
Dynamic Alterations to Hepatic MicroRNA-29a in Response to Long-Term High-Fat Diet and EtOH Feeding

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
New and Noteworthy
- Modeling human high-fat diet (HFD) and ethanol consumption from adolescence to old age.
- The short- to long-term feeding design (4 weeks, 13 weeks, 26 weeks, and 39 weeks), multiple feeding groups (CMD, HFD, HFD + 2%, 10%, or 20% v/v E), and data collection at multiple time points are the strengths of this study.
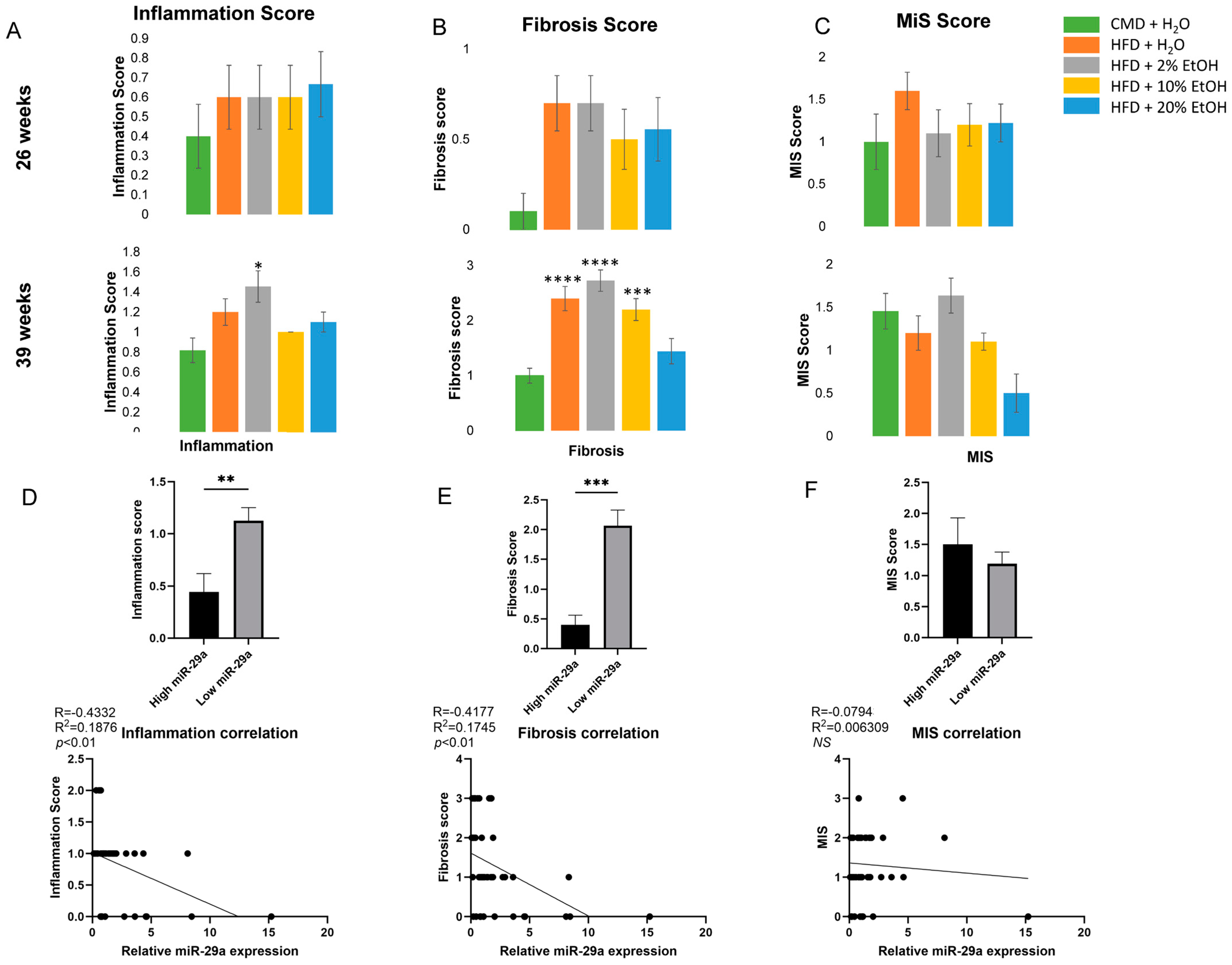
- Short-term feeding in 8-week-old mice demonstrated no significant changes, but longer feeding lengths resulted in significant physiological changes, including liver fibrosis.
- HFD feeding with chronic consumption of a high concentration of EtOH (20% E) was able to counteract weight gain, but increased mortality; a HFD with chronic consumption of a low concentration of EtOH (2% E) increased liver fibrosis.
- A HFD plus EtOH feeding drove significant initial up-regulation of miR-29a after 13 weeks but resulted in the down-regulation of miR-29a in association with liver injury after 26 weeks.
2. Results
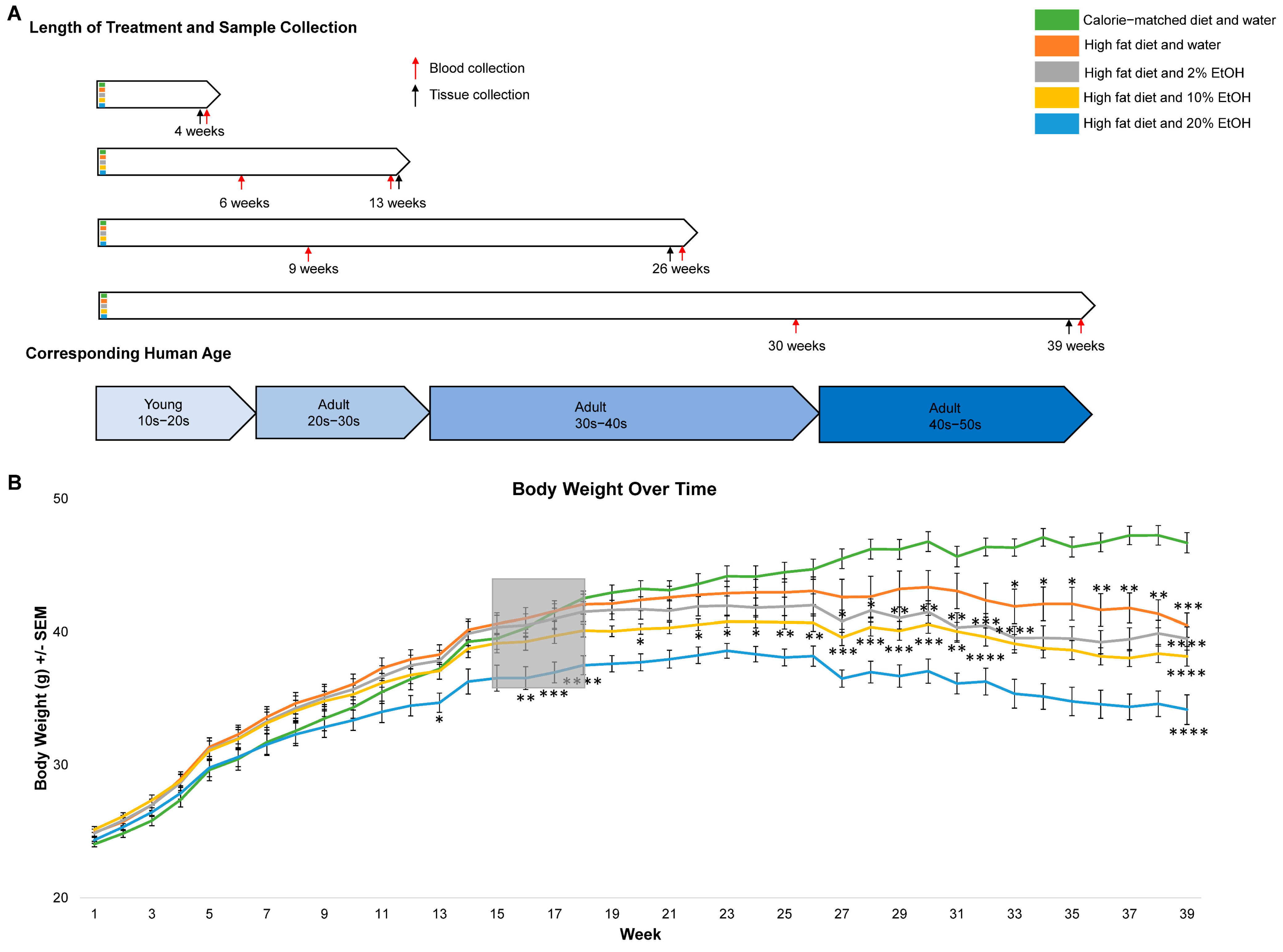
2.1. Long-Term Feeding of 20% EtOH Counters HFD-Induced Body Weight Gain
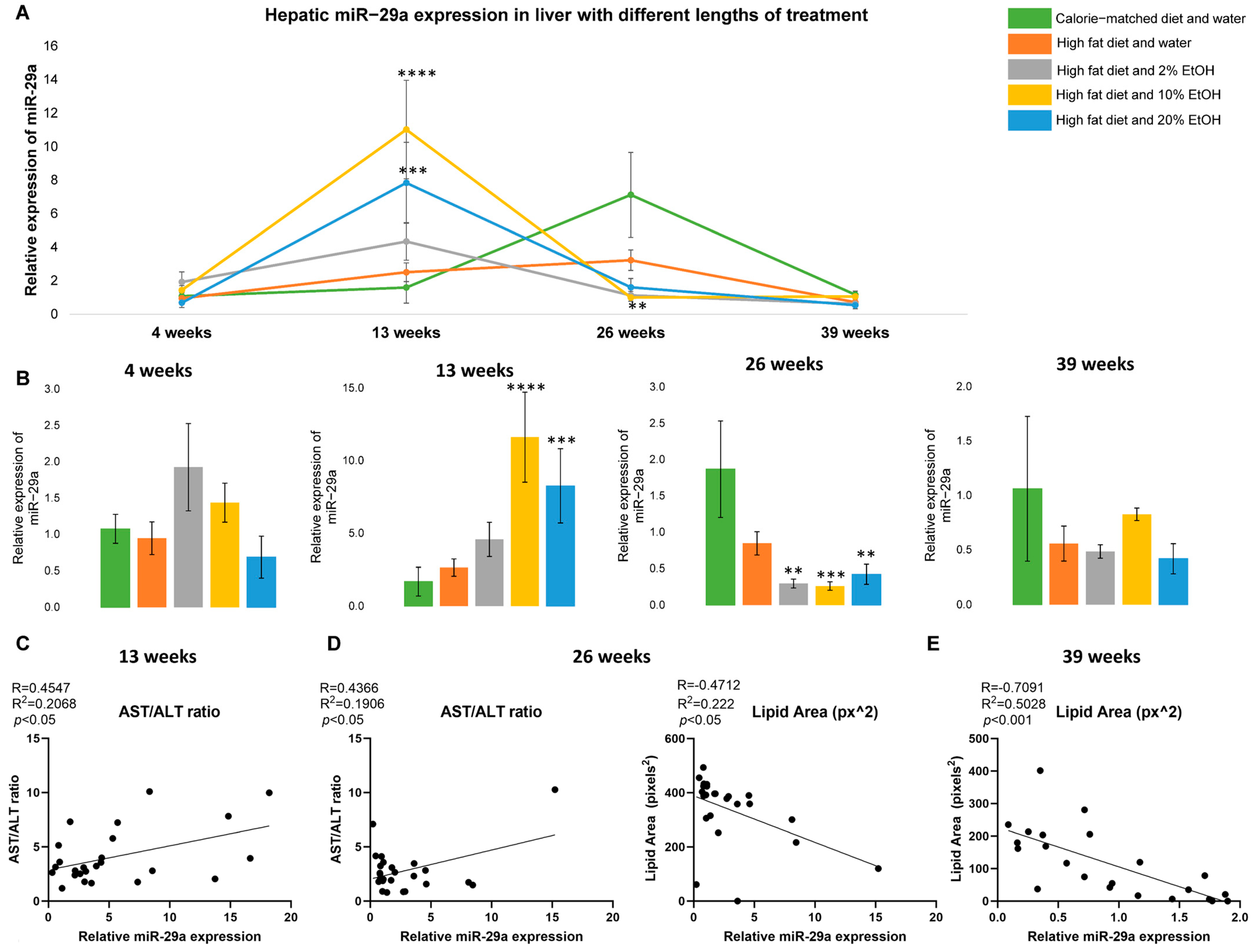
2.2. Dynamic Alteration of miR-29a Expression in Response to Feeding
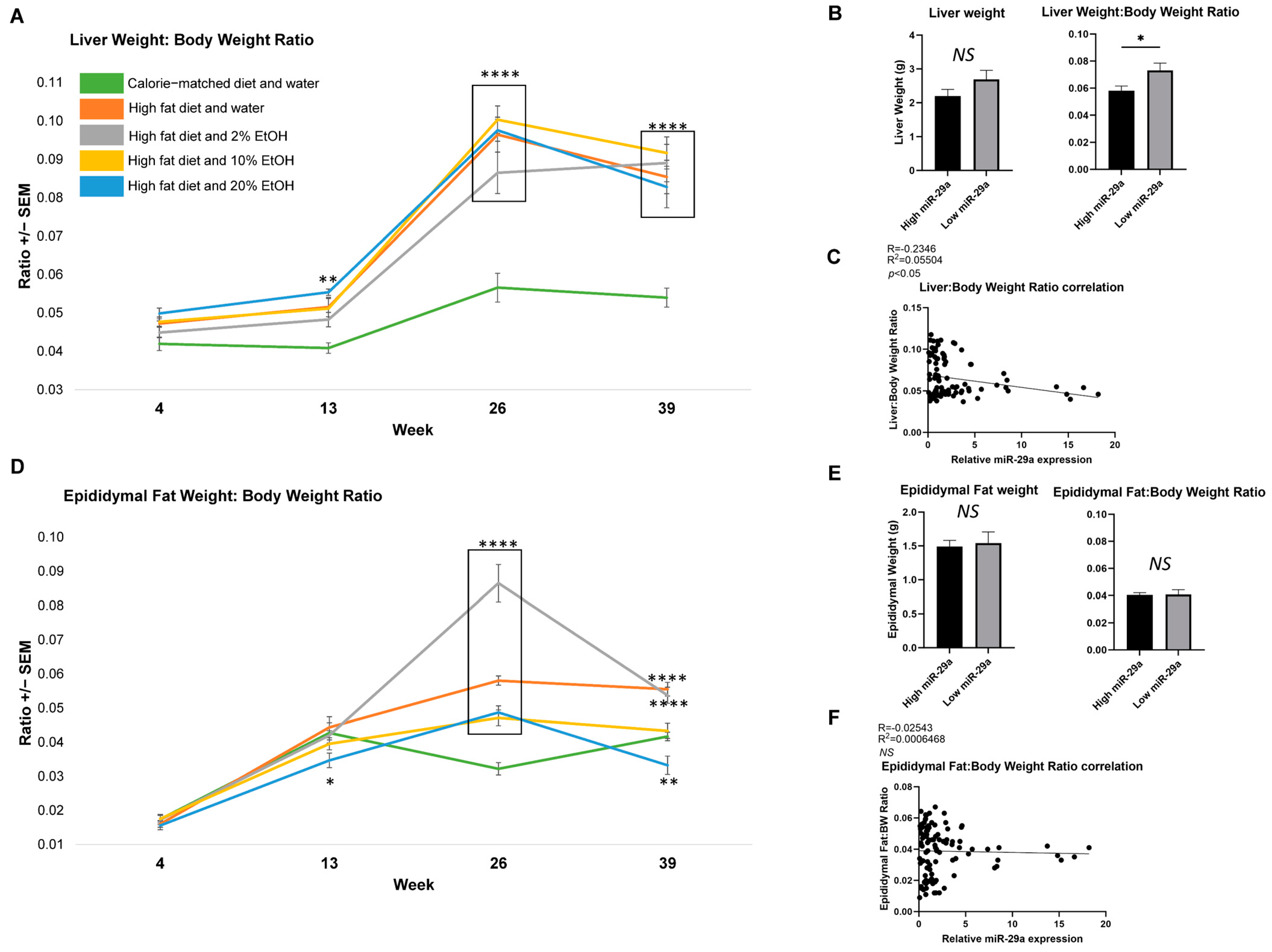
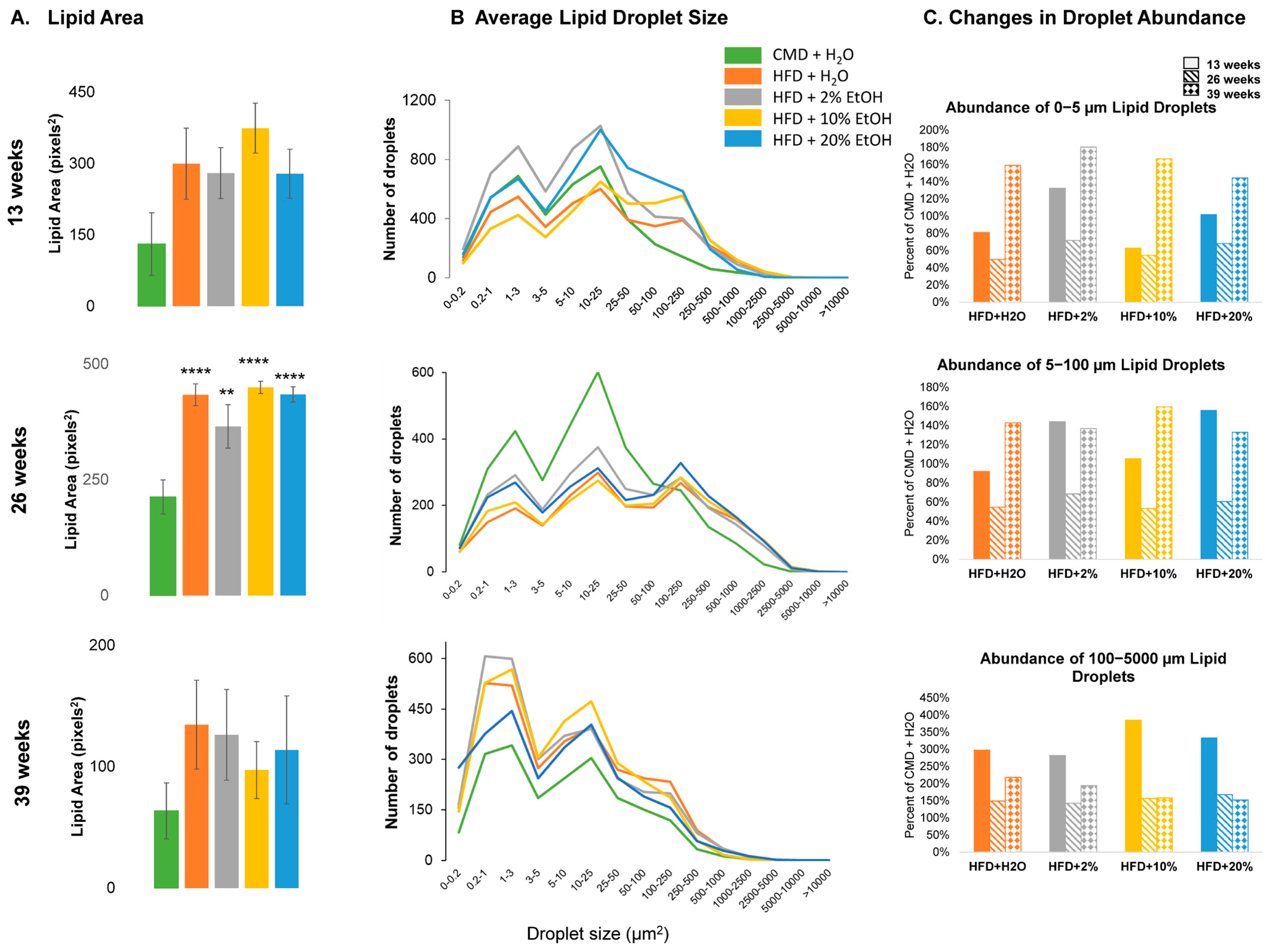
2.3. Feeding Length Results on Liver and Epididymal Fat Mass Alterations
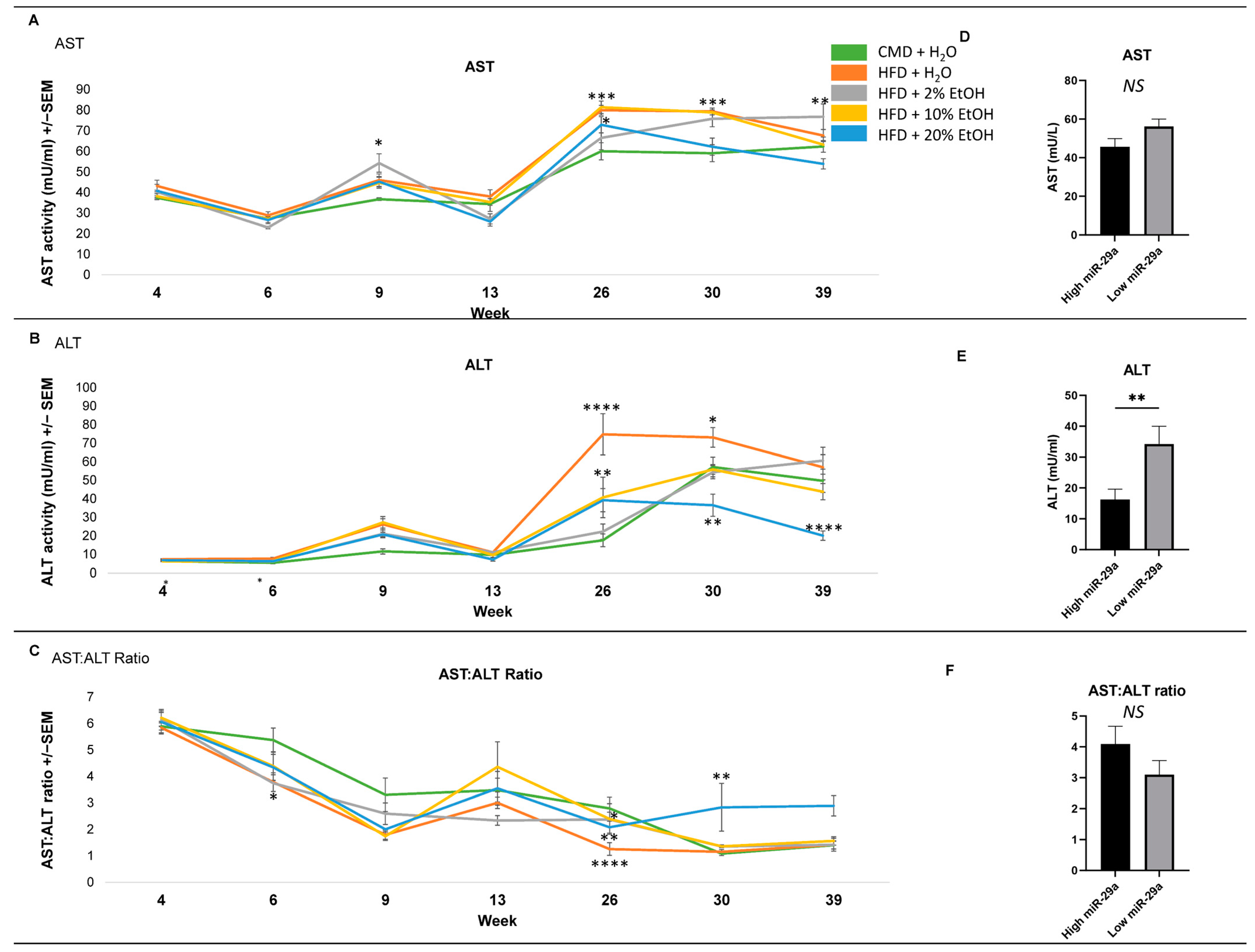
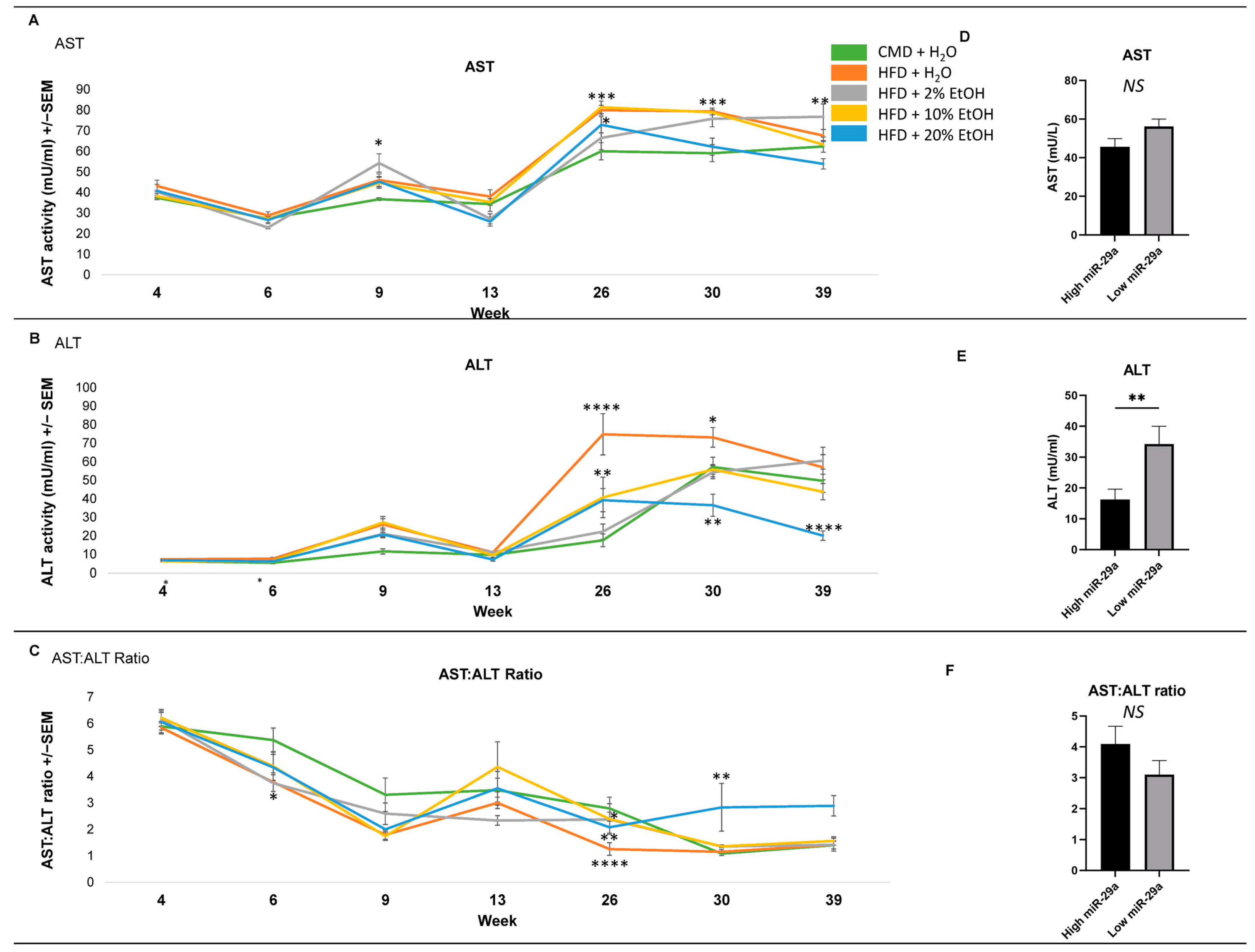
2.4. Serum Biochemical Parameters
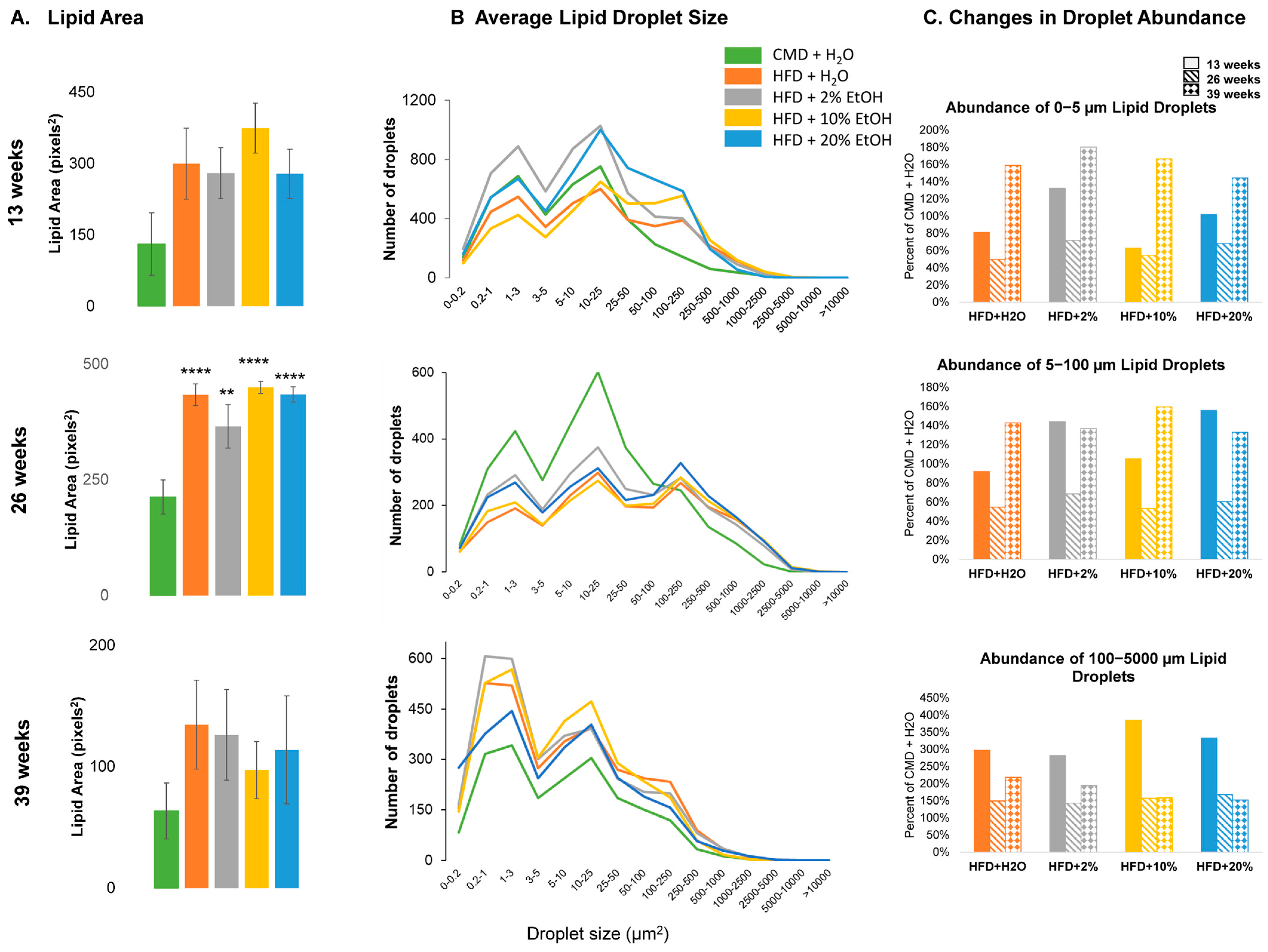
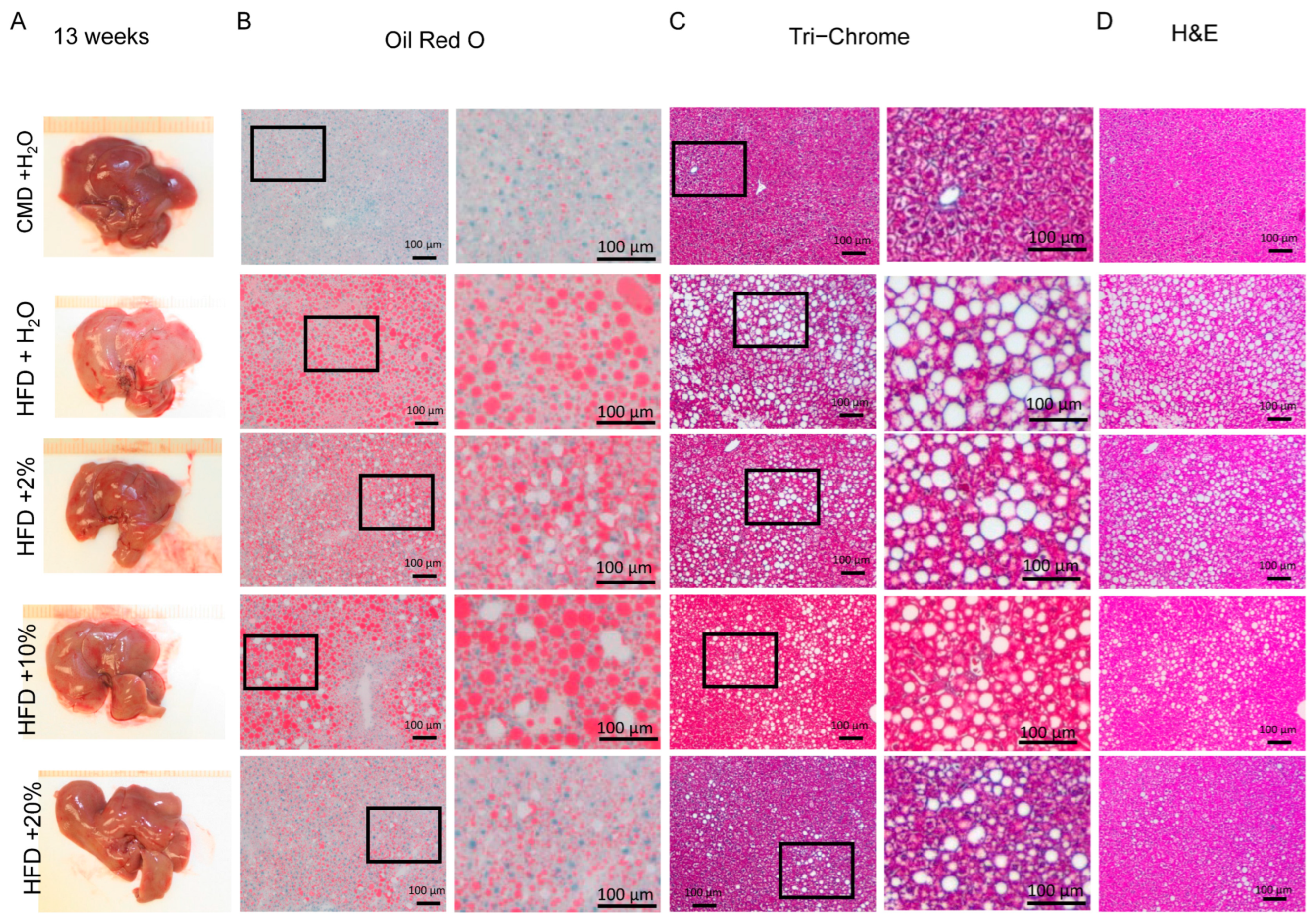
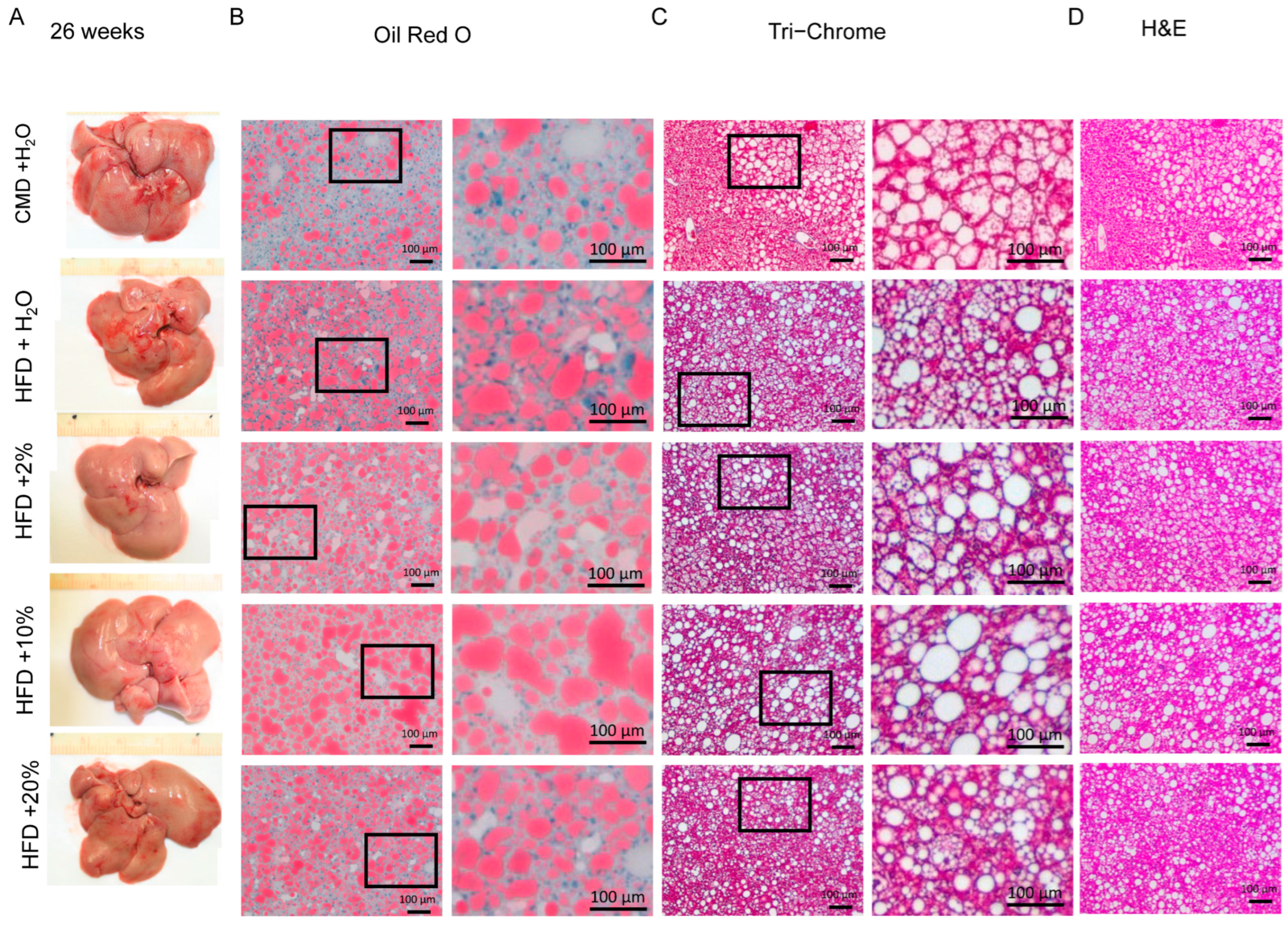
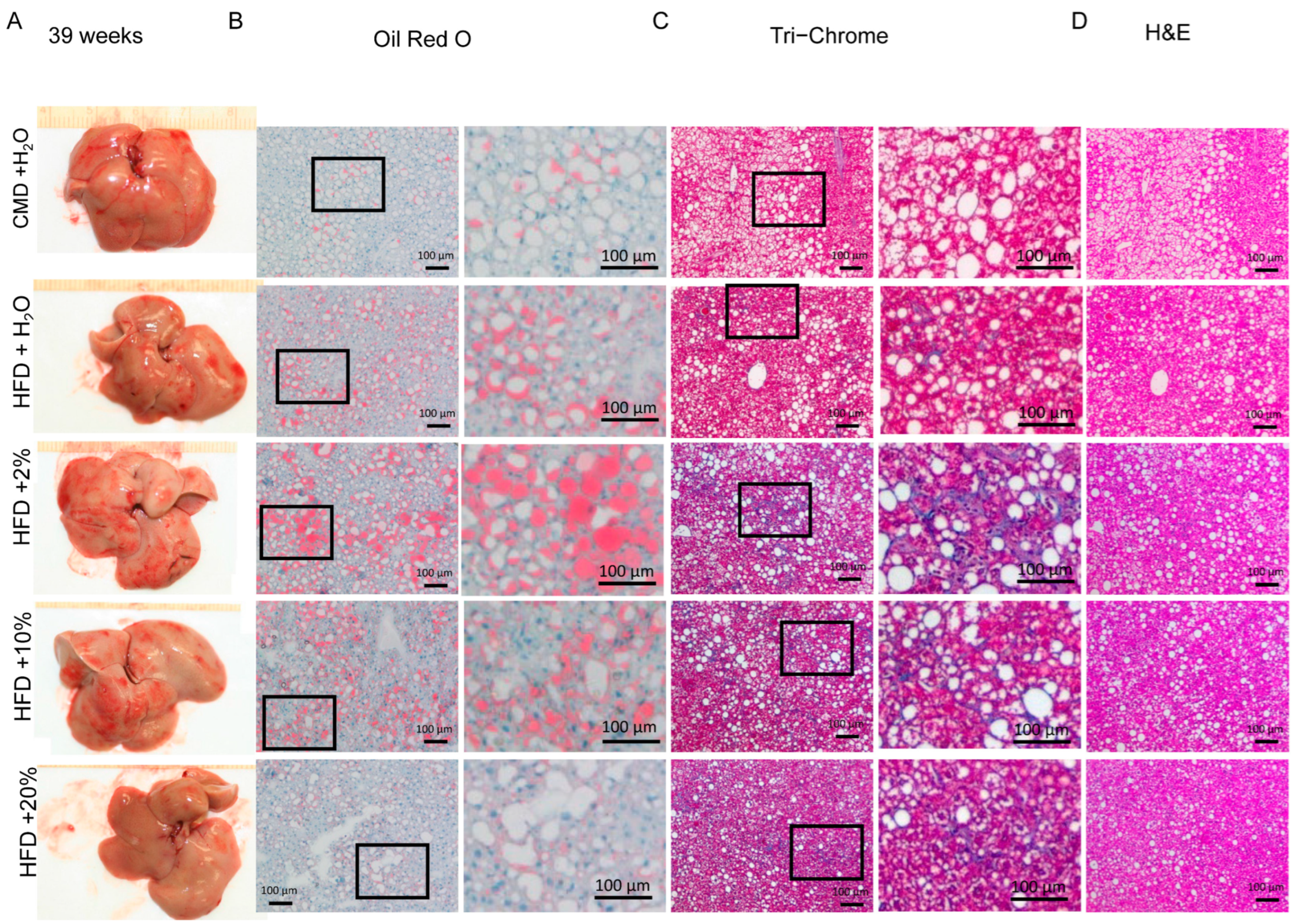
2.5. Histology Analysis
2.6. Overall Features
3. Discussion
4. Material and Methods
4.1. Animal and Experimental Design
4.2. Diet and Overall Health
4.3. Sample Collection and Histological Analysis
4.4. Serum Analysis
4.5. RNA Isolation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Mokdad, A.H.; Marks, J.S.; Stroup, D.F.; Gerberding, J.L. Actual causes of death in the United States, 2000. JAMA 2004, 291, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Pruett, S.B.; Szabo, G.; Arteel, G.E. Binge ethanol and liver: New molecular developments. Alcohol Clin. Exp. Res. 2013, 37, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, H.; Craig, D.; Barker, R.; Spiers, G.; Stow, D.; Anstee, Q.M.; Hanratty, B. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of population-based observational studies. PLoS Med. 2020, 17, e1003100. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2020, 158, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 2020, 111S, 154170. [Google Scholar] [CrossRef]
- Moreira, R.E., Jr.; de Carvalho, L.M.; Pedersen, A.S.B.; Damasceno, S.; Maioli, T.U.; de Faria, A.M.C.; Godard, A.L.B. Interaction between high-fat diet and ethanol intake leads to changes on the fecal microbiome. J. Nutr. Biochem. 2019, 72, 108215. [Google Scholar] [CrossRef]
- Chang, R.C.; Thomas, K.N.; Bedi, Y.S.; Golding, M.C. Programmed increases in LXRalpha induced by paternal alcohol use enhance offspring metabolic adaptation to high-fat diet induced obesity. Mol. Metab. 2019, 30, 161–172. [Google Scholar] [CrossRef]
- Daniels, S.J.; Leeming, D.J.; Detlefsen, S.; Bruun, M.F.; Hjuler, S.T.; Henriksen, K.; Hein, P.; Krag, A.; Karsdal, M.A.; Nielsen, M.J.; et al. Addition of trans fat and alcohol has divergent effects on atherogenic diet-induced liver injury in rodent models of steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G410–G418. [Google Scholar] [CrossRef]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Crocè, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar] [CrossRef]
- Seitz, H.K. Alcohol and breast cancer. Breast 2012, 21, 426–427. [Google Scholar] [CrossRef]
- Homann, N.; Konig, I.R.; Marks, M.; Benesova, M.; Stickel, F.; Millonig, G.; Mueller, S.; Seitz, H.K. Alcohol and colorectal cancer: The role of alcohol dehydrogenase 1C polymorphism. Alcohol. Clin. Exp. Res. 2009, 33, 551–556. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL clinical practical guidelines: Management of alcoholic liver disease. J. Hepatol. 2012, 57, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.L.; Rodd, Z.A.; Lumeng, L.; Murphy, J.M.; McBride, W.J. The alcohol-preferring P rat and animal models of excessive alcohol drinking. Addict. Biol. 2006, 11, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.N.; Lee, L.; Saxena, R.; Bemis, K.G.; Wang, M.; Theodorakis, J.L.; Vuppalanchi, R.; Alloosh, M.; Sturek, M.; Chalasani, N.; et al. Serum proteomic analysis of diet-induced steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G746–G754. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Darshi, M.; Ma, Y.; Perkins, G.A.; Shen, Z.; Haushalter, K.J.; Saito, R.; Chen, A.; Lee, Y.S.; Patel, H.H.; et al. Quantitative proteomic and functional analysis of liver mitochondria from high fat diet (HFD) diabetic mice. Mol. Cell Proteom. 2013, 12, 3744–3758. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Alloosh, M.; Bell, L.N.; Fullenkamp, A.; Saxena, R.; Van Alstine, W.; Bybee, P.; Werling, K.; Sturek, M.; Chalasani, N.; et al. Liver injury and fibrosis induced by dietary challenge in the Ossabaw miniature Swine. PLoS ONE 2015, 10, e0124173. [Google Scholar] [CrossRef]
- Reilly, M.E.; Mantle, D.; Salisbury, J.; Peters, T.J.; Preedy, V.R. Comparative effects of acute ethanol dosage on liver and muscle protein metabolism. Biochem. Pharmacol. 2000, 60, 1773–1785. [Google Scholar] [CrossRef]
- Lang, C.H.; Frost, R.A.; Kumar, V.; Wu, D.; Vary, T.C. Impaired protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4E in muscle and eIF2B in liver. Alcohol. Clin. Exp. Res. 2000, 24, 322–331. [Google Scholar] [CrossRef]
- Adachi, J.; Kudo, R.; Asano, M.; Ueno, Y.; Hunter, R.; Rajendram, R.; Martin, C.; Preedy, V.R. Skeletal muscle and liver oxysterols during fasting and alcohol exposure. Metabolism 2006, 55, 119–127. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Zhang, L.; Zhu, M.; Gao, L. A high-fat diet impairs mitochondrial biogenesis, mitochondrial dynamics, and the respiratory chain complex in rat myocardial tissues. J. Cell Biochem. 2018, 119, 9602. [Google Scholar] [CrossRef]
- Echeverria, F.; Valenzuela, R.; Bustamante, A.; Alvarez, D.; Ortiz, M.; Espinosa, A.; Illesca, P.; Gonzalez Manan, D.; Videla, L.A. High-fat diet induces mouse liver steatosis with a concomitant decline in energy metabolism: Attenuation by eicosapentaenoic acid (EPA) or hydroxytyrosol (HT) supplementation and the additive effects upon EPA and HT co-administration. Food Funct. 2019, 10, 6170–6183. [Google Scholar] [CrossRef]
- Howard, A.A.; Arnsten, J.H.; Gourevitch, M.N. Effect of alcohol consumption on diabetes mellitus: A systematic review. Ann. Intern. Med. 2004, 140, 211–219. [Google Scholar] [CrossRef]
- Haseeb, S.; Alexander, B.; Baranchuk, A. Wine and Cardiovascular Health: A Comprehensive Review. Circulation 2017, 136, 1434–1448. [Google Scholar] [CrossRef]
