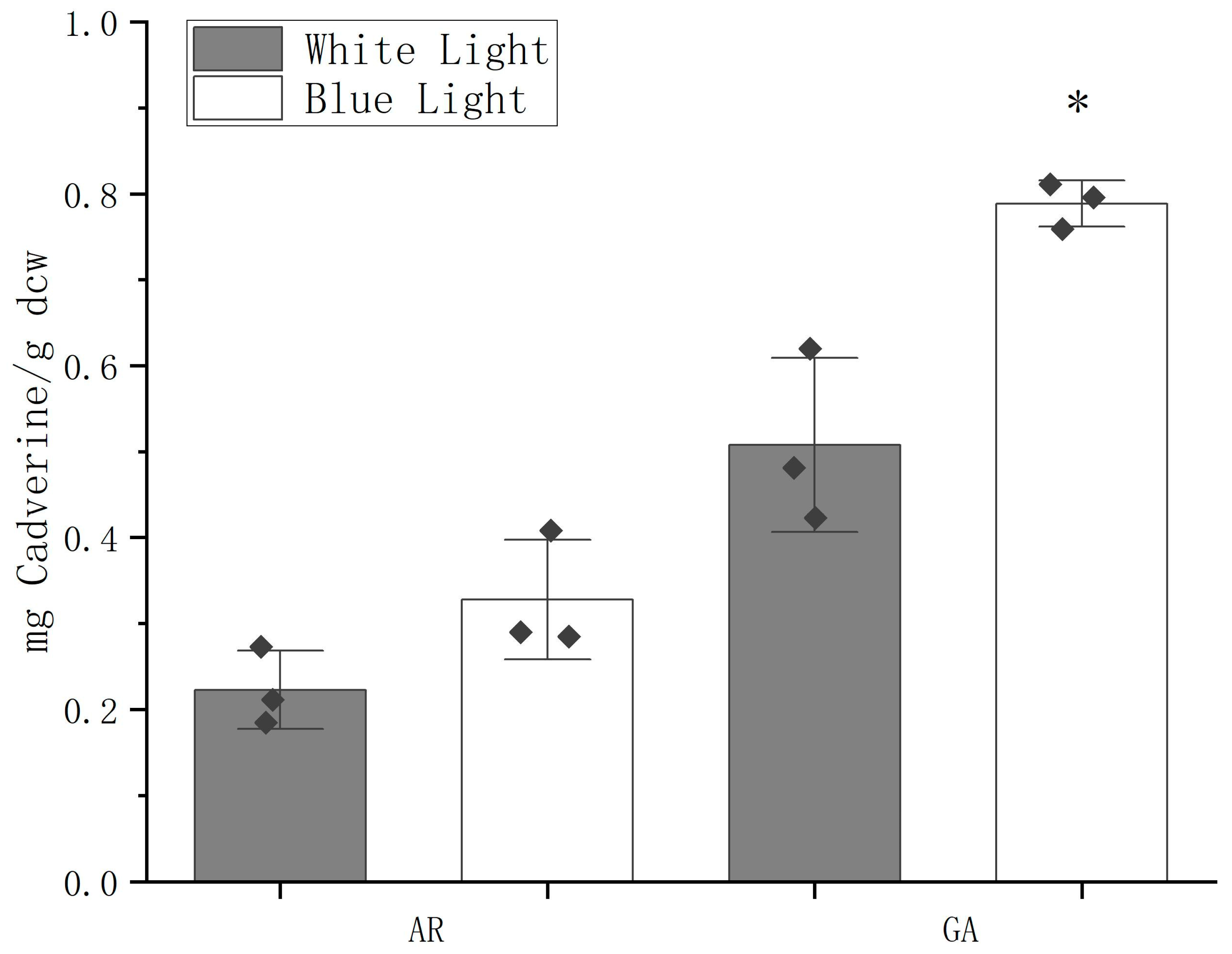
2.5. Production of Cadaverine Using GDH2-D1/ACP2-D1 Promoter
3. Discussion
4. Materials and Methods
4.1. C. reinhardtii Culture
4.2. Plasmid Construction
4.3. C. reinhardtii Transformation and Transgenic Strains
4.4. RNA Isolation and qRT-PCR
4.5. GUS Activity Quantitative Assay
4.6. Promoter Regulatory Sequence Analysis and Promoter Fragments
4.7. Cadaverine Measurement
4.8. RNA-Seq Transcriptomics
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, E.H. The Chlamydomonas Sourcebook: Introduction to Chlamydomonas and Its Laboratory Use: Volume 1; Academic Press: Cambridge, MA, USA, 2009; ISBN 978-0-08-091955-3. [Google Scholar]
- Salomé, P.A.; Merchant, S.S. A Series of Fortunate Events: Introducing Chlamydomonas as a Reference Organism. Plant Cell 2019, 31, 1682–1707. [Google Scholar] [CrossRef] [PubMed]
- León-Bañares, R.; González-Ballester, D.; Galván, A.; Fernández, E. Transgenic Microalgae as Green Cell-Factories. Trends Biotechnol. 2004, 22, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Rasala, B.A.; Mayfield, S.P. Photosynthetic Biomanufacturing in Green Algae; Production of Recombinant Proteins for Industrial, Nutritional, and Medical Uses. Photosynth. Res. 2015, 123, 227–239. [Google Scholar] [CrossRef]
- Nouemssi, S.B.; Ghribi, M.; Beauchemin, R.; Meddeb-Mouelhi, F.; Germain, H.; Desgagné-Penix, I. Rapid and Efficient Colony-PCR for High Throughput Screening of Genetically Transformed Chlamydomonas reinhardtii. Life 2020, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Almaraz-Delgado, A.L.; Flores-Uribe, J.; Pérez-España, V.H.; Salgado-Manjarrez, E.; Badillo-Corona, J.A. Production of Therapeutic Proteins in the Chloroplast of Chlamydomonas reinhardtii. AMB Express 2014, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Scranton, M.A.; Ostrand, J.T.; Fields, F.J.; Mayfield, S.P. Chlamydomonas as a Model for Biofuels and Bio-Products Production. Plant J. Cell Mol. Biol. 2015, 82, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-P.; Wang, M.; Wang, C. Nuclear Transformation of Chlamydomonas reinhardtii: A Review. Biochimie 2021, 181, 1–11. [Google Scholar] [CrossRef]
- Nour-Eldin, H.H.; Specht, E.A.; Ostrand, J.; Hoang, K.T.D.; Karunanithi, P.S.; Mayfield, S.P. High-Throughput System for Quantifying and Characterizing Homologous Recombination in Chlamydomonas reinhardtii. Algal Res. 2018, 31, 167–172. [Google Scholar] [CrossRef]
- Schroda, M. RNA Silencing in Chlamydomonas: Mechanisms and Tools. Curr. Genet. 2006, 49, 69–84. [Google Scholar] [CrossRef]
