
Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation?

 ,
,
Abstract
1. Introduction
2. Defective Regulation of the Alternative Complement Pathway in aHUS

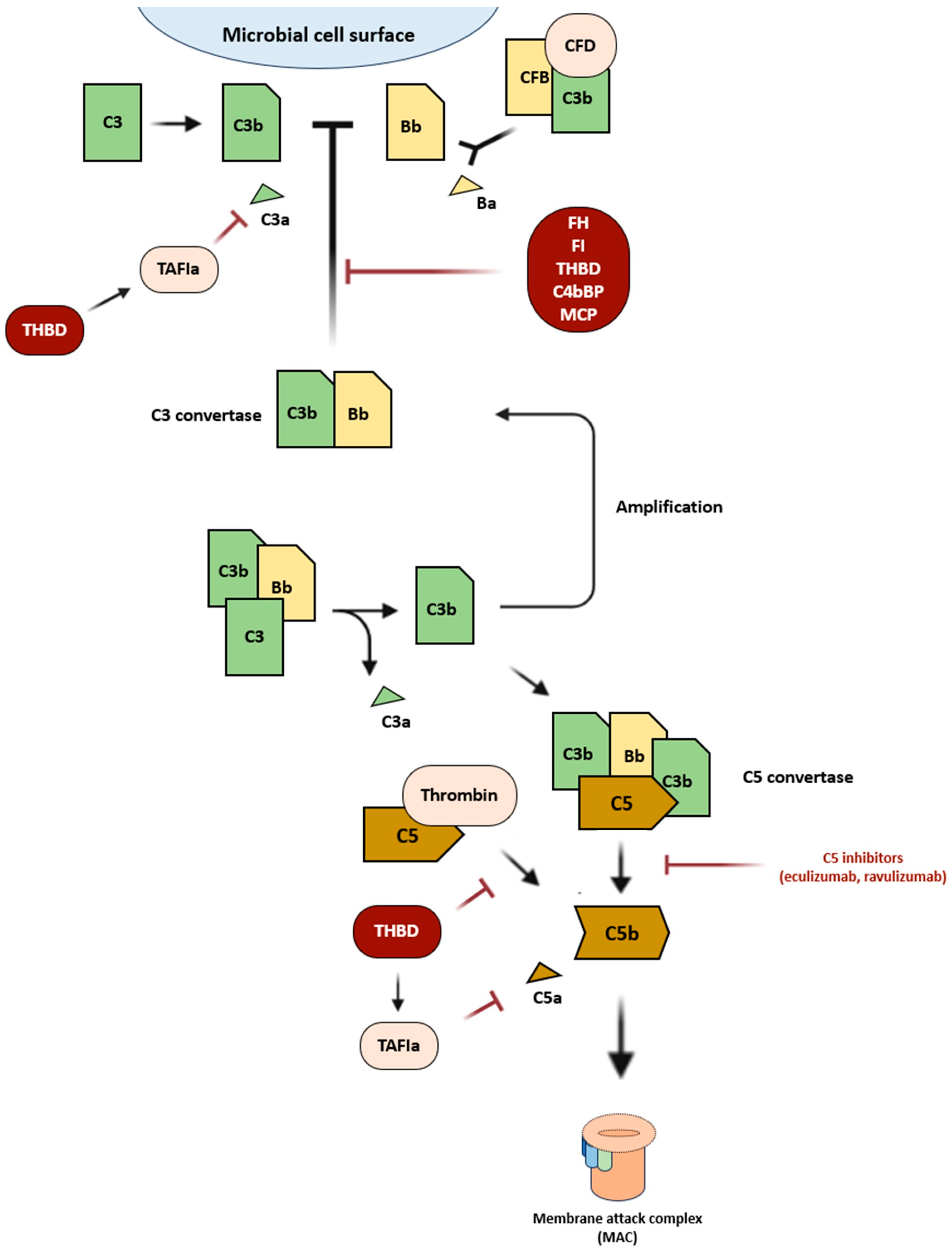{kind=link}
| Gene | Locus | Mechanism of Action | Mutation Frequency |
|---|---|---|---|
| CFH | 1q31.3 | CFH competitively binds to C3b to inhibit the C3 convertase production, accelerating the C3 convertase degradation, and as a cofactor for CFI in the cleavage of C3b. | >30% |
| MCP (CD46) | 1q32.2 | MCP is a cofactor for CFI-mediated C3b and C4b cleavage. | 8–10% |
| CFI | 4q25 | The CFI regulates both the classical and alternative pathways of the complement system by lysing C3b and C4b in a highly specific manner. | 4–10% |
| C3 | 19p13.3 | C3 is required for the formation of both the C3 convertase and C5 convertase. | 2–10% |
| CFB | 6p21.33 | Combines with C3b to create C3 convertase. | 0–3% |
| THBD | 20p11.21 | Encodes thrombomodulin, involved both in the coagulation cascade and the complement system. | 3–4% |