- Suarez, E.C.; Beckham, J.C.; Green, K.T. The Relation of Light-to-Moderate Alcohol Consumption to Glucose Metabolism and Insulin Resistance in Nondiabetic Adults: The Moderating Effects of Depressive Symptom Severity, Adiposity, and Sex. Int. J. Behav. Med. 2017, 24, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Camargo, C.A.; Manson, J.E., Jr.; Willett, W.C.; Rimm, E.B. Alcohol drinking patterns and risk of type 2 diabetes mellitus among younger women. Arch. Intern. Med. 2003, 163, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Kim, S.J.; Gao, B. Alcohol, adipose tissue and liver disease: Mechanistic links and clinical considerations. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 50–59. [Google Scholar]
- Hwang, S.; Ren, T.; Gao, B. Obesity and binge alcohol intake are deadly combination to induce steatohepatitis: A model of high-fat diet and binge ethanol intake. Clin. Mol. Hepatol. 2020, 26, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Kim, S.; Im, G.; Nahas, J.; Dhesi, B.; Vergis, N.; Sinha, A.; Ghezzi, A.; Rink, M.; McCune, A.; et al. Obesity in acute alcoholic hepatitis increases morbidity and mortality. EBioMedicine 2019, 45, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Jun, D.W.; Moon, J.H. Effects of Moderate Alcohol Drinking in Patients with Nonalcoholic Fatty Liver Disease. Gut Liver. 2019, 13, 308–314. [Google Scholar] [CrossRef]
- Bucher, S.; Begriche, K.; Catheline, D.; Trak-Smayra, V.; Tiaho, F.; Coulouarn, C.; Pinon, G.; Lagadic-Gossmann, D.; Rioux, V.; Fromenty, B. Moderate chronic ethanol consumption exerts beneficial effects on nonalcoholic fatty liver in mice fed a high-fat diet: Possible role of higher formation of triglycerides enriched in monounsaturated fatty acids. Eur. J. Nutr. 2020, 59, 1619–1632. [Google Scholar] [CrossRef]
- Sun, F.; Zhuang, Z.; Zhang, D.; Chen, Y.; Liu, S.; Gao, N.; Shi, J.; Wang, B. Chronic moderate alcohol consumption relieves high-fat high-cholesterol diet-induced liver fibrosis in a rat model. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Seitz, H.K.; Wang, X.D. Moderate alcohol consumption aggravates high-fat diet induced steatohepatitis in rats. Alcohol. Clin. Exp. Res. 2010, 34, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Rosa, D.F.; Sarandy, M.M.; Novaes, R.D.; Freitas, M.B.; do Carmo Gouveia Peluzio, M.; Goncalves, R.V. High-Fat Diet and Alcohol Intake Promotes Inflammation and Impairs Skin Wound Healing in Wistar Rats. Mediators Inflamm. 2018, 2018, 4658583. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chen, T.; Prough, R.A.; Cave, M.C.; McClain, C.J. Chronic Alcohol Consumption Causes Liver Injury in High-Fructose-Fed Male Mice Through Enhanced Hepatic Inflammatory Response. Alcohol Clin. Exp. Res. 2016, 40, 518–528. [Google Scholar] [CrossRef]