- Casas-Mollano, J.A.; Rohr, J.; Kim, E.-J.; Balassa, E.; van Dijk, K.; Cerutti, H. Diversification of the Core RNA Interference Machinery in Chlamydomonas reinhardtii and the Role of DCL1 in Transposon Silencing. Genetics 2008, 179, 69–81. [Google Scholar] [CrossRef]
- Wu-Scharf, D.; Jeong, B.; Zhang, C.; Cerutti, H. Transgene and Transposon Silencing in Chlamydomonas reinhardtii by a DEAH-Box RNA Helicase. Science 2000, 290, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu-Scharf, D.; Jeong, B.; Cerutti, H. A WD40-Repeat Containing Protein, Similar to a Fungal Co-Repressor, Is Required for Transcriptional Gene Silencing in Chlamydomonas. Plant J. 2002, 31, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Rombauts, S.; Florquin, K.; Lescot, M.; Marchal, K.; Rouzé, P.; van de Peer, Y. Computational Approaches to Identify Promoters and Cis-Regulatory Elements in Plant Genomes. Plant Physiol. 2003, 132, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J. What Have We Learned About Synthetic Promoter Construction? In Plant Synthetic Promoters: Methods and Protocols; Hehl, R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 1–13. ISBN 978-1-4939-6396-6. [Google Scholar]
- Venter, M. Synthetic Promoters: Genetic Control through Cis Engineering. Trends Plant Sci. 2007, 12, 118–124. [Google Scholar] [CrossRef]
- Sauret-Güeto, S.; Frangedakis, E.; Silvestri, L.; Rebmann, M.; Tomaselli, M.; Markel, K.; Delmans, M.; West, A.; Patron, N.J.; Haseloff, J. Systematic Tools for Reprogramming Plant Gene Expression in a Simple Model, Marchantia Polymorpha. ACS Synth. Biol. 2020, 9, 864–882. [Google Scholar] [CrossRef]
- Hou, L.; Chen, L.; Wang, J.; Xu, D.; Dai, L.; Zhang, H.; Zhao, Y. Construction of Stress Responsive Synthetic Promoters and Analysis of Their Activity in Transgenic Arabidopsis Thaliana. Plant Mol. Biol. Report. 2012, 30, 1496–1506. [Google Scholar] [CrossRef]
- Rosales-Mendoza, S.; Solís-Andrade, K.I.; Márquez-Escobar, V.A.; González-Ortega, O.; Bañuelos-Hernandez, B. Current Advances in the Algae-Made Biopharmaceuticals Field. Expert Opin. Biol. Ther. 2020, 20, 751–766. [Google Scholar] [CrossRef]
- Liu, W.; Yuan, J.S.; Stewart, C.N., Jr. Advanced Genetic Tools for Plant Biotechnology. Nat. Rev. Genet. 2013, 14, 781–793. [Google Scholar] [CrossRef]
- Mithra, S.V.A.; Kulkarni, K.; Srinivasan, R. Plant Promoters: Characterization and Applications in Transgenic Technology. In Plant Biotechnology: Principles and Applications; Abdin, M.Z., Kiran, U., Kamaluddin, Ali, A., Eds.; Springer: Singapore, 2017; pp. 117–172. ISBN 978-981-10-2961-5. [Google Scholar]
