
Disruption of Z-Disc Function Promotes Mechanical Dysfunction in Human Myocardium: Evidence for a Dual Myofilament Modulatory Role by Alpha-Actinin 2

 ,
,  , , ,
, , ,  , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Results
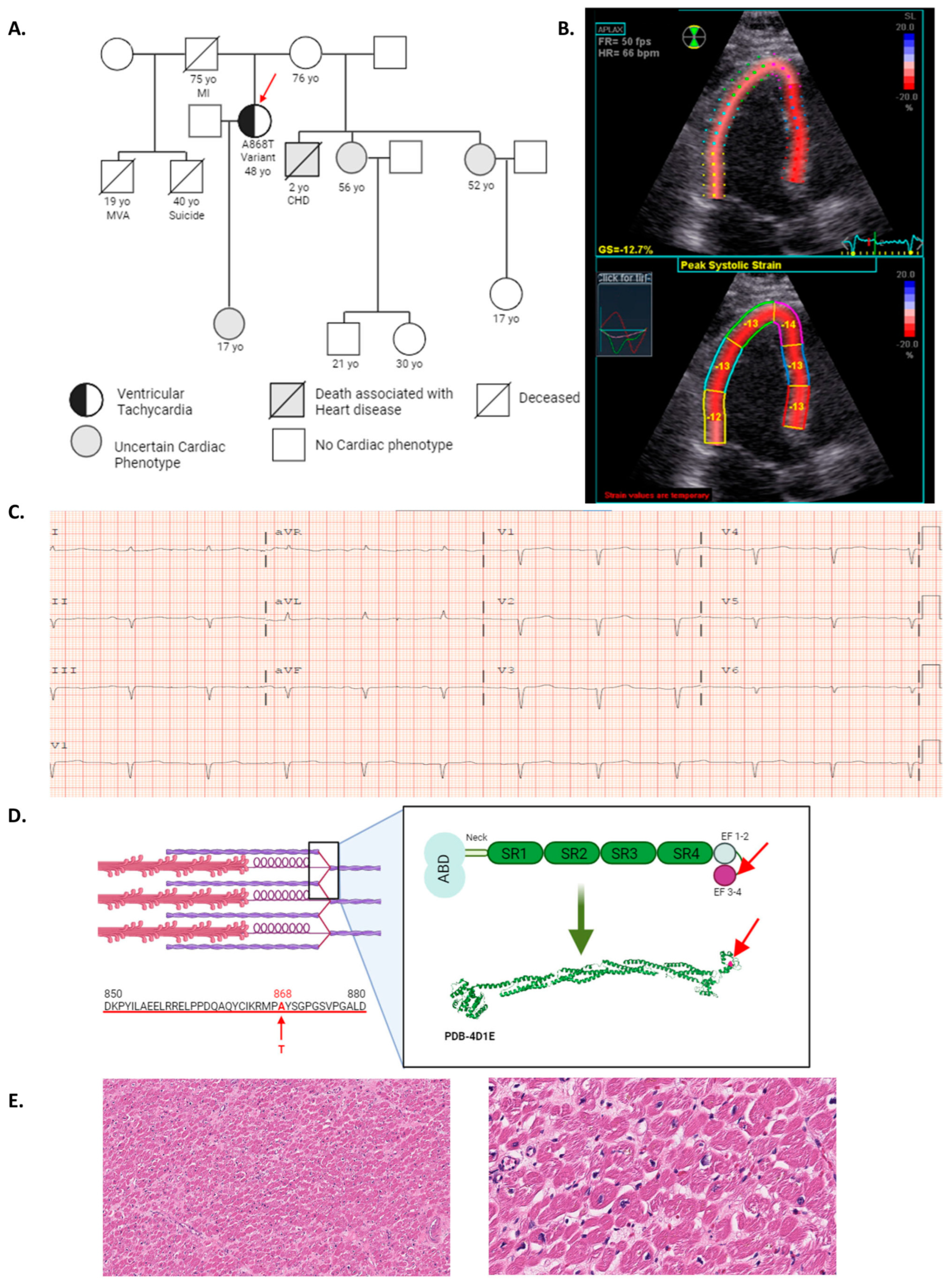
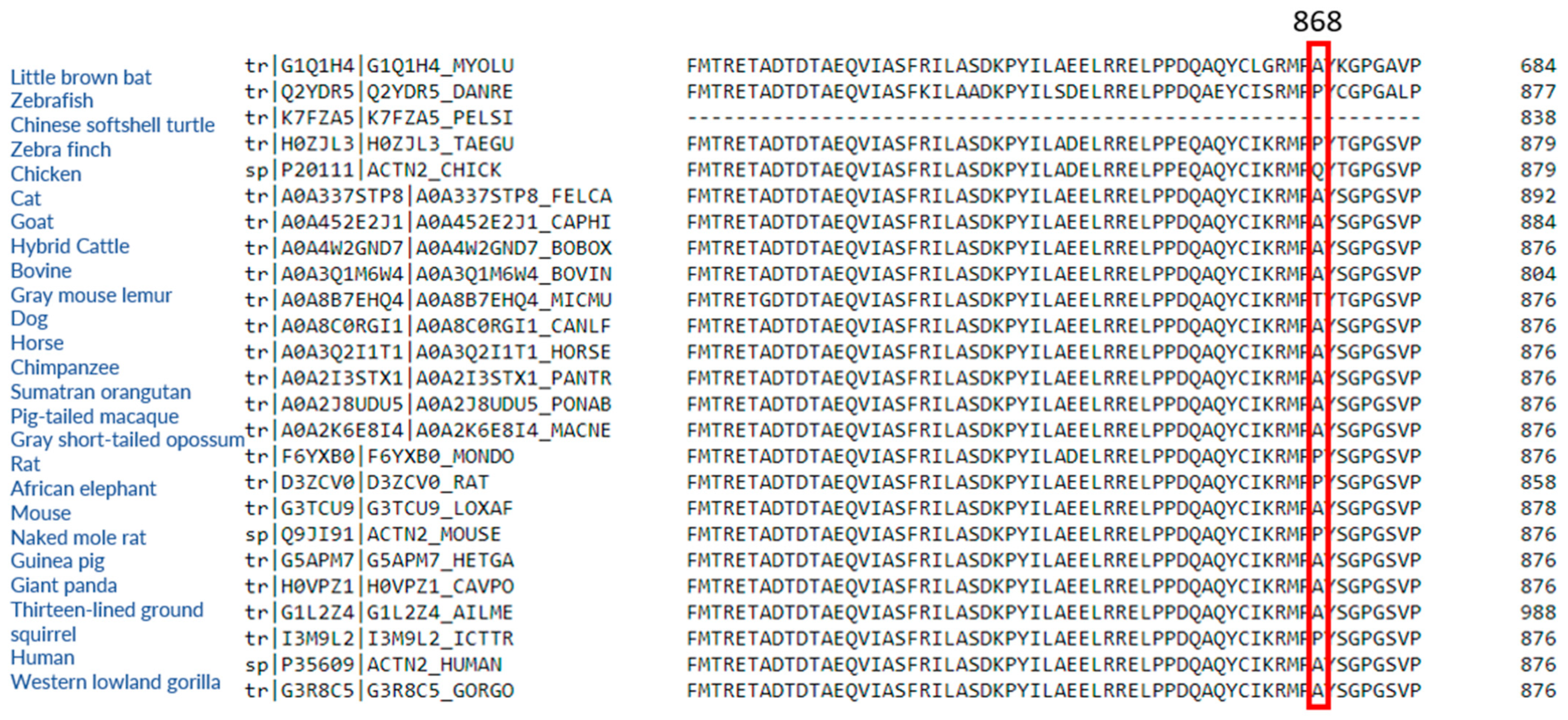
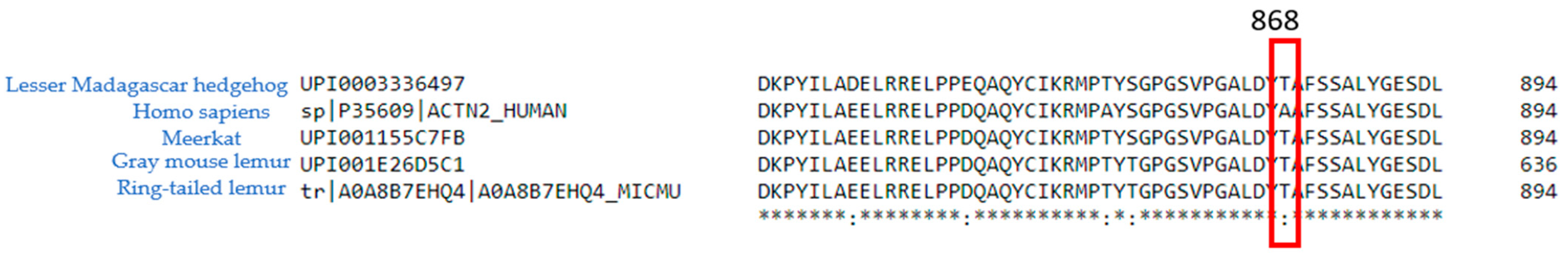
2.1. ACTN2 A868T Variant as a Candidate for Cardiac Disease Etiology
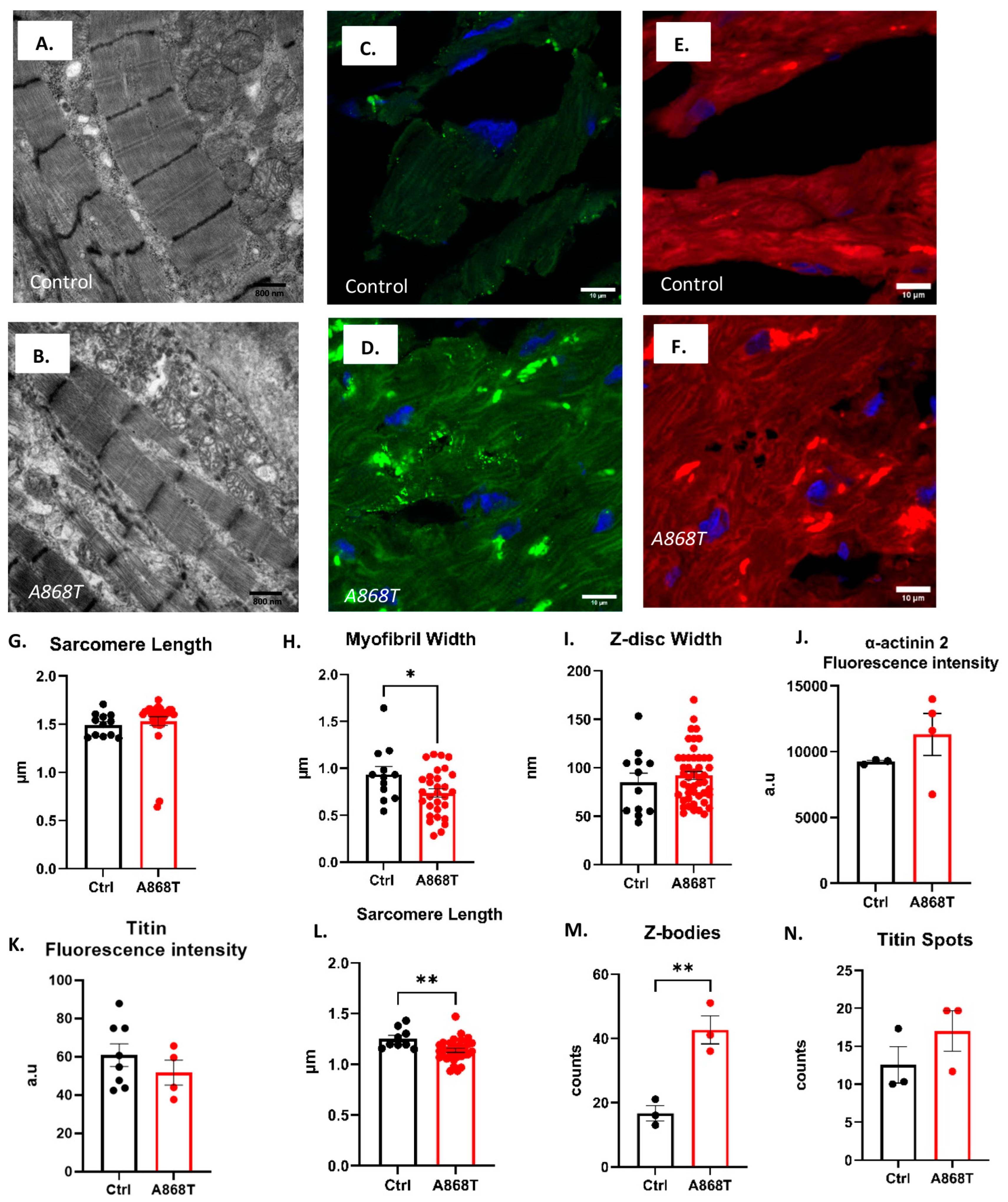
2.2. Characterization of Sarcomere Alterations in the Patient’s Heart
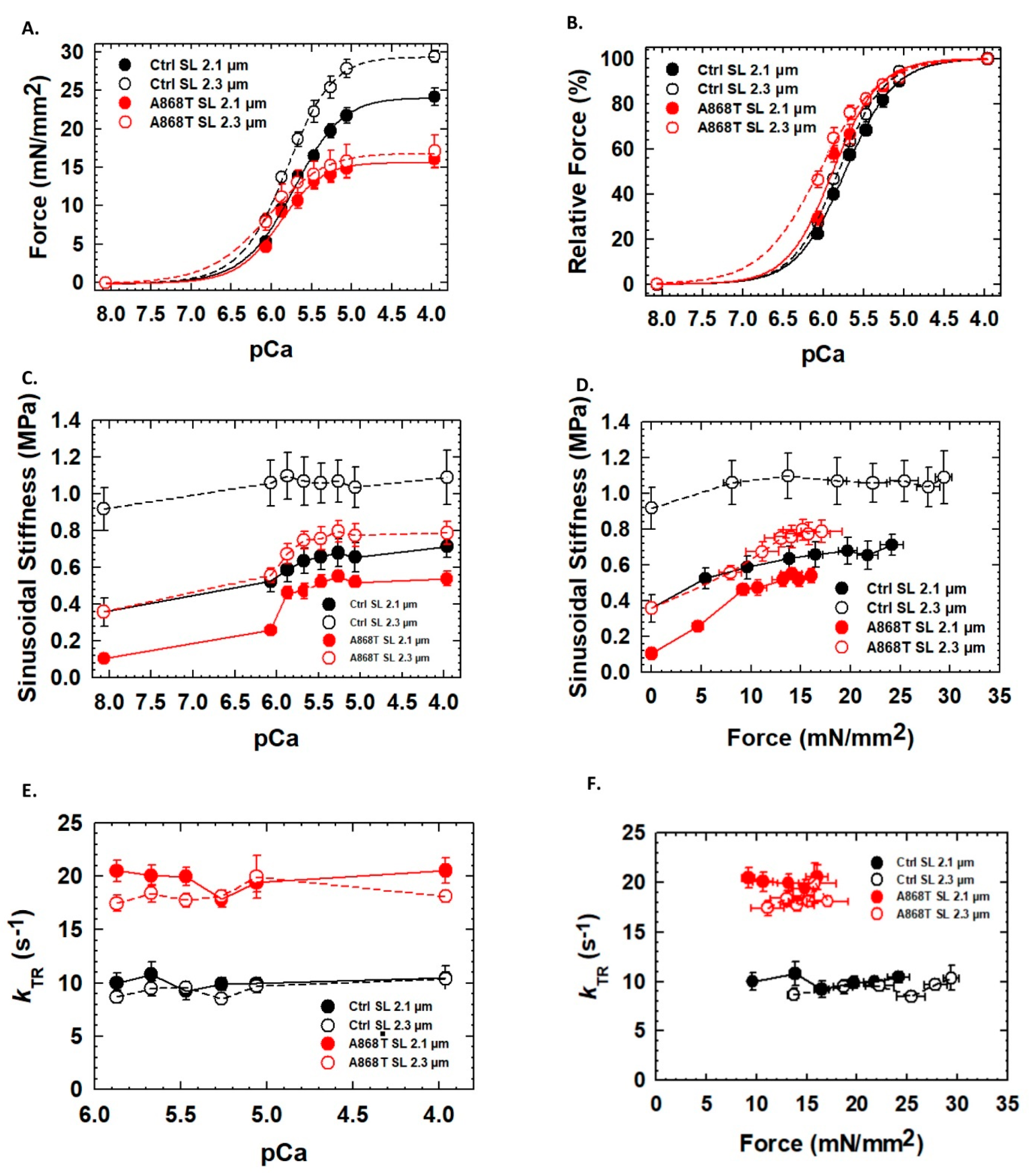
2.3. Abnormal Mechanics of Contraction in Cardiac Muscle Preparations Obtained from the Patient’s Heart
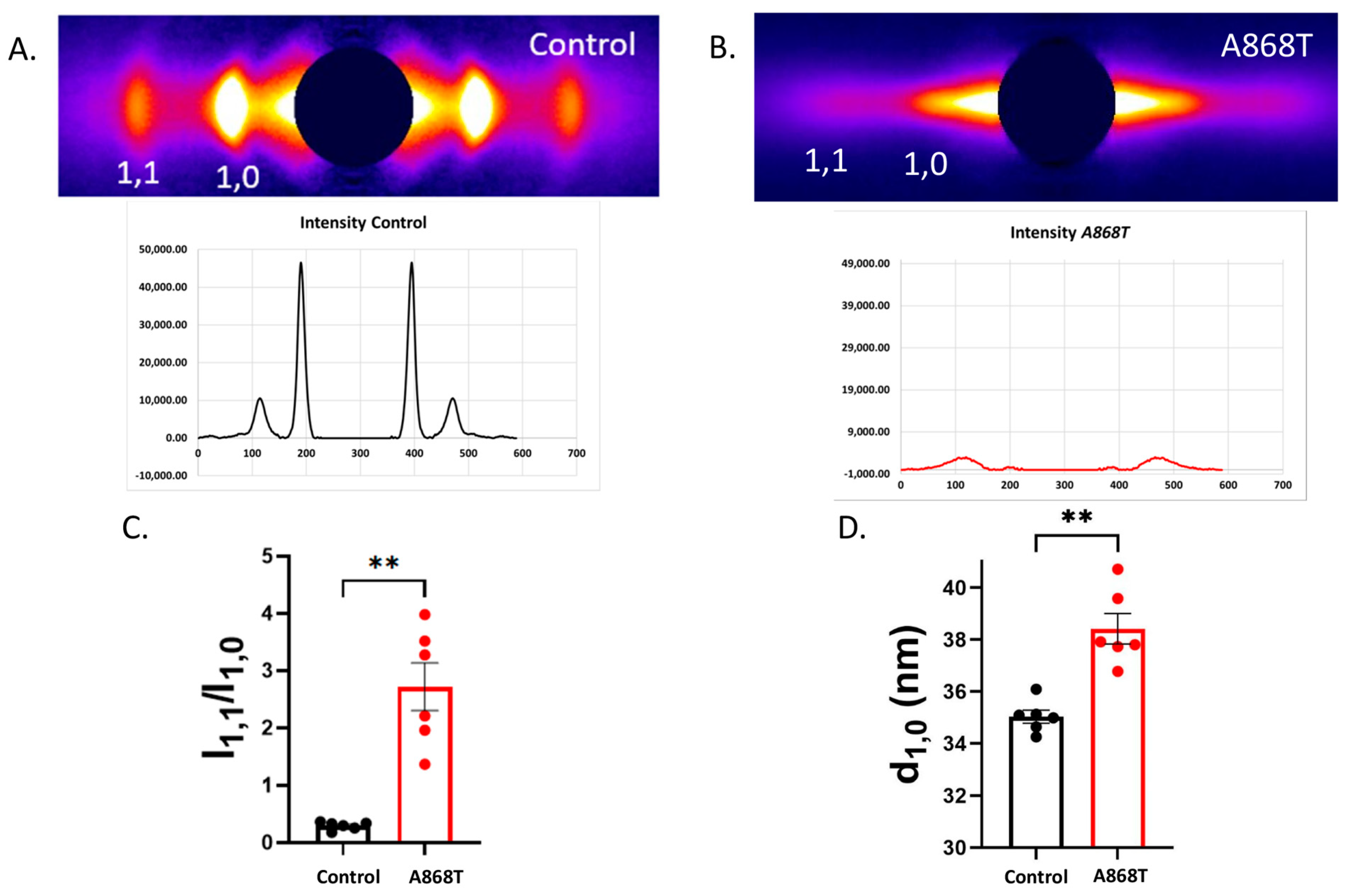
2.4. Cardiac Muscle Preparations Obtained from the Patient’s Heart Have Larger Inter-Filament Lattice Spacing
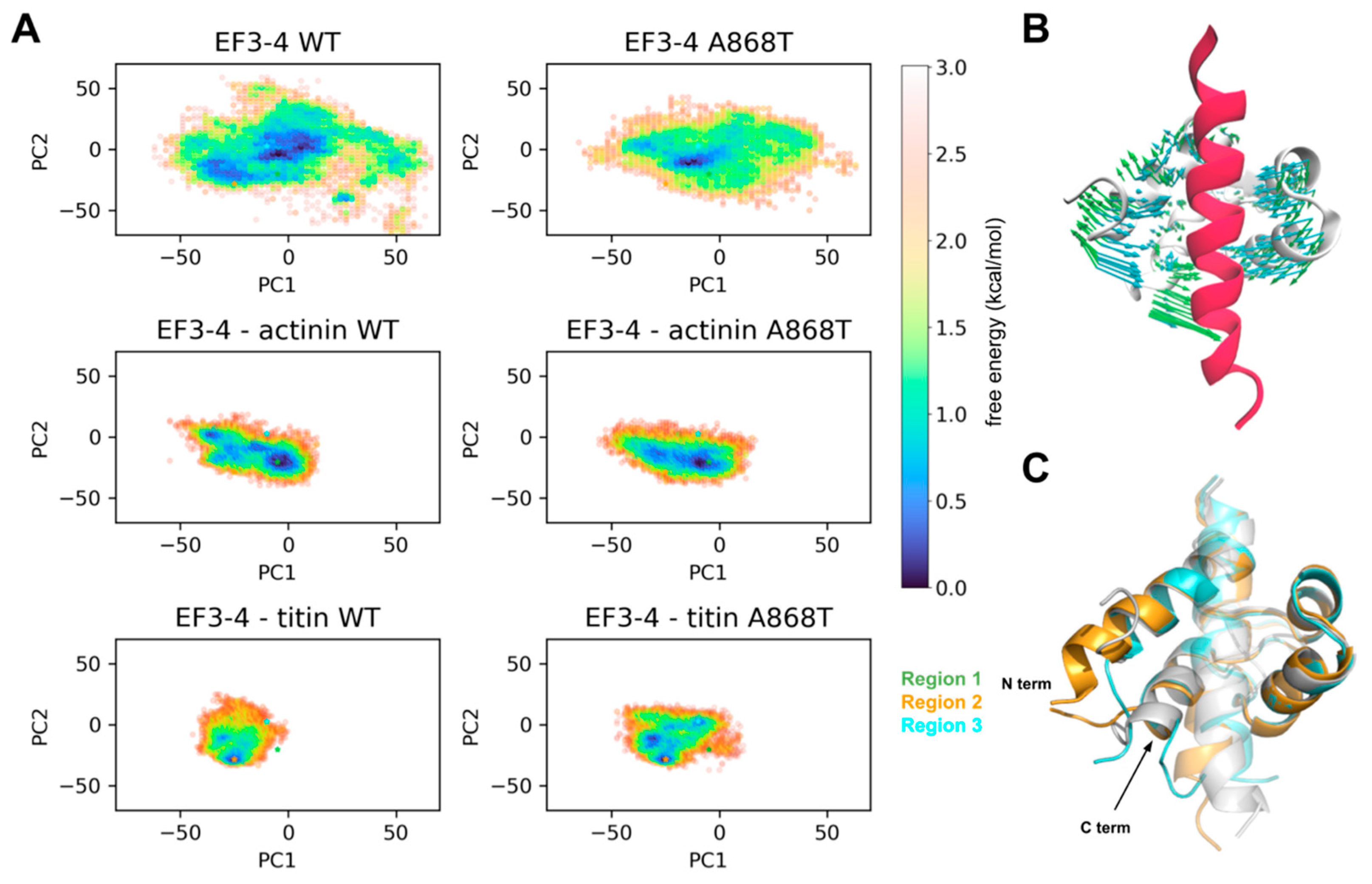
2.5. Molecular Interactions of ACTN2 A868T Variant with Surrounding Sarcomere Proteins
3. Discussion
3.1. Sarcomere-Based Ultrastructural and Functional Changes Are Observed in the A868T Variant
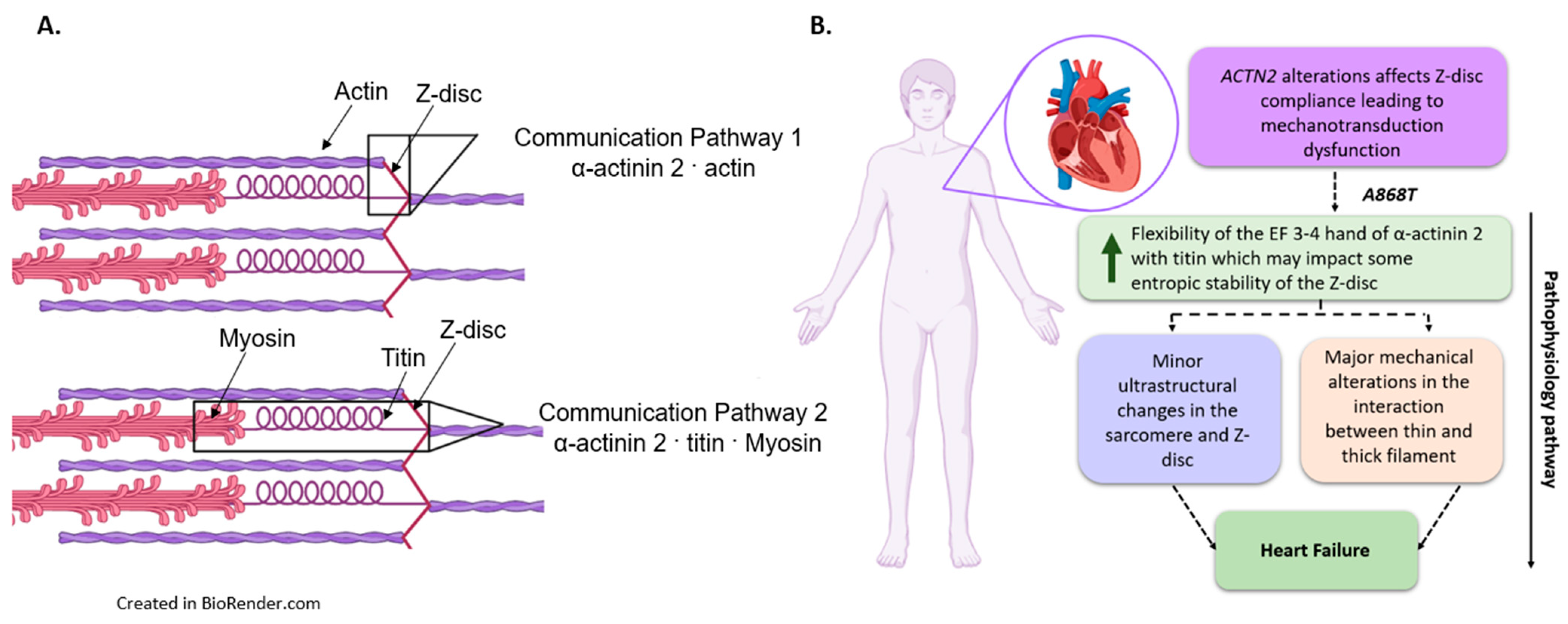
3.2. Interactions of Proteins Associated with the Z-Disc in the A868T Variant
3.3. Can Changes in Titin Flexibility Alter the Kinetics of Cardiac Muscle?
3.4. Implications of the A868T Variant as a Contributor to the Development of Cardiac Disease
4. Materials and Methods
4.1. Human Heart Muscle Samples
4.2. Electron Microscopy
4.3. Immunofluorescence
4.4. Cardiac Muscle Preparations (CMPs)
4.5. Muscle Mechanics
4.6. X-ray Diffraction
4.7. Small-Angle X-Diffraction
4.8. MD Simulations and Analysis
4.9. Statistical Analysis
5. Conclusions
Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Sjoblom, B.; Salmazo, A.; Djinovic-Carugo, K. Alpha-actinin structure and regulation. Cell. Mol. Life Sci. 2008, 65, 2688–2701. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.A.; Taylor, D.W.; Schachat, F. Isoforms of alpha-actinin from cardiac, smooth, and skeletal muscle form polar arrays of actin filaments. J. Cell Biol. 2000, 149, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.; Ohanian, V.; Critchley, D. The structure and function of alpha-actinin. J. Muscle Res. Cell Motil. 1989, 10, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Hampton, C.M.; Taylor, D.W.; Taylor, K.A. Novel structures for alpha-actinin: F-actin interactions and their implications for actin-membrane attachment and tension sensing in the cytoskeleton. J. Mol. Biol. 2007, 368, 92–104. [Google Scholar] [CrossRef]
- Masaki, T.; Endo, M.; Ebashi, S. Localization of 6S component of a alpha-actinin at Z-band. J. Biochem. 1967, 62, 630–632. [Google Scholar] [CrossRef]
- Squire, J.M. Architecture and function in the muscle sarcomere. Curr. Opin. Struct. Biol. 1997, 7, 247–257. [Google Scholar] [CrossRef]