3. Genetic Mutations and Outcome: Is There a Connection?
4. C5 Inhibitors Discontinuation: Is There a Path to Follow?
- -
- ≥1 variant reported as pathogenic/likely pathogenic in CFH, MCP/CD46, C3, CFI, and CFB;
- -
- positive anti-CFH titer despite immunosuppressive treatment;
- -
- ≥1 VUS in CFH, C3, or splice region variant in MCP/CD46;
- longer-term or lifelong therapy should be considered.
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Manrique-Caballero, C.L.; Peerapornratana, S.; Formeck, C.; Del Rio-Pertuz, G.; Gomez Danies, H.; Kellum, J.A. Typical and Atypical Hemolytic Uremic Syndrome in the Critically Ill. Crit. Care Clin. 2020, 36, 333–356. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic uraemic syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Viteri, B.; Saland, J.M. Hemolytic Uremic Syndrome. Pediatr. Rev. 2020, 41, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Loirat, C.; Fakhouri, F.; Ariceta, G.; Besbas, N.; Bitzan, M.; Bjerre, A.; Coppo, R.; Emma, F.; Johnson, S.; Karpman, D.; et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. Berl. Ger. 2016, 31, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Frémeaux-Bacchi, V. Thrombotic microangiopathy in aHUS and beyond: Clinical clues from complement genetics. Nat. Rev. Nephrol. 2021, 17, 543–553. [Google Scholar] [CrossRef]
- Raina, R.; Krishnappa, V.; Blaha, T.; Kann, T.; Hein, W.; Burke, L.; Bagga, A. Atypical Hemolytic-Uremic Syndrome: An Update on Pathophysiology, Diagnosis, and Treatment. Ther. Apher. Dial. 2019, 23, 4–21. [Google Scholar] [CrossRef]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. 2010, 5, 1844–1859. [Google Scholar] [CrossRef]
- Caprioli, J.; Noris, M.; Brioschi, S.; Pianetti, G.; Castelletti, F.; Bettinaglio, P.; Mele, C.; Bresin, E.; Cassis, L.; Gamba, S.; et al. Genetics of HUS: The impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006, 108, 1267–1279. [Google Scholar] [CrossRef]
- Laurence, J. Atypical hemolytic uremic syndrome (aHUS): Making the diagnosis. Clin. Adv. Hematol. Oncol. 2012, 10 (Suppl. S17), 1–12. [Google Scholar]
- Sellier-Leclerc, A.L.; Fremeaux-Bacchi, V.; Dragon-Durey, M.A.; Macher, M.A.; Niaudet, P.; Guest, G.; Boudailliez, B.; Bouissou, F.; Deschenes, G.; Gie, S.; et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2007, 18, 2392–2400. [Google Scholar] [CrossRef]
- Raina, R.; Vijayvargiya, N.; Khooblall, A.; Melachuri, M.; Deshpande, S.; Sharma, D.; Mathur, K.; Arora, M.; Sethi, S.K.; Sandhu, S. Pediatric Atypical Hemolytic Uremic Syndrome Advances. Cells 2021, 10, 3580. [Google Scholar] [CrossRef] [PubMed]
- Pangburn, M.K.; Müller-Eberhard, H.J. Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of C3. Ann. N. Y. Acad. Sci. 1983, 421, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R.; Tschoop, J.; Müller-Eberhard, H.J. Molecular organization of C9 within the membrane attack complex of complement. Induction of circular C9 polymerization by the C5b-8 assembly. J. Exp. Med. 1982, 156, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Maga, T.K.; Nishimura, C.J.; Weaver, A.E.; Frees, K.L.; Smith, R.J.H. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum. Mutat. 2010, 31, E1445–E1460. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Esparza-Gordillo, J.; de Jorge, E.G.; Garrido, C.A.; Carreras, L.; López-Trascasa, M.; Sánchez-Corral, P.; Rodríguez de Córdoba, S. Insights into hemolytic uremic syndrome: Segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol. Immunol. 2006, 43, 1769–1775. [Google Scholar] [CrossRef]