- Fischer, N.; Rochaix, J.-D. The Flanking Regions of PsaD Drive Efficient Gene Expression in the Nucleus of the Green Alga Chlamydomonas reinhardtii. Mol. Genet. Genom. 2001, 265, 888–894. [Google Scholar] [CrossRef]
- Schroda, M.; Blöcker, D.; Beck, C.F. The HSP70A Promoter as a Tool for the Improved Expression of Transgenes in Chlamydomonas. Plant J. 2000, 21, 121–131. [Google Scholar] [CrossRef]
- Wu, J.; Hu, Z.; Wang, C.; Li, S.; Lei, A. Efficient Expression of Green Fluorescent Protein (GFP) Mediated by a Chimeric Promoter in Chlamydomonas reinhardtii. Chin. J. Oceanol. Limnol. 2008, 26, 242–247. [Google Scholar] [CrossRef]
- López-Paz, C.; Liu, D.; Geng, S.; Umen, J.G. Identification of Chlamydomonas reinhardtii Endogenous Genic Flanking Sequences for Improved Transgene Expression. Plant J. Cell Mol. Biol. 2017, 92, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Milito, A.; Aschern, M.; McQuillan, J.L.; Yang, J.-S. Challenges and Advances towards the Rational Design of Microalgal Synthetic Promoters in Chlamydomonas reinhardtii. J. Exp. Bot. 2023, 74, erad100. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.-M.; Kallam, K.; Tidd, H.; Gendarini, G.; Salzman, A.; Patron, N.J. Rational Design of Minimal Synthetic Promoters for Plants. Nucleic Acids Res. 2020, 48, 11845–11856. [Google Scholar] [CrossRef] [PubMed]
- Kotopka, B.J.; Smolke, C.D. Model-Driven Generation of Artificial Yeast Promoters. Nat. Commun. 2020, 11, 2113. [Google Scholar] [CrossRef]
- Scranton, M.A.; Ostrand, J.T.; Georgianna, D.R.; Lofgren, S.M.; Li, D.; Ellis, R.C.; Carruthers, D.N.; Dräger, A.; Masica, D.L.; Mayfield, S.P. Synthetic Promoters Capable of Driving Robust Nuclear Gene Expression in the Green Alga Chlamydomonas reinhardtii. Algal Res. 2016, 15, 135–142. [Google Scholar] [CrossRef]
- Baek, K.; Lee, Y.; Nam, O.; Park, S.; Sim, S.J.; Jin, E. Introducing Dunaliella LIP Promoter Containing Light-Inducible Motifs Improves Transgenic Expression in Chlamydomonas reinhardtii. Biotechnol. J. 2016, 11, 384–392. [Google Scholar] [CrossRef]
- Yasmeen, E.; Wang, J.; Riaz, M.; Zhang, L.; Zuo, K. Designing Artificial Synthetic Promoters for Accurate, Smart, and Versatile Gene Expression in Plants. Plant Commun. 2023, 4, 100558. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Sebastian, A. FootprintDB: Analysis of Plant Cis-Regulatory Elements, Transcription Factors, and Binding Interfaces. Methods Mol. Biol. Clifton NJ 2016, 1482, 259–277. [Google Scholar] [CrossRef]