- Mills, M.; Yang, N.; Weinberger, R.; Vander Woude, D.L.; Beggs, A.H.; Easteal, S.; North, K. Differential expression of the actin-binding proteins, alpha-actinin-2 and -3, in different species: Implications for the evolution of functional redundancy. Hum. Mol. Genet. 2001, 10, 1335–1346. [Google Scholar] [CrossRef]
- Gregorich, Z.R.; Patel, J.R.; Cai, W.; Lin, Z.; Heurer, R.; Fitzsimons, D.P.; Moss, R.L.; Ge, Y. Deletion of Enigma Homologue from the Z-disc slows tension development kinetics in mouse myocardium. J. Gen. Physiol. 2019, 151, 670–679. [Google Scholar] [CrossRef]
- Burnette, D.T.; Hayes, J.B. The role of alpha actinin paralogs in cardiac hypertrophy and contractile force production. Biophys. J. 2023, 122, 7a. [Google Scholar] [CrossRef]
- Grum, V.L.; Li, D.; MacDonald, R.I.; Mondragon, A. Structures of two repeats of spectrin suggest models of flexibility. Cell 1999, 98, 523–535. [Google Scholar] [CrossRef]
- Rusu, M.; Hu, Z.; Taylor, K.A.; Trinick, J. Structure of isolated Z-disks from honeybee flight muscle. J. Muscle Res. Cell Motil. 2017, 38, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Grison, M.; Merkel, U.; Kostan, J.; Djinovic-Carugo, K.; Rief, M. Alpha-Actinin/titin interaction: A dynamic and mechanically stable cluster of bonds in the muscle Z-disk. Proc. Natl. Acad. Sci. USA 2017, 114, 1015–1020. [Google Scholar] [CrossRef]
- Gautel, M.; Goulding, D.; Bullard, B.; Weber, K.; Furst, D.O. The central Z-disk region of titin is assembled from a novel repeat in variable copy numbers. J. Cell Sci. 1996, 109 Pt 11, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Endo, T.; Imamura, M.; Takenawa, T. Alpha-Actinin and vinculin are PIP2-binding proteins involved in signaling by tyrosine kinase. J. Biol. Chem. 1994, 269, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Ede, A., Jr.; Pinotsis, N.; Ghisleni, A.; Salmazo, A.; Konarev, P.V.; Kostan, J.; Sjoblom, B.; Schreiner, C.; Polyansky, A.A.; Gkougkoulia, E.A.; et al. The structure and regulation of human muscle alpha-actinin. Cell 2014, 159, 1447–1460. [Google Scholar] [CrossRef]
- Lindholm, M.E.; Jimenez-Morales, D.; Zhu, H.; Seo, K.; Amar, D.; Zhao, C.; Raja, A.; Madhvani, R.; Abramowitz, S.; Espenel, C.; et al. Mono- and Biallelic Protein-Truncating Variants in Alpha-Actinin 2 Cause Cardiomyopathy Through Distinct Mechanisms. Circ. Genom. Precis. Med. 2021, 14, e003419. [Google Scholar] [CrossRef]
- Chiu, C.; Bagnall, R.D.; Ingles, J.; Yeates, L.; Kennerson, M.; Donald, J.A.; Jormakka, M.; Lind, J.M.; Semsarian, C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J. Am. Coll. Cardiol. 2010, 55, 1127–1135. [Google Scholar] [CrossRef]
- Zech, A.T.L.; Prondzynski, M.; Singh, S.R.; Pietsch, N.; Orthey, E.; Alizoti, E.; Busch, J.; Madsen, A.; Behrens, C.S.; Meyer-Jens, M.; et al. ACTN2 Mutant Causes Proteopathy in Human iPSC-Derived Cardiomyocytes. Cells 2022, 11, 2745. [Google Scholar] [CrossRef] [PubMed]