- Chang, B.; Xu, M.J.; Zhou, Z.; Cai, Y.; Li, M.; Wang, W.; Feng, D.; Bertola, A.; Wang, H.; Kunos, G.; et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology 2015, 62, 1070–1085. [Google Scholar] [CrossRef]
- Tadokoro, T.; Morishita, A.; Masaki, T. Diagnosis and Therapeutic Management of Liver Fibrosis by MicroRNA. Int. J. Mol. Sci. 2021, 22, 8139. [Google Scholar] [CrossRef]
- Hochreuter, M.Y.; Dall, M.; Treebak, J.T.; Barres, R. MicroRNAs in non-alcoholic fatty liver disease: Progress and perspectives. Mol. Metab. 2022, 65, 101581. [Google Scholar] [CrossRef]
- Wang, X.; He, Y.; Mackowiak, B.; Gao, B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut 2021, 70, 784–795. [Google Scholar] [CrossRef]
- Song, S.; Sun, K.; Dong, J.; Zhao, Y.; Liu, F.; Liu, H.; Sha, Z.; Mao, J.; Ding, G.; Guo, W.; et al. MicroRNA-29a regulates liver tumor-initiating cells expansion via Bcl-2 pathway. Exp. Cell Res. 2020, 387, 111781. [Google Scholar] [CrossRef]
- Jing, F.; Geng, Y.; Xu, X.Y.; Xu, H.Y.; Shi, J.S.; Xu, Z.H. MicroRNA29a Reverts the Activated Hepatic Stellate Cells in the Regression of Hepatic Fibrosis through Regulation of ATPase H+ Transporting V1 Subunit C1. Int. J. Mol. Sci. 2019, 20, 796. [Google Scholar] [CrossRef]
- Jampoka, K.; Muangpaisarn, P.; Khongnomnan, K.; Treeprasertsuk, S.; Tangkijvanich, P.; Payungporn, S. Serum miR-29a and miR-122 as Potential Biomarkers for Non-Alcoholic Fatty Liver Disease (NAFLD). Microrna 2018, 7, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA. 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, C.; Lin, X.; Li, J. MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie 2013, 95, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.J.; Factora, T.D.; Dey, S.; Kota, J. A Systematic Review of miR-29 in Cancer. Mol. Ther. Oncolytics 2019, 12, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, Z.; Tu, J.; Zhu, W.; Ge, J.; Zheng, X.; Yang, L.; Pa, X.; Yanm, H.; Zhu, J. MicroRNA-29a regulates pro-inflammatory cytokine secretion and scavenger receptor expression by targeting LPL in oxLDL-stimulated dendritic cells. FEBS Lett. 2011, 585, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, M.; Elfimova, N.; Noetel, A.; Töx, U.; Steffen, H.