- Tomazos, I.; Garlo, K.; Wang, Y.; Chen, P.; Laurence, J. Triggers in Patients with Atypical Hemolytic Uremic Syndrome: An Observational Cohort Study Using a US Claims Database. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- Bresin, E.; Rurali, E.; Caprioli, J.; Sanchez-Corral, P.; Fremeaux-Bacchi, V.; Rodriguez de Cordoba, S.; Pinto, S.; Goodship, T.H.J.; Alberti, M.; Ribes, D.; et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J. Am. Soc. Nephrol. 2013, 24, 475–486. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Wiech, T.; Stea, E.D.; Skerka, C. CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J. Am. Soc. Nephrol. 2020, 31, 241–256. [Google Scholar] [CrossRef]
- Piras, R.; Breno, M.; Valoti, E.; Alberti, M.; Iatropoulos, P.; Mele, C.; Bresin, E.; Donadelli, R.; Cuccarolo, P.; Smith, R.J.H.; et al. CFH and CFHR Copy Number Variations in C3 Glomerulopathy and Immune Complex-Mediated Membranoproliferative Glomerulonephritis. Front. Genet. 2021, 12, 670727. [Google Scholar] [CrossRef]
- Pérez-Caballero, D.; González-Rubio, C.; Gallardo, M.E.; Vera, M.; López-Trascasa, M.; Rodríguez de Córdoba, S.; Sánchez-Corral, P. Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am. J. Hum. Genet. 2001, 68, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Hellwage, J.; Friese, M.A.; Hegasy, G.; Jokiranta, S.T.; Meri, S. Factor H and disease: A complement regulator affects vital body functions. Mol. Immunol. 1999, 36, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez de Córdoba, S.; Esparza-Gordillo, J.; Goicoechea de Jorge, E.; Lopez-Trascasa, M.; Sánchez-Corral, P. The human complement factor H: Functional roles, genetic variations and disease associations. Mol. Immunol. 2004, 41, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Vaziri-Sani, F.; Heinen, S.; Kristoffersson, A.C.; Gydell, K.H.; Raafat, R.; Gutierrez, A.; Beringer, O.; Zipfel, P.F.; Karpman, D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 2008, 111, 5307–5315. [Google Scholar] [CrossRef]
- Ferreira, V.P.; Herbert, A.P.; Cortés, C.; McKee, K.A.; Blaum, B.S.; Esswein, S.T.; Uhrín, D.; Barlow, P.N.; Pangburn, M.K.; Kavanagh, D. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J. Immunol. 2009, 182, 7009–7018. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.Y.; Song, D.; Liu, X.R.; Chen, Z.; Xiao, H.J.; Ding, J.; Sun, S.Z.; Liu, H.Y.; Wang, S.X.; Yu, F.; et al. Immunological features and functional analysis of anti-CFH autoantibodies in patients with atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2019, 34, 269–281. [Google Scholar] [CrossRef]
- Manuelian, T.; Hellwage, J.; Meri, S.; Caprioli, J.; Noris, M.; Heinen, S.; Jozsi, M.; Neumann, H.P.H.; Remuzzi, G.; Zipfel, P.F. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J. Clin. Investig. 2003, 111, 1181–1190. [Google Scholar] [CrossRef]
- Sánchez-Corral, P.; González-Rubio, C.; Rodríguez de Córdoba, S.; López-Trascasa, M. Functional analysis in serum from atypical Hemolytic Uremic Syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol. Immunol. 2004, 41, 81–84. [Google Scholar] [CrossRef]