- Broadway-Stringer, S.; Jiang, H.; Wadmore, K.; Hooper, C.; Douglas, G.; Steeples, V.; Azad, A.J.; Singer, E.; Reyat, J.S.; Galatik, F.; et al. Insights into the Role of a Cardiomyopathy-Causing Genetic Variant in ACTN2. Cells. 2023, 12, 721. [Google Scholar] [CrossRef]
- Kosaraju, A.; Goyal, A.; Grigorova, Y.; Makaryus, A.N. Left Ventricular Ejection Fraction. In StatPearls [Internet]; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Wu, V.C.; Takeuchi, M. Echocardiographic assessment of right ventricular systolic function. Cardiovasc. Diagn. Ther. 2018, 8, 70–79. [Google Scholar] [CrossRef]
- Kong, W.K.F.; Vollema, E.M.; Prevedello, F.; Perry, R.; Ng, A.C.T.; Poh, K.K.; Almeida, A.G.; Gonzalez, A.; Shen, M.; Yeo, T.C.; et al. Prognostic implications of left ventricular global longitudinal strain in patients with bicuspid aortic valve disease and preserved left ventricular ejection fraction. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Larson, L.W.; Gerbert, D.A.; Herman, L.M.; Leger, M.M.; McNellis, R.; O’Donoghue, D.L.; Ulshafer, C.; Van Dyke, E.M.; American College of Cardiology; American Heart Association. ACC/AHA 2005 guideline update: Chronic heart failure in the adult. JAAPA 2006, 19, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Reant, P.; Mirabel, M.; Lloyd, G.; Peyrou, J.; Lopez Ayala, J.M.; Dickie, S.; Bulluck, H.; Captur, G.; Rosmini, S.; Guttmann, O.; et al. Global longitudinal strain is associated with heart failure outcomes in hypertrophic cardiomyopathy. Heart 2016, 102, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Mignot, A.; Donal, E.; Zaroui, A.; Reant, P.; Salem, A.; Hamon, C.; Monzy, S.; Roudaut, R.; Habib, G.; Lafitte, S. Global longitudinal strain as a major predictor of cardiac events in patients with depressed left ventricular function: A multicenter study. J. Am. Soc. Echocardiogr. 2010, 23, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horvath, A.; Di Mauro, V.; Koivumaki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Kramer, E.; et al. Disease modeling of a mutation in alpha-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Saetersdal, T.S.; Myklebust, R.; Skagseth, E.; Engedal, H. Ultrastructural studies on the growth of filaments and sarcomeres in mechanically overloaded human hearts. Virchows Arch. B Cell Pathol. 1976, 21, 91–112. [Google Scholar] [CrossRef]
- Russell, B.; Curtis, M.W.; Koshman, Y.E.; Samarel, A.M. Mechanical stress-induced sarcomere assembly for cardiac muscle growth in length and width. J. Mol. Cell Cardiol. 2010, 48, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.A.; Thibodeau, F.; Stranahan, A. Electron microscopic studies of human myocardium. JAMA 1962, 182, 537–540. [Google Scholar] [CrossRef]