- Hong, C.K.Y.; Cohen, B.A. Genomic Environments Scale the Activities of Diverse Core Promoters. Genome Res. 2022, 32, 85–96. [Google Scholar] [CrossRef]
- Brückner, K.; Schäfer, P.; Weber, E.; Grützner, R.; Marillonnet, S.; Tissier, A. A Library of Synthetic Transcription Activator-like Effector-Activated Promoters for Coordinated Orthogonal Gene Expression in Plants. Plant J. 2015, 82, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, E.; Komatsu, K.R.; Saito, H. Large-Scale Analysis of RNA-Protein Interactions for Functional RNA Motif Discovery Using FOREST. Methods Mol. Biol. Clifton NJ 2022, 2509, 279–290. [Google Scholar] [CrossRef]
- Zhao, M.; Yuan, Z.; Wu, L.; Zhou, S.; Deng, Y. Precise Prediction of Promoter Strength Based on a De Novo Synthetic Promoter Library Coupled with Machine Learning. ACS Synth. Biol. 2022, 11, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Riethoven, J.-J.M. Regulatory Regions in DNA: Promoters, Enhancers, Silencers, and Insulators. Methods Mol. Biol. Clifton NJ 2010, 674, 33–42. [Google Scholar] [CrossRef]
- Aysha, J.; Noman, M.; Wang, F.; Liu, W.; Zhou, Y.; Li, H.; Li, X. Synthetic Promoters: Designing the Cis Regulatory Modules for Controlled Gene Expression. Mol. Biotechnol. 2018, 60, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and Validation of Promoters and Cis-Acting Regulatory Elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef]
- Sawyer, A.L.; Hankamer, B.D.; Ross, I.L. Sulphur Responsiveness of the Chlamydomonas reinhardtii LHCBM9 Promoter. Planta 2015, 241, 1287–1302. [Google Scholar] [CrossRef]
- Iwai, M.; Ikeda, K.; Shimojima, M.; Ohta, H. Enhancement of Extraplastidic Oil Synthesis in Chlamydomonas reinhardtii Using a Type-2 Diacylglycerol Acyltransferase with a Phosphorus Starvation–Inducible Promoter. Plant Biotechnol. J. 2014, 12, 808–819. [Google Scholar] [CrossRef]
- Abe, K.; Miyake, K.; Nakamura, M.; Kojima, K.; Ferri, S.; Ikebukuro, K.; Sode, K. Engineering of a Green-Light Inducible Gene Expression System in Synechocystis Sp. PCC6803. Microb. Biotechnol. 2014, 7, 177–183. [Google Scholar] [CrossRef]
- Jefferson, R.A.; Burgess, S.M.; Hirsh, D. Beta-Glucuronidase from Escherichia Coli as a Gene-Fusion Marker. Proc. Natl. Acad. Sci. USA 1986, 83, 8447–8451. [Google Scholar] [CrossRef]