- Sugawara, Y.; Kato, H.; Nagasaki, M.; Yoshida, Y.; Fujisawa, M.; Minegishi, N.; Yamamoto, M.; Nangaku, M. CFH-CFHR1 hybrid genes in two cases of atypical hemolytic uremic syndrome. J. Hum. Genet. 2023, 68, 427–430. [Google Scholar] [CrossRef]
- Eyler, S.J.; Meyer, N.C.; Zhang, Y.; Xiao, X.; Nester, C.M.; Smith, R.J.H. A novel hybrid CFHR1/CFH gene causes atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2013, 28, 2221–2225. [Google Scholar] [CrossRef][Green Version]
- Moore, I.; Strain, L.; Pappworth, I.; Kavanagh, D.; Barlow, P.N.; Herbert, A.P.; Schmidt, C.Q.; Staniforth, S.J.; Holmes, L.V.; Ward, R.; et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 2010, 115, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Tsiftsoglou, S.A.; Willis, A.C.; Li, P.; Chen, X.; Mitchell, D.A.; Rao, Z.; Sim, R.B. The catalytically active serine protease domain of human complement factor I. Biochemistry 2005, 44, 6239–6249. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, D.; Kemp, E.J.; Mayland, E.; Winney, R.J.; Duffield, J.S.; Warwick, G.; Richards, A.; Ward, R.; Goodship, J.A.; Goodship, T.H.J. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2005, 16, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, M.K.; Leung, M.; Cui, W.; Subramanian, V.B.; Parkinson, J.; Barlow, P.N.; Manchester, M.; Atkinson, J.P. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46). J. Biol. Chem. 2000, 275, 37692–37701. [Google Scholar] [CrossRef] [PubMed]
- Fremeaux-Bacchi, V.; Sanlaville, D.; Menouer, S.; Blouin, J.; Dragon-Durey, M.A.; Fischbach, M.; Vekemans, M.; Fridman, W.H. Unusual clinical severity of complement membrane cofactor protein-associated hemolytic-uremic syndrome and uniparental isodisomy. Am. J. Kidney Dis. 2007, 49, 323–329. [Google Scholar] [CrossRef]
- Funato, M.; Uemura, O.; Ushijima, K.; Ohnishi, H.; Orii, K.; Kato, Z.; Yamakawa, S.; Nagai, T.; Ohara, O.; Kaneko, H.; et al. A complement factor B mutation in a large kindred with atypical hemolytic uremic syndrome. J. Clin. Immunol. 2014, 34, 691–695. [Google Scholar] [CrossRef]
- Delvaeye, M.; Noris, M.; De Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar] [CrossRef]
- Schaefer, F.; Ardissino, G.; Ariceta, G.; Fakhouri, F.; Scully, M.; Isbel, N.; Lommelé, Å.; Kupelian, V.; Gasteyger, C.; Greenbaum, L.A.; et al. Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int. 2018, 94, 408–418. [Google Scholar] [CrossRef]
- Piras, R.; Valoti, E.; Alberti, M.; Bresin, E.; Mele, C.; Breno, M.; Liguori, L.; Donadelli, R.; Rigoldi, M.; Benigni, A.; et al. CFH and CFHR structural variants in atypical Hemolytic Uremic Syndrome: Prevalence, genomic characterization and impact on outcome. Front. Immunol. 2022, 13, 1011580. [Google Scholar] [CrossRef]
- Merinero, H.M.; García, S.P.; García-Fernández, J.; Arjona, E.; Tortajada, A.; Rodríguez de Córdoba, S. Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int. 2018, 93, 470–481. [Google Scholar] [CrossRef]