- Solaro, R.J.; Leinwand, L.A. Role of Sarcomeres in Cellular Tension, Shortening, and Signaling in Cardiac Muscle. In Muscle Fundamental Biology and Mechanisms of Disease; Academic Press: Cambridge, MA, USA, 2012; pp. 161–172. [Google Scholar]
- Coscarella, I.L.; Landim-Vieira, M.; Rastegarpouyani, H.; Chase, P.B.; Irianto, J.; Pinto, J.R. Nucleus Mechanosensing in Cardiomyocytes. Int. J. Mol. Sci. 2023, 24, 13341. [Google Scholar] [CrossRef] [PubMed]
- Pioner, J.M.; Racca, A.W.; Klaiman, J.M.; Yang, K.C.; Guan, X.; Pabon, L.; Muskheli, V.; Zaunbrecher, R.; Macadangdang, J.; Jeong, M.Y.; et al. Isolation and Mechanical Measurements of Myofibrils from Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2016, 6, 885–896. [Google Scholar] [CrossRef]
- Solaro, R.J. Why we need to understand the mechanics of developing cardiac sarcomeres in humans. J. Physiol. 2016, 594, 255. [Google Scholar] [CrossRef] [PubMed]
- Knoll, R.; Buyandelger, B.; Lab, M. The sarcomeric Z-disc and Z-discopathies. J. Biomed. Biotechnol. 2011, 2011, 569628. [Google Scholar] [CrossRef]
- Crocini, C.; Gotthardt, M. Cardiac sarcomere mechanics in health and disease. Biophys. Rev. 2021, 13, 637–652. [Google Scholar] [CrossRef]
- Chechenova, M.B.; Bryantsev, A.L.; Cripps, R.M. The Drosophila Z-disc protein Z(210) is an adult muscle isoform of Zasp52, which is required for normal myofibril organization in indirect flight muscles. J. Biol. Chem. 2013, 288, 3718–3726. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.X.; Tanada, Y.; Bello, G.D.; Fleming, J.C.; Alkassis, F.F.; Ladd, T.; Golde, T.; Koh, J.; Chen, S.; Kasahara, H. Cardiac MLC2 kinase is localized to the Z-disc and interacts with alpha-actinin2. Sci. Rep. 2019, 9, 12580. [Google Scholar] [CrossRef]
- Dabiri, G.A.; Turnacioglu, K.K.; Sanger, J.M.; Sanger, J.W. Myofibrillogenesis visualized in living embryonic cardiomyocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 9493–9498. [Google Scholar] [CrossRef] [PubMed]
- Sanger, J.W.; Chowrashi, P.; Shaner, N.C.; Spalthoff, S.; Wang, J.; Freeman, N.L.; Sanger, J.M. Myofibrillogenesis in skeletal muscle cells. Clin. Orthop. Relat. Res. 2002, 403, S153–S162. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, C.C.; Antin, P.B. To the heart of myofibril assembly. Trends Cell Biol. 2000, 10, 355–362. [Google Scholar] [CrossRef]
- van der Ven, P.F.; Schaart, G.; Croes, H.J.; Jap, P.H.; Ginsel, L.A.; Ramaekers, F.C. Titin aggregates associated with intermediate filaments align along stress fiber-like structures during human skeletal muscle cell differentiation. J. Cell Sci. 1993, 106 Pt 3, 749–759. [Google Scholar]
- Tokuyasu, K.T.; Maher, P.A. Immunocytochemical studies of cardiac myofibrillogenesis in early chick embryos. II. Generation of alpha-actinin dots within titin spots at the time of the first myofibril formation. J. Cell Biol. 1987, 105 Pt 1, 2795–2801. [Google Scholar] [CrossRef]