-M.; Hacker, U.; Nischt, R.; Dienes, H.P.; Odenthal, M. Expression of platelet-derived growth factor-C and insulin-like growth factor I in hepatic stellate cells is inhibited by miR-29. Lab. Investig. 2012, 92, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.Y.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef]
- Zhao, Y.; Srivastava, D. A developmental view of microRNA function. Trends Biochem. Sci. 2007, 32, 189–197. [Google Scholar] [CrossRef]
- Almiron-Roig, E.; Palla, L.; Guest, K.; Ricchiuti, C.; Vint, N.A.; Jebb, S.; Drewnowski, A. Factors that determine energy compensation: A systematic review of preload studies. Nutr. Rev. 2013, 71, 458–473. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Doyle, C.J.; McKillop, I.H.; Wentz, S.C.; Brandon-Warner, E.; Matos, J.M.; Sandrasegaran, K.; Saxena, R.; Hennig, M.E.; Wu, H.; et al. Alcohol induces liver neoplasia in a novel alcohol-preferring rat model. Alcohol Clin. Exp. Res. 2011, 35, 2216–2225. [Google Scholar] [CrossRef] [PubMed]
- De Vloo, C.; Nevens, F. Cholestatic pruritus: An update. Acta Gastroenterol. Belg. 2019, 82, 75–82. [Google Scholar] [PubMed]
- Kastenmayer, R.J.; Fain, M.A.; Perdue, K.A. A retrospective study of idiopathic ulcerative dermatitis in mice with a C57BL/6 background. J. Am. Assoc. Lab. Anim. Sci. 2006, 45, 8–12. [Google Scholar] [PubMed]
- Oberlin, B.; Best, C.; Matson, L.; Henderson, A.; Grahame, N. Derivation and characterization of replicate high- and low-alcohol preferring lines of mice and a high-drinking crossed HAP line. Behav. Genet. 2011, 41, 288–302. [Google Scholar] [CrossRef]
- Thompson, K.J.; Nazari, S.S.; Jacobs, W.C.; Grahame, N.J.; McKillop, I.H. Use of a crossed high alcohol preferring (cHAP) mouse model with the NIAAA-model of chronic-binge ethanol intake to study liver injury. Alcohol Alcohol. 2017, 52, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Addolorato, G.; Di Campli, C.; Simoncini, M.; Pasini, P.; Nardo, B.; Cavallari, A.; Pola, P.; Roda, A.; Gasbarrini, G.; Gasbarrini, A. Oxygen free radical production in rat liver: Dose-related effect of ethanol on reperfusion injury. Dig. Dis. Sci. 2001, 46, 1057–1066. [Google Scholar] [CrossRef]