- Martín Merinero, H.; Zhang, Y.; Arjona, E.; Del Angel, G.; Goodfellow, R.; Gomez-Rubio, E.; Ji, R.R.; Michelena, M.; Smith, R.J.H.; Rodríguez de Córdoba, S. Functional characterization of 105 factor H variants associated with aHUS: Lessons for variant classification. Blood 2021, 138, 2185–2201. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, M.; Kato, H.; Yoshida, Y.; Usui, T.; Takata, M.; Fujimoto, M.; Wada, H.; Uchida, Y.; Kokame, K.; Matsumoto, M.; et al. Clinical characteristics and genetic backgrounds of Japanese patients with atypical hemolytic uremic syndrome. Clin. Exp. Nephrol. 2018, 22, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Fila, M.; Provôt, F.; Delmas, Y.; Barbet, C.; Châtelet, V.; Rafat, C.; Cailliez, M.; Hogan, J.; Servais, A.; et al. Pathogenic Variants in Complement Genes and Risk of Atypical Hemolytic Uremic Syndrome Relapse after Eculizumab Discontinuation. Clin. J. Am. Soc. Nephrol. 2017, 12, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Fila, M.; Hummel, A.; Ribes, D.; Sellier-Leclerc, A.L.; Ville, S.; Pouteil-Noble, C.; Coindre, J.P.; Le Quintrec, M.; Rondeau, E.; et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: A prospective multicenter study. Blood 2021, 137, 2438–2449. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, R.A. Eculizumab and aHUS: To stop or not. Blood 2021, 137, 2419–2420. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, R.N.; van de Kar, N.C.A.J.; Wetzels, J.F.M. Enough is enough: Targeted eculizumab withdrawal in atypical hemolytic uremic syndrome. Kidney Int. 2021, 100, 265–268. [Google Scholar] [CrossRef]
- Acosta-Medina, A.A.; Moyer, A.M.; Go, R.S.; Willrich, M.A.V.; Fervenza, F.C.; Leung, N.; Bourlon, C.; Winters, J.L.; Spears, G.M.; Bryant, S.C.; et al. Complement gene variant effect on relapse of complement-mediated thrombotic microangiopathy after eculizumab cessation. Blood Adv. 2023, 7, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, R.N.; Duineveld, C.; Wijnsma, K.L.; Bemelman, F.J.; van der Heijden, J.W.; van Wijk, J.A.E.; Bouts, A.H.M.; van de Wetering, J.; Dorresteijn, E.; Berger, S.P.; et al. Early Eculizumab Withdrawal in Patients With Atypical Hemolytic Uremic Syndrome in Native Kidneys Is Safe and Cost-Effective: Results of the CUREiHUS Study. Kidney Int. Rep. 2022, 8, 91–102. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Every Fifteen Days Forever? Kidney Int. Rep. 2022, 8, 4–7. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spasiano, A.; Palazzetti, D.; Dimartino, L.; Bruno, F.; Baccaro, R.; Pesce, F.; Grandaliano, G. Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation? Int. J. Mol. Sci. 2023, 24, 14496. https://doi.org/10.3390/ijms241914496
Spasiano A, Palazzetti D, Dimartino L, Bruno F, Baccaro R, Pesce F, Grandaliano G. Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation? International Journal of Molecular Sciences. 2023; 24(19):14496. https://doi.org/10.3390/ijms241914496
Chicago/Turabian StyleSpasiano, Andrea, Daniela Palazzetti, Lucrezia Dimartino, Francesca Bruno, Rocco Baccaro, Francesco Pesce, and Giuseppe Grandaliano. 2023. "Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation?" International Journal of Molecular Sciences 24, no. 19: 14496. https://doi.org/10.3390/ijms241914496
APA StyleSpasiano, A., Palazzetti, D., Dimartino, L., Bruno, F., Baccaro, R., Pesce, F., & Grandaliano, G. (2023). Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation? International Journal of Molecular Sciences, 24(19), 14496. https://doi.org/10.3390/ijms241914496