- Landim-Vieira, M.; Johnston, J.R.; Ji, W.; Mis, E.K.; Tijerino, J.; Spencer-Manzon, M.; Jeffries, L.; Hall, E.K.; Panisello-Manterola, D.; Khokha, M.K.; et al. Familial Dilated Cardiomyopathy Associated With a Novel Combination of Compound Heterozygous TNNC1 Variants. Front. Physiol. 2019, 10, 1612. [Google Scholar] [CrossRef]
- Gonzalez-Martinez, D.; Johnston, J.R.; Landim-Vieira, M.; Ma, W.; Antipova, O.; Awan, O.; Irving, T.C.; Bryant Chase, P.; Pinto, J.R. Structural and functional impact of troponin C-mediated Ca(2+) sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell Cardiol. 2018, 123, 26–37. [Google Scholar] [CrossRef]
- Ma, W.; Gong, H.; Jani, V.; Lee, K.H.; Landim-Vieira, M.; Papadaki, M.; Pinto, J.R.; Aslam, M.I.; Cammarato, A.; Irving, T. Myofibril orientation as a metric for characterizing heart disease. Biophys. J. 2022, 121, 565–574. [Google Scholar] [CrossRef]
- Landim-Vieira, M.; Ma, W.; Song, T.; Rastegarpouyani, H.; Gong, H.; Coscarella, I.L.; Bogaards, S.J.P.; Conijn, S.P.; Ottenheijm, C.A.C.; Hwang, H.S.; et al. Cardiac troponin T N-domain variant destabilizes the actin interface resulting in disturbed myofilament function. Proc. Natl. Acad. Sci. USA 2023, 120, e2221244120. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; McNamara, J.W.; Ma, W.; Landim-Vieira, M.; Lee, K.H.; Martin, L.A.; Heiny, J.A.; Lorenz, J.N.; Craig, R.; Pinto, J.R.; et al. Fast skeletal myosin-binding protein-C regulates fast skeletal muscle contraction. Proc. Natl. Acad. Sci. USA 2021, 118, 17. [Google Scholar] [CrossRef]
- van de Locht, M.; Donkervoort, S.; de Winter, J.M.; Conijn, S.; Begthel, L.; Kusters, B.; Mohassel, P.; Hu, Y.; Medne, L.; Quinn, C.; et al. Pathogenic variants in TNNC2 cause congenital myopathy due to an impaired force response to calcium. J. Clin. Investig. 2021, 131, 9. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gong, H.; Irving, T. Myosin Head Configurations in Resting and Contracting Murine Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 2643. [Google Scholar] [CrossRef] [PubMed]
- Reconditi, M. Recent Improvements in Small Angle X-ray Diffraction for the Study of Muscle Physiology. Rep. Prog. Phys. 2006, 69, 2709–2759. [Google Scholar] [CrossRef]
- Squire, J. The Structural Basis of Muscular Contraction; Plenum Press: New York, NY, USA, 1981; Volume xvii. [Google Scholar]
- Ma, W.; Henze, M.; Anderson, R.L.; Gong, H.; Wong, F.L.; Del Rio, C.L.; Irving, T. The Super-Relaxed State and Length Dependent Activation in Porcine Myocardium. Circ. Res. 2021, 129, 617–630. [Google Scholar] [CrossRef]
- Elkins, J.M.; Gileadi, C.; Shrestha, L.; Phillips, C.; Wang, J.; Muniz, J.R.; Doyle, D.A. Unusual binding interactions in PDZ domain crystal structures help explain binding mechanisms. Protein Sci. 2010, 19, 731–741. [Google Scholar] [CrossRef]