- Addolorato, G.; Capristo, E.; Greco, A.V.; Stefanini, G.F.; Gasbarrini, G. Influence of chronic alcohol abuse on body weight and energy metabolism: Is excess ethanol consumption a risk factor for obesity or malnutrition? J. Intern. Med. 1998, 244, 387–395. [Google Scholar] [CrossRef]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Stärkel, P.; van Pijkeren, J.-P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 2015, 148, 203–214.e16. [Google Scholar] [CrossRef]
- Chen, Y.L.; Peng, H.C.; Wang, X.D.; Yang, S.C. Dietary saturated fatty acids reduce hepatic lipid accumulation but induce fibrotic change in alcohol-fed rats. Hepatobiliary Surg. Nutr. 2015, 4, 172–183. [Google Scholar]
- Kirpich, I.A.; Solovieva, N.V.; Leikhter, S.N.; Shidakova, N.A.; Lebedeva, O.V.; Sidorov, P.I.; Bazhukova, T.A.; Soloviev, A.G.; Barve, S.S.; McClain, C.J.; et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: A pilot study. Alcohol 2008, 42, 675–682. [Google Scholar] [CrossRef]
- Hulbert, A.J. Explaining longevity of different animals: Is membrane fatty acid composition the missing link? Age 2008, 30, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Andersen, O.S. Alcohol’s effects on lipid bilayer properties. Biophys. J. 2011, 101, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Dopico, A.M.; Lovinger, D.M. Acute alcohol action and desensitization of ligand-gated ion channels. Pharmacol. Rev. 2009, 61, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, L.; Genheden, S.; Faria-Oliveira, F.; Allard, S.; Eriksson, L.A.; Olsson, L.; Bettiga, M. Alcohols enhance the rate of acetic acid diffusion in S. cerevisiae: Biophysical mechanisms and implications for acetic acid tolerance. Microb. Cell. 2017, 5, 42–55. [Google Scholar] [CrossRef]
- Rasineni, K.; McVicker, B.L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Rab GTPases associate with isolated lipid droplets (LDs) and show altered content after ethanol administration: Potential role in alcohol-impaired LD metabolism. Alcohol Clin. Exp. Res. 2014, 38, 327–335. [Google Scholar] [CrossRef]
- Zhao, L.; Ouyang, H.; Zhang, N.; Wang, C.; Ji, B.; Zhou, F. Effects of Huangjiu, Baijiu and Red Wine Combined With High-Fat Diet on Glucose and Lipid Metabolism: Aggravate or Alleviate? Alcohol Alcohol. 2021, 56, 334–347. [Google Scholar] [CrossRef]
- Dionisi, T.; Addolorato, G. Effect of Alcohol Combined With High Fat Diet: Two Wrongs Don’t Make a Right but They Could Make a Good Excuse. Alcohol Alcohol. 2021, 56, 348–350. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yang, Y.L.; Wang, F.S. The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis. Int. J. Mol. Sci. 2018, 19, 1889. [Google Scholar] [CrossRef]