- Freudenberg, R.A. High Cell Density Cultivation Enables Efficient and Sustainable Recombinant Polyamine Production in the Microalga Chlamydomonas reinhardtii. Bioresour. Technol. 2021, 323, 124542. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, R.A.; Kavanagh, T.A.; Bevan, M.W. GUS Fusions: Beta-Glucuronidase as a Sensitive and Versatile Gene Fusion Marker in Higher Plants. EMBO J. 1987, 6, 3901–3907. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Code | Promoter Length (bp) | Name | Symbol |
|---|---|---|---|
| CHLRE_02g077800v5 | 1083 | Flagellar Associated Protein 310 | FAP310 |
| CHLRE_09g390615v5 | 2143 | Flagellar Associated Protein 12 | FAP12 |
| 461 | Hybrid of HSP70A and RBCS2 promoter from C. reinhardtii | AR | |
| CHLRE_13g577100v5 | 1460 | Acyl-carrier protein 2 | ACP2 |
| CHLRE_05g238332v5 | 822 | Photosystem I reaction center subunit II, 20 kDa | PSAD |
| CHLRE_12g546150v5 | 1088 | Chloroplast cytochrome b6f PetM subunit | PETM |
| CHLRE_04g232104v5 | 1542 | Light-harvesting complex II chlorophyll a/b binding protein M3 | LHCBM3 |
| CHLRE_05g232150v5 | 1246 | Glutamate dehydrogenase | GDH2 |
| Promoter Name | Length (bp) |
|---|---|
| ACP2-D1 | 120 |
| ACP2-D2 | 473 |
| ACP2-D3 | 756 |
| GDH2-D1 | 138 |
| GDH2-D2 | 213 |
| GDH2-D1 + ACP2-D1 (GA) | 258 |
| GDH2-D2 + ACP2-D1 | 333 |
| HSP70A + ACP2-D1 | 383 |
| PSAD + ACP2-D1 | 942 |
| GDH2-D1 + PSAD | 960 |
| GDH2-D1 + ACP2-D2 | 611 |
| Time (min) | A% (Water) | B% (Methanol) |
|---|---|---|
| 0 | 45 | 55 |
| 3 | 38 | 62 |
| 5 | 32 | 68 |
| 9 | 15 | 85 |
| 12 | 12 | 88 |
| 13 | 0 | 100 |
| 15 | 0 | 100 |
| 17 | 45 | 55 |
| 19 | 45 | 55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Chen, J.; Wu, G.; Li, L.; Hu, Z.; Li, X. A Blue Light-Responsive Strong Synthetic Promoter Based on Rational Design in Chlamydomonas reinhardtii. Int. J. Mol. Sci. 2023, 24, 14596. https://doi.org/10.3390/ijms241914596
Chen C, Chen J, Wu G, Li L, Hu Z, Li X. A Blue Light-Responsive Strong Synthetic Promoter Based on Rational Design in Chlamydomonas reinhardtii. International Journal of Molecular Sciences. 2023; 24(19):14596. https://doi.org/10.3390/ijms241914596
Chicago/Turabian StyleChen, Chen, Jun Chen, Guangxi Wu, Liling Li, Zhangli Hu, and Xiaozheng Li. 2023. "A Blue Light-Responsive Strong Synthetic Promoter Based on Rational Design in Chlamydomonas reinhardtii" International Journal of Molecular Sciences 24, no. 19: 14596. https://doi.org/10.3390/ijms241914596
APA StyleChen, C., Chen, J., Wu, G., Li, L., Hu, Z., & Li, X. (2023). A Blue Light-Responsive Strong Synthetic Promoter Based on Rational Design in Chlamydomonas reinhardtii. International Journal of Molecular Sciences, 24(19), 14596. https://doi.org/10.3390/ijms241914596