- Moulder, G.L.; Cremona, G.H.; Duerr, J.; Stirman, J.N.; Fields, S.D.; Martin, W.; Qadota, H.; Benian, G.M.; Lu, H.; Barstead, R.J. Alpha-actinin is required for the proper assembly of Z-disk/focal-adhesion-like structures and for efficient locomotion in Caenorhabditis elegans. J. Mol. Biol. 2010, 403, 516–528. [Google Scholar] [CrossRef][Green Version]
- Fyrberg, C.; Ketchum, A.; Ball, E.; Fyrberg, E. Characterization of lethal Drosophila melanogaster alpha-actinin mutants. Biochem. Genet. 1998, 36, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, X. Alpha-Actinin2 is required for the lateral alignment of Z discs and ventricular chamber enlargement during zebrafish cardiogenesis. FASEB J. 2012, 26, 4230–4242. [Google Scholar] [CrossRef]
- Tanner, B.C.; Farman, G.P.; Irving, T.C.; Maughan, D.W.; Palmer, B.M.; Miller, M.S. Thick-to-thin filament surface distance modulates cross-bridge kinetics in Drosophila flight muscle. Biophys. J. 2012, 103, 1275–1284. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martyn, D.; Chase, P.; Regnier, M.; Gordon, A. A simple model with myofilament compliance predicts activation-dependent crossbridge kinetics in skinned skeletal fibers. Biophys. J. 2002, 83, 3425–3434. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Salcedo, M.K.; Irving, T.C.; Regnier, M.; Daniel, T.L. The length-tension curve in muscle depends on lattice spacing. Proc. Biol. Sci. 2013, 280, 20130697. [Google Scholar] [CrossRef]
- Foley, K.S.; Young, P.W. The non-muscle functions of actinins: An update. Biochem. J. 2014, 459, 1–13. [Google Scholar] [CrossRef]
- Sun, B.; Kekenes-Huskey, P.M. Myofilament-associated proteins with intrinsic disorder (MAPIDs) and their resolution by computational modeling. Q. Rev. Biophys. 2023, 56, e2. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, C.C.; Trombitás, K.; Centner, T.; Kolmerer, B.; Stier, G.; Kunke, K.; Suzuki, K.; Obermayr, F.; Herrmann, B.; Granzier, H. The NH2 terminus of titin spans the Z-disc: Its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J. Cell Biol. 1998, 143, 1013–1027. [Google Scholar] [CrossRef]
- Swist, S.; Unger, A.; Li, Y.; Vöge, A.; von Frieling-Salewsky, M.; Skärlén, Å.; Cacciani, N.; Braun, T.; Larsson, L.; Linke, W.A. Maintenance of sarcomeric integrity in adult muscle cells crucially depends on Z-disc anchored titin. Nat. Commun. 2020, 11, 4479. [Google Scholar] [CrossRef]