- Xu, X.Y.; Du, Y.; Liu, X.; Ren, Y.; Dong, Y.; Xu, H.Y.; Shi, J.-S.; Jiang, D.; Xu, X.; Li, L.; et al. Targeting Follistatin like 1 ameliorates liver fibrosis induced by carbon tetrachloride through TGF-beta1-miR29a in mice. Cell Commun. Signal. 2020, 18, 151. [Google Scholar] [CrossRef]
- Lin, H.Y.; Wang, F.S.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a Suppresses CD36 to Ameliorate High Fat Diet-Induced Steatohepatitis and Liver Fibrosis in Mice. Cells 2019, 8, 1298. [Google Scholar] [CrossRef]
- Alfaifi, M.; Verma, A.K.; Alshahrani, M.Y.; Joshi, P.C.; Alkhathami, A.G.; Ahmad, I.; Hakami, A.R.; Beg, M.M.A. Assessment of Cell-Free Long Non-Coding RNA-H19 and miRNA-29a, miRNA-29b Expression and Severity of Diabetes. Diabetes Metab. Syndr. Obes. 2020, 13, 3727–3737. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.J.; Schriever, S.C.; Roscher, A.A.; Ensenauer, R. Digital image analysis approach for lipid droplet size quantitation of Oil Red O-stained cultured cells. Anal. Biochem. 2014, 445, 87–89. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, T.; Kota, J.; Williams, K.E.; Saxena, R.; Gawrieh, S.; Zhong, X.; Zimmers, T.A.; Chalasani, N. Dynamic Alterations to Hepatic MicroRNA-29a in Response to Long-Term High-Fat Diet and EtOH Feeding. Int. J. Mol. Sci. 2023, 24, 14564. https://doi.org/10.3390/ijms241914564
Liang T, Kota J, Williams KE, Saxena R, Gawrieh S, Zhong X, Zimmers TA, Chalasani N. Dynamic Alterations to Hepatic MicroRNA-29a in Response to Long-Term High-Fat Diet and EtOH Feeding. International Journal of Molecular Sciences. 2023; 24(19):14564. https://doi.org/10.3390/ijms241914564
Chicago/Turabian StyleLiang, Tiebing, Janaiah Kota, Kent E. Williams, Romil Saxena, Samer Gawrieh, Xiaoling Zhong, Teresa A. Zimmers, and Naga Chalasani. 2023. "Dynamic Alterations to Hepatic MicroRNA-29a in Response to Long-Term High-Fat Diet and EtOH Feeding" International Journal of Molecular Sciences 24, no. 19: 14564. https://doi.org/10.3390/ijms241914564
APA StyleLiang, T., Kota, J., Williams, K. E., Saxena, R., Gawrieh, S., Zhong, X., Zimmers, T. A., & Chalasani, N. (2023). Dynamic Alterations to Hepatic MicroRNA-29a in Response to Long-Term High-Fat Diet and EtOH Feeding. International Journal of Molecular Sciences, 24(19), 14564. https://doi.org/10.3390/ijms241914564