- Hessel, A.L.; Ma, W.; Mazara, N.; Rice, P.E.; Nissen, D.; Gong, H.; Kuehn, M.; Irving, T.; Linke, W.A. Titin force in muscle cells alters lattice order, thick and thin filament protein formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2209441119. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.M.; Pepper, I.; Belknap, B.; Landim-Vieira, M.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. The structure of the native cardiac thin filament at systolic Ca(2+) levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2024288118. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.M.; Belknap, B.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. High-resolution cryo-EM structure of the junction region of the native cardiac thin filament in relaxed state. PNAS Nexus 2023, 2, pgac298. [Google Scholar] [CrossRef]
- Veltri, T.; de Oliveira, G.A.P.; Bienkiewicz, E.A.; Palhano, F.L.; Marques, M.A.; Moraes, A.H.; Silva, J.L.; Sorenson, M.M.; Pinto, J.R. Amide hydrogens reveal a temperature-dependent structural transition that enhances site-II Ca(2+)-binding affinity in a C-domain mutant of cardiac troponin C. Sci. Rep. 2017, 7, 691. [Google Scholar] [CrossRef]
- Hancock, W.O.; Huntsman, L.L.; Gordon, A.M. Models of calcium activation account for differences between skeletal and cardiac force redevelopment kinetics. J. Muscle Res. Cell Motil. 1997, 18, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.R.; Yang, S.W.; Hitz, M.P.; Parvatiyar, M.S.; Jones, M.A.; Liang, J.; Kokta, V.; Talajic, M.; Tremblay, N.; Jaeggi, M.; et al. Fetal cardiac troponin isoforms rescue the increased Ca2+ sensitivity produced by a novel double deletion in cardiac troponin T linked to restrictive cardiomyopathy: A clinical, genetic, and functional approach. J. Biol. Chem. 2011, 286, 20901–20912. [Google Scholar] [CrossRef]
- Johnston, J.R.; Landim-Vieira, M.; Marques, M.A.; de Oliveira, G.A.P.; Gonzalez-Martinez, D.; Moraes, A.H.; He, H.; Iqbal, A.; Wilnai, Y.; Birk, E.; et al. The intrinsically disordered C terminus of troponin T binds to troponin C to modulate myocardial force generation. J. Biol. Chem. 2019, 294, 20054–20069. [Google Scholar] [CrossRef]
- Fischetti, R.; Stepanov, S.; Rosenbaum, G.; Barrea, R.; Black, E.; Gore, D.; Heurich, R.; Kondrashkina, E.; Kropf, A.J.; Wang, S.; et al. The BioCAT undulator beamline 18ID: A facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J. Synchrotron. Radiat. 2004, 11 Pt 5, 399–405. [Google Scholar] [CrossRef]
- Jiratrakanvong, J.; Shao, J.; Menendez, M.; Li, X.; Li, J.; Ma, W.; Agam, G.; Irving, T. MuscleX: Software Suite for Diffraction X-ray Imaging, V1.13.1; Zenodo: Geneva, Switzerland, 2018. [Google Scholar] [CrossRef]
- Atkinson, R.A.; Joseph, C.; Kelly, G.; Muskett, F.W.; Frenkiel, T.A.; Nietlispach, D.; Pastore, A. Ca2+-independent binding of an EF-hand domain to a novel motif in the alpha-actinin-titin complex. Nat. Struct. Biol. 2001, 8, 853–857. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sun, B.; Fang, X.; Johnson, C.N.; Hauck, G.; Kou, Y.; Davis, J.P.; Kekenes-Huskey, P.M. Non-Canonical Interaction between Calmodulin and Calcineurin Contributes to the Differential Regulation of Plant-Derived Calmodulins on Calcineurin. J. Chem. Inf. Model 2021, 61, 5223–5233. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, J.C.W.L.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jean-Paul Ryckaert, G.C.; Berendsen, H.J. C Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (Ctrl) | α-actinin 2 Mutant (A868T) | |||
|---|---|---|---|---|
| Ctrl 2.1 µm | Ctrl 2.3 µm | A868T 2.1 µm | A868T 2.3 µm | |
| Fmax (mN/mm2) | 24.16 ± 1.13 | 29.38 ± 0.80 * | 16.02 ± 1.13 # | 17.08 ± 2.06 # |
| Fpass (mN/mm2) | 1.57 ± 0.31 | 2.94 ± 0.47 ** | 0.78 ± 0.32 # | 0.91 ± 0.11 ### |
| FpCa50 | 5.72 ± 0.01 | 5.81 ± 0.02 * | 5.88 ± 0.03 ## | 6.06 ± 0.05 **,### |
| FnHill | 1.43 ± 0.06 | 1.54 ± 0.13 | 1.56 ± 0.06 | 1.22 ± 0.08 |
| kTRmax(s−1) | 10.47 ± 0.55 | 10.37 ± 1.25 | 20.54 ± 1.22 # | 18.10 ± 0.51 # |
| SSpass (MPa) | 0.36 ± 0.08 | 0.91 ± 0.12 * | 0.10 ± 0.01 # | 0.35 ± 0.01 *,# |
| SSmax (MPa) | 0.71 ± 0.06 | 1.09 ± 0.14 * | 0.54 ± 0.04 | 0.79 ± 0.06 # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez Garcia, M.; Schmeckpeper, J.; Landim-Vieira, M.; Coscarella, I.L.; Fang, X.; Ma, W.; Spran, P.A.; Yuan, S.; Qi, L.; Kahmini, A.R.; et al. Disruption of Z-Disc Function Promotes Mechanical Dysfunction in Human Myocardium: Evidence for a Dual Myofilament Modulatory Role by Alpha-Actinin 2. Int. J. Mol. Sci. 2023, 24, 14572. https://doi.org/10.3390/ijms241914572
Rodriguez Garcia M, Schmeckpeper J, Landim-Vieira M, Coscarella IL, Fang X, Ma W, Spran PA, Yuan S, Qi L, Kahmini AR, et al. Disruption of Z-Disc Function Promotes Mechanical Dysfunction in Human Myocardium: Evidence for a Dual Myofilament Modulatory Role by Alpha-Actinin 2. International Journal of Molecular Sciences. 2023; 24(19):14572. https://doi.org/10.3390/ijms241914572
Chicago/Turabian StyleRodriguez Garcia, Michelle, Jeffrey Schmeckpeper, Maicon Landim-Vieira, Isabella Leite Coscarella, Xuan Fang, Weikang Ma, Payton A. Spran, Shengyao Yuan, Lin Qi, Aida Rahimi Kahmini, and et al. 2023. "Disruption of Z-Disc Function Promotes Mechanical Dysfunction in Human Myocardium: Evidence for a Dual Myofilament Modulatory Role by Alpha-Actinin 2" International Journal of Molecular Sciences 24, no. 19: 14572. https://doi.org/10.3390/ijms241914572
APA StyleRodriguez Garcia, M., Schmeckpeper, J., Landim-Vieira, M., Coscarella, I. L., Fang, X., Ma, W., Spran, P. A., Yuan, S., Qi, L., Kahmini, A. R., Shoemaker, M. B., Atkinson, J. B., Kekenes-Huskey, P. M., Irving, T. C., Chase, P. B., Knollmann, B. C., & Pinto, J. R. (2023). Disruption of Z-Disc Function Promotes Mechanical Dysfunction in Human Myocardium: Evidence for a Dual Myofilament Modulatory Role by Alpha-Actinin 2. International Journal of Molecular Sciences, 24(19), 14572. https://doi.org/10.3390/ijms241914572